

Abstract
Histone deacetylase (HDAC) inhibitors have shown cytotoxicity as single agents in preclinical studies for multiple myeloma (MM) cells. LBH589 is a novel hydroxamic acid derivative that at low nanomolar concentrations induces apoptosis in MM cells resistant to conventional therapies via caspase activation and poly-(ADP-ribose) polymerase (PARP) cleavage. Significant synergistic cytotoxicity was observed with LBH589 in combination with bortezomib against MM cells that were sensitive and resistant to dexamethasone (Dex), as well as primary patient MM cells. LBH589 at low nanomolar concentrations also induced α-tubulin hyperacetylation. Aggresome formation was observed in the presence of bortezomib, and the combination of LBH589 plus bortezomib induced the formation of abnormal bundles of hyeracetylated α-tubulin but with diminished aggresome size and apoptotic nuclei. These data confirm the potential clinical benefit of combining HDAC inhibitors with proteasome inhibitors, and provide insight into the mechanisms of synergistic anti-MM activity of bortezomib in combination with LBH589.
Introduction
Gene expression, cellular differentiation, and survival are regulated by the opposing activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). HDAC inhibitors are a new treatment strategy in early clinical development against multiple myeloma (MM).1-6 HDAC inhibition results in accumulation of acetylated nuclear histones, as well as nonhistone proteins such as α-tubulin, resulting in caspase-dependent and caspase-independent apoptosis of transformed cells. In preclinical studies, HDAC inhibitors sensitize MM cells to death receptor-mediated apoptosis and inhibit the secretion of interleukin-6 (IL-6) induced in bone marrow stromal cells (BMSCs) by binding of MM cells, thereby overcoming cell-adhesion-mediated drug resistance (CAMDR) in the BM microenvironment. They also inhibit tumor growth in xenograft animal models of human myeloma. HDAC inhibitors with dexamethasone (Dex), bortezomib, and insulin-like growth factor-1 (IGF-1) inhibitors have synergistic cytotoxicity against MM,4,5,7 as have tyrosine kinase inhibitors.6-10 Phase 1 and 2 clinical trials of hydroxamic acid-derived HDAC inhibitors suberoylanilide hydroxamic acid (SAHA) and LBH589 for the treatment of MM are now under way in the United States.
The 26S proteasome is a large ubiquitous protein complex with multiple nonlysosomal proteolytic functions essential in regulating diverse cellular functions, including cell cycle, apoptosis, and resistance to drug-induced cell death; antigen presentation by major histocompatibility complex (MHC) class I molecules; degradation of intracellular proteins regulating cell adhesion, transcription, and angiogenesis; and rapid elimination of abnormally folded or oxidized proteins.11-19 Misfolded proteins are nonfunctional and prone to forming aggregates that can interfere with normal cellular function.20 The mammalian unfolded protein response (UPR) protects the cell against the stress of misfolded proteins.21,22 Importantly, the increased protein turnover in plasma cells, and in particular MM cells, suggests the importance of the UPR in conferring sensitivity of MM cells to proteasome inhibition.
When the production of unfolded and misfolded ubiquitinated proteins exceeds the capacity of cellular proteasomes to degrade them, they accumulate in aggresomes. Aggresomes are microtubule (MT)-based inclusion bodies formed by the retrograde transport of aggregated protein on MTs, and represent a general cellular response to the presence of misfolded proteins.23,24 Aggresomes form initially as small protein aggregates that coalesce at dispersed sites throughout the periphery of the cytoplasm. They then travel on MTs to the MT organizing center (MTOC) region, where they remain as distinct but closely apposed stable particulate structures entangled with collapsed intermediate filaments.23,25 Tubulin acetylation plays an important role in the differentiation of MT structure and function. Acetylation of α-tubulin is a posttranslational modification consisting of the addition of an acetyl group to lysine 40, which is reversed by tubulin deacetylase (TDAC, or HDAC6).
TDAC is an enzyme with deacetylase and ubiquitin binding activity, and both activities are required for TDAC to regulate aggresome formation.26 TDAC binds both polyubiquitinated mis-folded proteins and dynein, recruiting misfolded protein to dynein motors for transport to aggresomes along MTs. Aggresome formation is paralleled by the redistribution of the intermediate filament protein vimentin and the recruitment of proteasomes and chaperonins, a process dependent specifically on TDAC activity.26 There are 3 main classes of HDACs. TDAC belongs to class II HDACs, which all show homology to yeast Hda1. TDAC is unique among the class II members in that it has 2 catalytically active HDAC domains and also a C-terminal zinc finger domain that binds ubiquitin.27 The second catalytic domain in TDAC confers resistance to the HDAC inhibitors trapoxin B and sodium butyrate.28,29 TDAC is also resistant to HDAC inhibitors depsipeptide FR9012208 and the hybrid compound cyclic hydroxamic acid-containing peptide CHAP1,30 whereas trichostatin A and SAHA inhibit TDAC to a similar extent as other HDACs.31,32
In this study, we evaluated the activity of the novel hydroxamic acid-derived HDAC inhibitor LBH589 against MM cells; delineated LBH589-induced apoptotic signaling; determined whether synergistic cytotoxicity is induced with bortezomib; and defined effects of LBH589, alone or in combination with bortezomib, on TDAC activity and patterns of acetylated forms of α-tubulin and aggresome formation in MM cells.
Materials and methods
TDAC/HDAC inhibitor; proteasome inhibitor
LBH589 (Novartis Pharmaceuticals, East Hanover, NJ) and bortezomib (Millennium, Cambridge, MA) were dissolved in deiodinized water and stored at -20°C, then thawed and diluted in media for cell-culture experiments.
MM-derived cell lines and patient cells
Dexamethasone (Dex)-sensitive (MM.1S) and Dex-resistant (MM.1R) human MM cell lines, as well as RPMI8226 cells resistant to doxorubicin (Dox40), mitoxantrone (MR20), and melphalan (LR5), were cultured in RPMI1640 media (Cellgro, Mediatech, VA) with 10% fetal bovine serum, 2 mM l-glutamine (GIBCO, Grand Island, NY), 100 U/mL penicillin, and 100 mg/mL streptomycin (GIBCO). Drug-resistant cell lines were cultured with doxorubicin, mitoxantrone, melphalan, or Dex to confirm their lack of drug sensitivity. MM patient cells were obtained from bone marrow samples taken with permission for research purposes. Bone marrow mononuclear cells were separated using Ficoll-Hipaque density sedimentation, and plasma cells were purified (> 95% CD138+) by positive selection with anti-CD138 MACS Microbeads (Miltenyi, San Diego, CA). Peripheral-blood mononuclear cells were obtained by Ficoll-Hipaque density sedimentation of healthy donor peripheral blood. Cells were incubated in 96-well plates with or without LBH589 and/or bortezomib.
Bone marrow stromal-cell (BMSC) cultures
Bone marrow specimens were obtained from patients with MM. Mononuclear cells, separated by Ficoll-Hipaque density sedimentation, were used to establish long-term BMSC cultures. When an adherent cell monolayer had developed, cells were harvested in Hanks balanced salt solution (HBSS) containing 0.25% trypsin and 0.02% EDTA, and collected by centrifugation. The BMSCs were plated onto flat-bottom 96-well plates (Costar, Cambridge, MA) overnight and 3 × 104 MM.1S cells were then added for 48 hours. Supernatant (150 μL) was removed, and cells were pulsed with 3H-thymidine (18.5 KBq/well; NEN Products, Boston, MA) during the last 8 hours of 48-hour cultures. Duoset enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN) was used to measure IL-6 in supernatants of 48-hour cultures of BMSCs with or without MM.1S cells, in the presence or absence of LBH589.
Growth inhibition assay
The inhibitory effect of LBH589 on MM-cell growth was assessed by measuring 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium, inner salt (MTS) (Promega, Madison, WI) dye absorbance of cells. Cells in 200 μL media plus drug were pulsed with 40 μL of 5 mg/mL MTS to each well for the last 4 hours of 48-hour cultures. Absorbance was measured at 490 nm using a spectrophotometer (Molecular Devices, Sunnyvale, CA).
Immunoblotting
MM.1S cells were cultured with up to 100 nM LBH589; harvested; washed; and lysed using lysis buffer: RIPA buffer, 2 mM Na3VO4, 5 mM NaF, 1 mM phenylmethyl sulfonyl fluoride (PMSF), 5 mg/mL leupeptin, and 5 mg/mL aprotinin. Cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF) membrane, and immunoblotted with anti-acetylated histone (Upstate Biotechnology, Lake Placid, NY), anti-acetyl-α-tubulin (clone 6-11B-1) (Sigma, St Louis, MO), anti-p21 antibody (Santa Cruz Biotech, Santa Cruz, CA), anti-poly(ADP-ribose) polymerase (PARP); anti-caspase-8, as well as anti-caspase-9 and anti-caspase-3 (Cell Signaling, Beverly, MA). Membranes were stripped and reprobed with antitubulin (Sigma) to ensure equivalent protein loading.
Immunofluorescence
RPMI8226 cells were treated for 24 hours with LBH589, bortezomib, or both together. The cells were then spun onto glass slides and fixed. The slides were then stained with anti-acetyl-α-tubulin antibody (Sigma), and visualized using an Olympus BX60 inverted microscope (Olympus, Lake Success, NY) equipped with Hoffman objective lenses (10×/0.3 NA; 20×/0.5 NA; 40×/1.0 NA oil; Diagnostic Instruments, Sterling Heights, MI) connected to a SPOT One Digital Camera (Diagnostic Instruments). Images were then processed using Adobe Photoshop software (Adobe, San Jose, CA). Tubulin patterns were examined and classified according to the following categories: reticulated, linear latticed appearance; segmented, linear tubulin with interruption of the lattice; diffuse, loss of linear tubulin; reticulated aggresome, aggresome formation with lattice; focal aggresome, aggresome without lattice; diffuse hyperacetylated tubulin, diffuse but hyperacetylated. Representative counts from 3 separate experiments are presented in graph form.
Statistical analysis
Statistical significance of differences observed in drug-treated versus control cultures was determined using Student t test. The minimal level of significance was P below .05. For drug combination studies, cell viability measurements as determined by MTS assay were converted into values representing the fraction of growth affected (FA) of drug-treated cells compared with control cells, and data were analyzed by Calcusyn software (Biosoft, Ferguson, MO and Cambridge, United Kingdom) based upon the Chou-Talalay method.33 Briefly, the Chou-Talalay method uses the following equation: CI = (D)1 / (Dx)1 + (D)2 / (Dx)2, where (D)1 and (D)2 are the doses of drug 1 and drug 2 that have x effect when used in combination, and (Dx)1 and (Dx)2 are the doses of drug 1 and drug 2 that have the same x effect when used alone. When the combination index (CI) is equal to 1.0, this equation represents the conservation isobologram and indicates additive effects. Synergy is present when the CI is less than 1.0. The combination is additive when CI equals 1.0, and antagonistic when it is more than 1.0.
Results
LBH589 inhibits growth viability of MM cell lines resistant to conventional chemotherapy
LBH589 inhibited in vitro growth of human MM cell lines at 48 hours with an inhibitory concentration (IC50) of less than 20 nM against RPMI8226, MM.1S, U266, OPM1, and KMS11 cell lines; IC50 less than 50 nM against MM.1R, Dox40 cells or MR20 cells; and IC50 less than 100 nM against LR5 cells (Figure 1A-B). The inhibition of cell lines was significant (P < .01) in all situations except Dox40 at 20 nM, which was not significant.
Figure 1.

LBH589 induces cytotoxicity in human MM cell lines resistant to conventional chemotherapy. (A) Viability of MM.1S, RPMI8226, U266, OPM1, and KMS11 at 48 hours was inhibited by LBH589 in a dose-dependent manner as demonstrated by MTS assay. (B) Growth of Dex-resistant MM.1R cells and RPMI8226 MM cells resistant to Dox (Dox40 cells), Mit (MR20 cells), or Mel (LR5 cells) was inhibited in a dose-dependent manner, as demonstrated by MTS assay. (C) Adhesion of MM.1S cells to BMSCs induced a significant increase in 3H-thymidine uptake by MM.1S cells at 48 hours, which was completely inhibited by LBH589. Supernatants from the experiment (C) were examined for cytokine secretion (D). LBH589 inhibited adhesion-induced up-regulation of IL-6 secretion in BMSCs triggered by coculture with MM.1S. (E) LBH589 up to 1 μM decreased BMSC viability, with 61% viability at 48 hours and 59% viability at 72 hours. Data represent mean ± SD of quadruplicate cultures.
Dex-sensitive MM.lS cells express high levels of the glucocorticoid receptor (GR), whereas Dex-resistant MM.IR cells express low to undetectable levels of a functional GR.34 In contrast, glucocorticoid resistance of the myeloma line OPM1 is believed to occur via an alternative mechanism downstream.35 LR5 cells have acquired melphalan resistance through enhanced DNA repair via the Fanconi anemia/BRCA pathway,36 and show reduced melphalan-induced DNA crosslinks and elevated glutathione levels.37 These results therefore indicate that LBH589 acts independently of the glucocorticoid receptor or DNA cross-linking. MR20 cells overexpress breast cancer resistance protein (BCRP), an adenosine triphosphate-binding cassette that reduces intracellular drug accumulation without increased expression of the P-glycoprotein drug resistance transporter.38 Dox40 cells possess the MDR phenotype.39-42 Therefore, LBH589 is not a substrate for these drug efflux transporter mechanisms of drug resistance.
Effect of LBH589 on patient MM-derived BMSCs, and the interaction between BMSCs and MM cells
MM cells are localized in the BM microenvironment where they adhere to extracellular matrix proteins and to BMSCs. This interaction triggers the production of cytokines mediating autocrine and paracrine growth and survival of MM cells, as well as protection against drug-induced apoptosis.43-46 We therefore next examined the effect of LBH589 on BMSC-induced MM-cell proliferation (Figure 1C). At 48 hours there was a 2.8-fold increase in the proliferation of MM.1S cells cocultured with BMSCs, evidenced by thymidine uptake (P = .007). However, thymidine uptake of MM.1S cells was reduced to 11% and 6% of control by 10 nM and 20 nM LBH589, respectively (P = .075 and .007). Therefore, LBH589 inhibits growth even in the presence of BMSCs. The effect of LBH589 on BMSC thymidine uptake at 10 nM was not significant (P = .2; Figure 1C), but at 20 nM there was a statistically significant reduction (P = .03). In addition, at 48 hours LBH589 inhibited adhesion-induced up-regulation of IL-6 secretion from BMSCs triggered by coculture with MM.1S cells (Figure 1D). The coculture-induced IL-6 increase was significant (P = .04), and was significantly inhibited by LBH589 (P = .03). Treatment of BMSCs with LBH589 (1 μM) reduced viability of BMSCs to 61% and 60% at 48 and 72 hours, respectively (P = .02 and .05; Figure 1E). The reduction in BMSC viability at 24 hours was significant (P = .02), but the later time points were not different from this reduction at 24 hours.
LBH589 induces histone and tubulin hyperacetylation, caspase and PARP cleavage, up-regulation of p21, and down-regulation of c-myc in MM.1S cells
The effect of LBH589 on histone acetylation status was next examined. MM.1S cells were incubated with 20 nM to 100 nM LBH589 for 24 hours, and whole-cell extracts were then analyzed by Western blot. LBH589 induced a dose-dependent increase in histone acetylation (Figure 2A). Tubulin hyperacetylation was observed at low nanomolar LBH589 concentrations, indicating potent inhibition of TDAC.28,47-49 To confirm apoptosis in MM cells, MM.1S cells were incubated with LBH589 for 24 hours, and then examined for caspase activation (Figure 2B). At 24 hours, LBH589 induced cleavage of caspase-8, -9, and -3 as well as PARP. The promoter for p21 transcription is regulated by histone acetylation status,50 and up-regulation of p21 has been widely reported with HDAC inhibitors.51,52 As we previously reported with NVP-LAQ824,5 p21 up-regulation was triggered at low nanomolar concentrations of LBH589. In contrast, the oncogene c-myc was down-regulated.
Figure 2.

LBH589 potently induces histone and α-tubulin hyperacetylation as well as apoptotic signaling. MM.1S cells were incubated with up to 100 nM LBH589 for 24 hours, and whole-cell extracts were analyzed by Western blot. (A) There was a dose-dependent increase in both histone acetylation (top panel) and α-tubulin acetylation (bottom panel). (B) Cleavage of caspase-8, -9, -3 and PARP was induced in a dose-dependent manner. In addition, p21 was up-regulated and c-myc was down-regulated. FL indicates full length; CL, cleavage fragment.
Synergistic effects of LBH589 in combination with bortezomib on cell viability and cytoskeletal architecture
Given the TDAC inhibitory effects of LBH589 and the role of TDAC and proteasome inhibition in aggresome formation,23,26,28 we next investigated the effects of combining LBH589 with bortezomib on both cell viability and aggresome formation. LBH589 and bortezomib induced synergistic cytotoxicity against RPMI8226 cells, demonstrated by MTS assay (Figure 3A), which was, at least in part, caspase-dependent (Figure 3B). Synergy was confirmed by isobologram analysis (Figure 3C-D). Synergistic activity is present when the combination index (CI) is less than 1.0, versus antagonistic activity when CI is more than 1.0; strong synergy is indicated when the CI is less than 0.3. As seen in Figure 3D, strong synergy was observed with CI indices of 0.404, 0.054, and 0.067 at 12, 16, and 20 nM of LBH589 and bortezomib. At concentrations of 4 nM the combination of LBH589 and bortezomib was statistically more potent than bortezomib alone but not LBH589 alone (Figure 3A). Although this was reported as antagonism according to the method of Chou-Talalay (Figure 3D), this is unlikely to be clinically relevant. At concentrations of 8 nM and above the combination was significantly more potent than either drug alone (P < .01) and was very strongly synergistic (Figure 3D). Cell rescue achieved by ZVAD was significant (P < .01) in every situation (Figure 3B). In contrast, both LBH589 and bortezomib were significantly less toxic to normal donor peripheral-blood mononuclear cells, and the combination was not even additive (P ranged between 0.1 to 0.9; Figure 3E).
Figure 3.
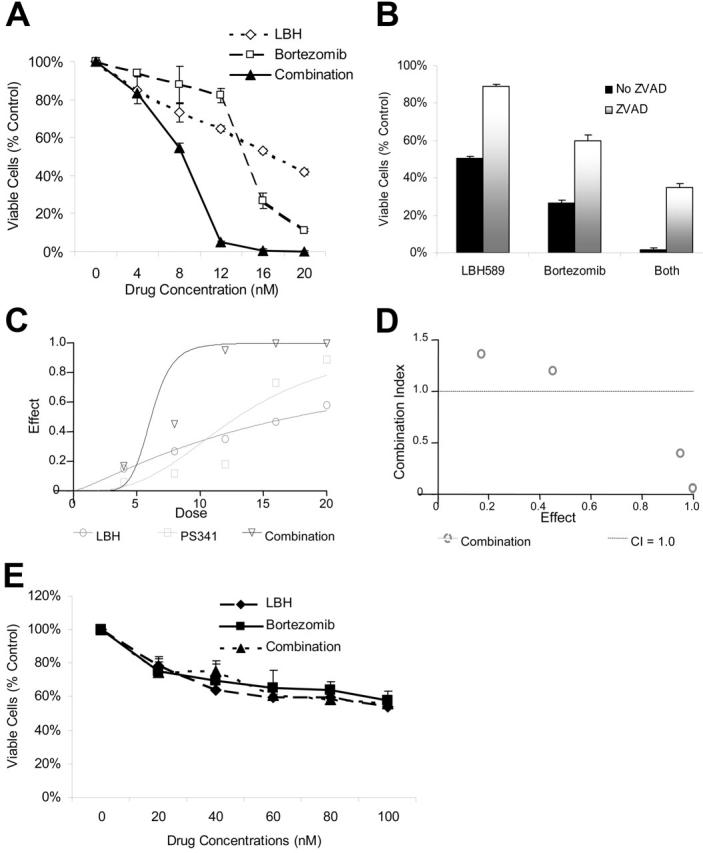
LBH589 and bortezomib trigger synergistic cytotoxicity in RPMI8226 MM cell lines. (A) RPMI8226 MM cells were incubated with LBH589 (0-20 nM), bortezomib (0-20 nM), or both for 48 hours. Synergistic cytotoxicity was demonstrated by MTS assay. (B) Cytotoxicity of LBH589 and bortezomib was partly inhibited by pan-caspase inhibitor ZVAD-FMK. Data represent mean ± SD of quadruplicate cultures. The viability, as determined by MTS assay and shown in panel A, was converted into effect (1-viability), and shown as an isobologram graph (C), as well as converted to combination index (CI) (D) according to the Chou-Talalay equation (“Materials and methods”). Synergy is present when the CI is less than 1.0. The combination is additive when CI equals 1.0, and antagonistic when it is more than 1.0. Synergy was observed with a CI of 0.404, 0.054, and 0.067 at 12, 16, and 20 nM of LBH589 and bortezomib. (E) Healthy donor peripheral-blood mononuclear cells (PBMCs) were incubated with LBH589 (0-100 nM), bortezomib (0-100 nM), or both for 48 hours. No synergy was observed against PBMCs. Data represent mean ± SD of quadruplicate cultures.
Synergy was also observed against CD138-selected patient MM cells, MM.1S, MM.1R, and OPM1 cells (Figure 4A-D). Synergy was observed against patient cells for all concentrations of bortezomib versus combination (P < .04), and for all concentrations of LBH589 versus combination (P < .03). Against MM.1S cells at 4 nM, the combination effect was significantly different from bortezomib alone (P = .03), but LBH589 was not significantly different from the combination (P = .15). Therefore, most of the effects against MM.1S at this low concentration were due to LBH589. As a result, the combination indices indicate antagonism, but this reflects a lack of effect of either drug at lower concentrations rather than any biologically significant antagonism. At all higher concentrations up to 20 nM there was a significant difference between LBH589 and combination (P < .01). However, the dose-response curve was quite steep for bortezomib, and the combination was significantly different from bortezomib only at 4, 8, and 12 nM but not at 16 and 20 nM because of the strong single agent activity of bortezomib at these concentrations. For MM.1R cells, the effect of both drugs in combination was significantly different from either LBH589 or bortezomib alone (P < .01) at all concentrations except for between LBH589 and combination at 4 and 8 nM, which was not different from LBH589 alone. Therefore, only weak synergy was observed. For OPM1 cells, neither LBH589 nor bortezomib was statistically different at 4 nM, and at 8 nM bortezomib was not different from combination. Therefore, like MM.1S, the combination indices for OPM1 at lower concentrations did not indicate synergy. However, at 12 nM to 20 nM concentrations of both drugs, the combinations were statistically different from either drug alone, which was reflected as synergy in the combination indices.
Figure 4.
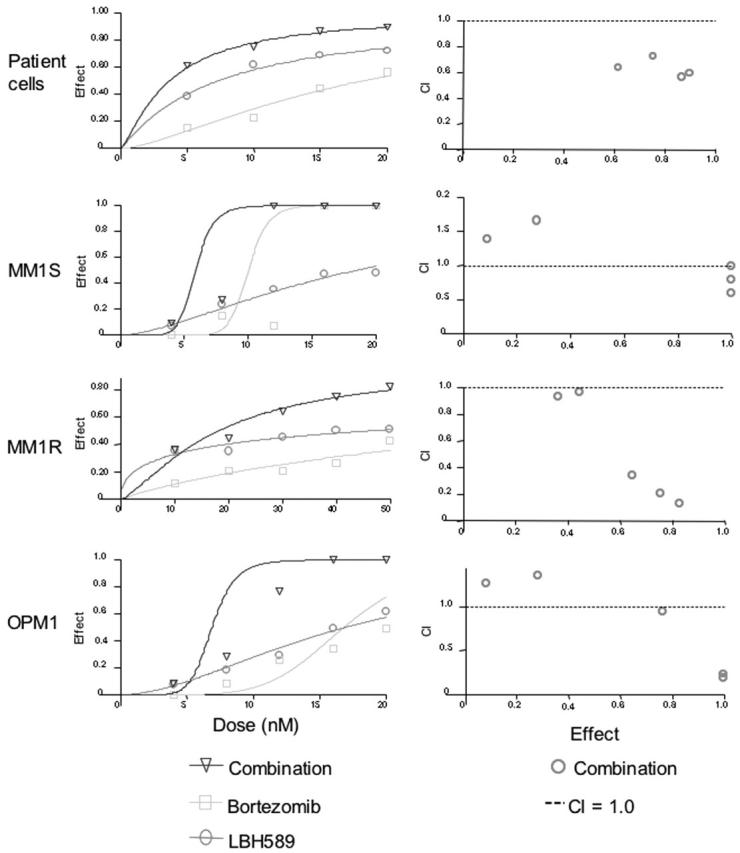
LBH589 and bortezomib induce synergistic cytotoxicity against MM cell lines sensitive and resistant to Dex, as well as freshly isolated CD138+ patient MM cells. (A-D) Synergistic cytotoxicity of LBH589 and bortezomib was observed in RPMI8226 and MM.1S, patient cells, and OPM1 (4-20 nM), as well as in MM.1R cells (10-50 nM), as determined by MTS assay and presented as a fraction of cells affected (left panels) and combination indices (right panels).
Alpha-tubulin acetylation and aggresome formation were examined using fluorescent microscopic examination of acetyl-α-tubulin at concentrations of LBH589 and bortezomib that demonstrated synergistic cytotoxicity against RPMI8226 cells (Figure 5A-D). Figure 5A shows the appearance of untreated RPMI cells. The left panel displays a reticulated pattern of acetyl-α-tubulin, consisting of connecting fibrils forming a latticed network. The normal heterochromatin pattern is demonstrated by the DAPI stain (center panel), and the merged image is on the right. A normal mitotic figure (arrow) is shown with other nondividing cells. In Figure 5, panels B, C, and D are all merged acetyl-α-tubulin/DAPI images demonstrating patterns of α-tubulin (see “Materials and methods”). Figure 5B shows the effects of LBH589 (16 nM) alone. The left panel shows hyperacetylated acetyl-α-tubulin (arrow), and an apoptotic cell with diffuse acetyl-α-tubulin (arrowhead). The cytoplasmic appearance of diffuse acetyl-α-tubulin (arrowhead) is a nonspecific feature of apoptosis that was also observed when cells were treated with topoisomerase inhibitors, which induce cell death in MM cells in a caspase-dependent manner.53 The Figure 5B center panel shows hyperacetylated segmented α-tubulin, and the right panel shows a mitotic figure (arrow) with marked disruption of equatorial plate formation versus the normal mitotic figure featured in Figure 5A, confirming the disruptive effects of HDAC inhibition on chromosomal alignment and the MTOC during mitosis. The Figure 5C left and center panels show aggresome formation associated with reticulated acetyl-α-tubulin, whereas the right panel shows focal aggresome formation (arrow); the middle and right panel DAPI stains also demonstrate the condensed lobulated nuclear patterns characteristic of apoptosis (arrowheads).
Figure 5.
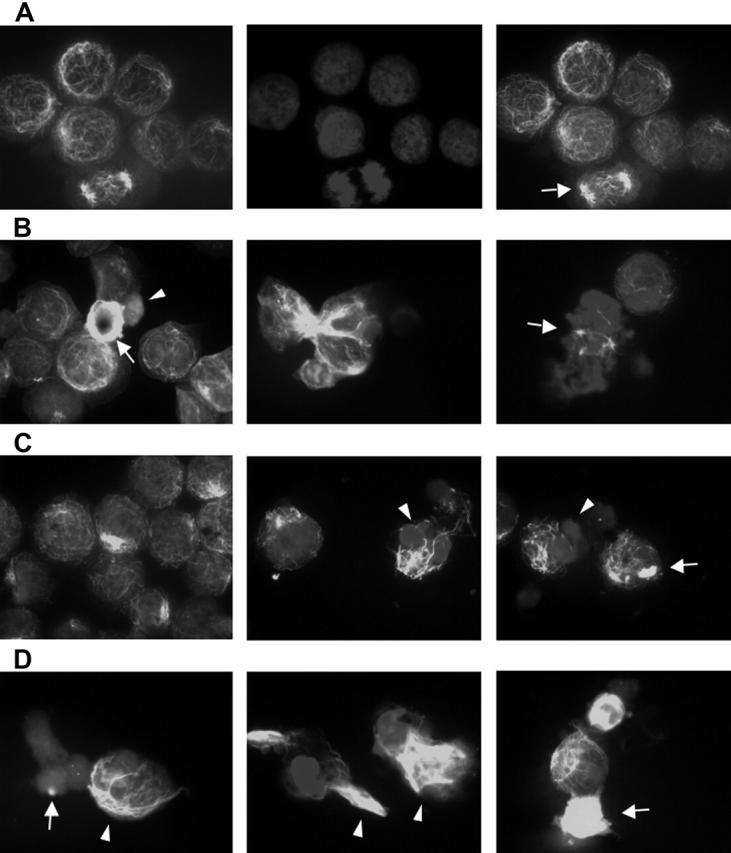
Bortezomib-induced aggresome formation and LBH589-induced α-tubulin hyperacetylation. RPMI8226 cells were treated for 24 hours with 16 nM LBH589, 16 nM bortezomib, or both, stained with anti-acetyl-α-tubulin and DAPI, then examined by inverted microscopy. (A) In untreated RPMI8226 cells, acetylated α-tubulin showed a reticulated appearance (left panel). The center panel shows the DAPI stain. In the merged image in the right panel, a normal mitotic figure (arrow, right panel) is shown, as well as other nondividing cells. (B) HDAC inhibitor LBH589 induced diffuse or segmented α-tubulin hyperacetylation. Hyperacetylated-α-tubulin (arrow, left panel); diffuse acetyl-α-tubulin (arrowhead, left panel); segmented α-tubulin hyperacetylation (center panel); mitotic figure (arrow, right panel). (C) Proteasome inhibitor bortezomib induced aggresomes in the presence of normal reticulated α-tubulin (left panel). Focal aggresome (arrow, right panel); apoptotic nuclei (arrowheads, center and right panels). (D) LBH589 and bortezomib induced large bundles of hyperacetylated α-tubulin (arrowheads, left and center panels) that were not seen in cells treated with LBH589 alone. Diffuse hyperacetylated α-tubulin was also observed (arrow, right panel). Although aggresome formation was observed (arrow, left panel), here they were smaller than when treated with bortezomib alone, and were associated with a diffuse cytoplasmic background and apoptotic nuclei.
Figure 5D demonstrates the effects of LBH589 in combination with bortezomib on acetyl-α-tubulin. In general, the qualitative effects of either drug alone were enhanced by the presence of the other. The most striking feature was a marked accumulation of bundles of abnormally displaced hyperacetylated α-tubulin (arrow-heads). Aggresomes were more frequent, but were smaller in size and often associated with apoptotic diffuse cytoplasmic α-tubulin (arrow, left panel). Diffuse hyperacetylated α-tubulin was also observed (arrow, right panel). In addition to these qualitative changes, the percentage of cells displaying such characteristics was increased more than would be expected if the effect were purely additive. The DAPI stain revealed a synergistic increase in apoptotic nuclei at 24 hours. Bundles of hyperacetylated α-tubulin were not observed after incubation of cells with a topoisomerase-1 inhibitor, nor were they inhibited by the caspase inhibitor ZVAD, and were therefore not induced by caspase activation.
Patterns of α-tubulin acetylation and aggresome formation were classified (as in “Materials and methods”), cell counts were performed on at least 150 cells in each treatment group, and representative counts from 3 separate experiments are shown in Figure 6. Viable untreated RPMI8226 cells displayed fine reticulated lattice of acetyl-α-tubulin in 88%, and in 5% there was some α-tubulin segmentation, possibly associated with motility or mitotic functions. Apoptotic nuclei associated with diffuse α-tubulin were observed in 7% of cells. Cells treated with 16 nM LBH589 alone for 24 hours displayed normal reticulated appearance in 36%; 8% of cells were viable with hyperacetylated α-tubulin (Figure 5B, left panel); 13% of cells were viable with segmented hyperacetylated α-tubulin (Figure 5B, center panel); 40% of cells were apoptotic with diffuse α-tubulin. No aggresomes were observed when cells were treated with low-dose LBH589 alone. Cells treated with bortezomib 16 nM displayed normal appearance in 40%; 8% displayed aggresome formation associated with normal reticulated acetylated α-tubulin (Figure 5C, left panel); 28% were apoptotic with diffuse α-tubulin; 17% were apoptotic with aggresome formation associated with reticulated acetylated α-tubulin, although reticulated acetylated α-tubulin in association with apoptosis was more segmented with a tendency to polarize within the cell (Figure 5C, center and right panels).
Figure 6.

Bortezomib-induced aggresome formation and LBH589-induced α-tubulin hyperacetylation are augmented in combination. Patterns of α-tubulin acetylation and aggresome formation were classified as described in “Materials and methods.” Cell counts were performed on at least 150 cells in each treatment group. Morphologic patterns defined by acetyated α-tubulin were further classified based on nuclear appearance to be either normal or apoptotic. A representative set of counts from 3 separate experiments is shown. Data represent means ± SD.
Cells treated with both LBH589 and bortezomib displayed features of either drug alone, with important qualitative differences. The nuclei were apoptotic in 96% of cells; 39% displayed diffuse α-tubulin, similar to that seen with either drug. Large eccentric bundles of hyperacetylated α-tubulin were observed in 22% of cells. Diminished focal aggresomes associated with diffuse α-tubulin were observed in 31% of cells. The apoptotic diffuse cells were not significantly different between LBH589, bortezomib, and the combination of both drugs. However, there was a significant increase in hyperacetylated apoptotic cells in the combination compared with either LBH589 or bortezomib (P < .01). Therefore, although bortezomib did not induce α-tubulin hyperacetylation alone, it did augment LBH589-induced α-tubulin hyperacetylation. The focal aggresomes were more numerous in the combination compared with LBH589 but not bortezomib, consistent with aggresome induction by bortezomib but diminished size in the presence of LBH589. These data are represented graphically in Figure 6.
The overall combination effect of LBH589 and bortezomib on the cytoskeleton is illustrated diagrammatically in Figure 7.
Figure 7.

Schematic representation of upstream cytoskeletal events when bortezomib and LBH589 are combined. (A) Viable plasma cell with reticulated acetylated α-tubulin. (B) Aggresome formation in the presence of bortezomib. Aggregates of misfolded proteins are directed along the acetylated α-tubulin fibrils toward a single perinuclear region, forming the aggresome. This process requires TDAC activity. (C) In the presence of bortezomib in combination with LBH589, aggresome formation is diminished, bundles of hyperacetylated α-tubulin form, and the cell undergoes apoptosis.
These results are consistent with the combination of proteasome inhibition and TDAC inhibition interacting in a synergistic mechanism on α-tubulin hyperacetylation, aggresome formation, and apoptosis. Bortezomib with LBH589 triggers an overwhelming demand on the cellular UPR-aggresome pathway manifesting as large accumulations of unipolar hyperacetylated α-tubulin up-stream, but small aggresome aggregates downstream, of the UPR-ubiquitin-proteasome-TDAC-aggresome pathway.
Discussion
This study has demonstrated in vitro activity of the novel hydroxamic acid-derived HDAC inhibitor LBH589 against MM cell lines both sensitive and resistant to conventional chemotherapy, as well as freshly isolated MM patient cells. Proliferation of MM.1S cells cocultured with BMSCs was also inhibited, with minimal toxicity against BMSCs. Dose-dependent inhibition of HDAC, including TDAC, was observed in MM.1S cells at low nanomolar concentrations of LBH589, evidenced by histone- and α-tubulin-hyperacetylation. LBH589 induced apoptotic signaling with up-regulation of p21; down-regulation of c-myc; activation of caspase-8, -9, and -3; and PARP cleavage. Similar to NVP-LAQ824, LBH589 is therefore an hydroxamic acid-derived HDAC inhibitor that in nanomolar range induces caspase-mediated apoptotic activity.
The current experiments demonstrate striking synergistic cytotoxic activity induced by LBH589 in combination with bortezomib, which is partially caspase-dependent. In general, caspase-mediated apoptosis is triggered via initiator caspases (caspase-8 and -9) and downstream effector caspases (caspase-3, -6, and -7), and occurs when the activity of proapoptotic molecules such as reactive oxygen species (ROS) overcome antiapoptotic mechanisms, such as activation of nuclear factor κ-B. Synergy has been observed between bortezomib and the anthracycline antibiotic doxorubicin via inhibition of DNA repair enzymes,54 and between bortezomib and the topoisomerase-1 inhibitor SN38 via inhibition of nuclear factor κ-B.53,55 Synergistic apoptosis induced by bortezomib in combination with SAHA and sodium butyrate is mediated via mitochondrial dysfunction and ROS-dependent mechanisms, and downstream events include nuclear factor κ-B inactivation, c-Jun NH terminal kinase activation, p53 induction, caspase activation, PARP cleavage, and caspase-dependent cleavage of p21, p27, bcl-2, mcl-1, X-linked inhibitor of apoptosis, and down-regulation of cyclin D1.6,56 Therefore, a number of potential molecular mechanisms may contribute to mediating the synergy between bortezomib and LBH589. In the current studies, we investigated a potential novel interaction between upstream apoptotic triggers specific to both bortezomib and LBH589. Given the roles of proteasome inhibition and TDAC in the misfolded protein response and aggresome formation, we explored the effects of bortezomib and LBH589 on α-tubulin cytoarchitecture and aggresome formation. Untreated interphase cells were observed to form a branching cytoskeletal network of acetyl-α-tubulin extending to the cell periphery. Exposure to low concentrations of LBH589 induced α-tubulin hyperacetylation that was either diffuse or segmented, and mostly associated with normal nuclei. Apoptotic nuclei were associated with nonspecific cytoplasmic α-tubulin architecture, and visible aggresomes were not induced at low concentrations of LBH589. Exposure to bortezomib induced aggresome formation with preservation of the cytoskeletal architecture in cells with normal nuclei, whereas focal discrete aggresomes were associated with apoptotic nuclei. After exposure to LBH589 in combination with bortezomib, the effects of each drug alone were significantly augmented. Aggresomes and hyperacetylated α-tubulin aggregates were significantly increased in apoptotic cells, with a marked overall increase in apoptotic nuclei. Although TDAC inhibition would be expected to inhibit aggresome formation,26 the addition of bortezomib induced a slight increase in aggresome numbers, but these aggresomes were smaller, and the cells were apoptotic. The most striking feature was the development of large bundles of hyperacetylated α-tubulin, which were abnormally displaced from their usual extended cytoplasmic distribution. These changes have been previously described with the intermediate filament vimentin in cells overexpressing the inefficiently folded integral membrane cystic fibrosis transmembrane conductance regulator (CFTR) that were exposed to the MT inhibitor nocodozole, and suggest increased demand on the aggresome pathway coupled with inhibition of MT cytodynamics. These findings are consistent with the TDAC inhibitory properties of LBH589 in combination with aggresome induction by bortezomib.23,24
The aggresome pathway prevents a widespread intracellular accumulation of misfolded proteins that escape the other protective processes within the cell, in particular proteasome inhibition. Prominent aggresomes or inclusion bodies are not normally evident in unstressed cells. The life spans of cells containing aggresomes induced by overexpression of misfolded proteins are similar to normal cells.25 However, if the pathway is overwhelmed, the continuing accumulation of protein aggregates is detrimental.24 Cells deficient in TDAC cannot form aggresomes properly, fail to clear misfolded protein aggregates from the cytoplasm, and are hypersensitive to the accumulation of misfolded proteins. In TDAC knockdown cells that do form visible aggresomes in response to proteasome inhibition, aggresome size is significantly smaller than in normal cells.26 TDAC is therefore very important in the cellular management of misfolded protein-induced stress.
Like LBH589, the more selective TDAC inhibitor tubacin is also synergistic with bortezomib against MM cells.57 One notable difference between tubacin and LBH589 is that tubacin did not induce p21 up-regulation, which is an almost universal feature of HDAC inhibitors.51 This difference is consistent with regulation of the promoter for p21 transcription by histone acetylation status.50 LBH589 is cytotoxic in low nanomolar concentrations (IC50 < 100 nM) for all myeloma cells tested, whereas the cytotoxic IC50 for tubacin is over 5 micromoles for most cells tested. However, the different potencies also apply to TDAC inhibitory activity, and may not necessarily reflect differences in selectivity. When more potent selective TDAC inhibitors have been developed there will be better understanding of the contribution of TDAC inhibition to the cytotoxic effects of HDAC inhibitors.
LBH589 is not a selective TDAC inhibitor, but rather a broad spectrum HDAC inhibitor with potent single agent apoptosis-inducing properties in MM cells. Therefore, other factors are likely to contribute to the cytoarchitectural disturbances observed in cells exposed to LBH589 in combination with bortezomib. For example, macromolecular crowding could stimulate aggregation of aggresome precursors throughout the cell.25,58 HDAC/TDAC inhibition may induce an increase in misfolded proteins that would normally be degraded by the proteaseome, but in the presence of bortezomib form diffuse microaggregates that are redirected toward an inhibited aggresome pathway, resulting in abnormal redistribution of hyperacetylated acetyl-α-tubulin bundles. The drug combination-induced augmentation of aggresome formation in the current study was observed in apoptotic cells, and may therefore represent an inadequate response to an increased cellular demand due to proteasome inhibition. Thus the increase in aggresomes was accompanied by a more striking increase in aggregated bundles of hyperacetylated α-tubulin in apoptotic cells.
These data provide insights into the cytoarchitectural dynamics induced by the combination of bortezomib with LBH589 on MM cells. Other upstream molecular mechanisms may contribute to the synergistic interaction between LBH589 and bortezomib. For example, Hsp90 is up-regulated in response to bortezomib treatment, and Hsp90 inhibition sensitizes cells to bortezomib.59 Hsp90 and α-tubulin hyperacetylation are induced by LBH589 simultaneously because both are mediated by TDAC inhibition.60 Similarly, bortezomib inhibits the unfolded protein response in myeloma, which may contribute to aggresome formation.21 It will be important to determine the contribution of each mechanism to the synergistic interactions between bortezomib and LBH589. Ongoing studies are also delineating the role of MT-dependent retrograde transport of aggregated protein during MM-cell apoptosis. Resolution of these issues may identify key targets involved in cellular trafficking pathways in MM, and reveal insights into the selective activity of bortezomib against MM cells.
In conclusion, LBH589 represents a novel hydroxamic acid-derived HDAC inhibitor with activity against MM in nanomolar concentrations, which should be readily achievable in patients. Strong synergy against MM cells was observed with bortezomib, which was associated with marked abnormalities in the cellular cytoskeletal architecture due to aggresome induction and TDAC inhibition. These results provide insight into the mechanisms of synergistic anti-MM activity of bortezomib in combination with LBH589, and provide the rationale for clinical protocols using LBH589, either alone or in combination with bortezomib, to overcome drug resistance and improve patient outcome in MM.
Prepublished online as Blood First Edition Paper, June 13, 2006; DOI 10.1182/blood-2006-04-016055.
Supported by Multiple Myeloma Research Foundation Awards (Y.-T.T., T.H., D.C.), National Institutes of Health (NIH) grants ROI50 947 and POI78 378, and the Doris Duke Distinguished Clinical Research Scientist Award (K.C.A.).
P.A. is an employee of Novartis Pharmaceuticals, which holds a patent on LBH589.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
References
- 1.Remiszewski SW. Recent advances in the discovery of small molecule histone deacetylase inhibitors. Curr Opin Drug Discov Devel. 2002;5: 487-499. [PubMed] [Google Scholar]
- 2.Remiszewski SW. The discovery of NVP-LAQ824: from concept to clinic. Curr Med Chem. 2003;10: 2393-2402. [DOI] [PubMed] [Google Scholar]
- 3.Marks PA, Rifkind RA, Richon VM. Histone deacetylase inhibitors: development of suberoylanilide hydroxamic acid (SAHA) for the treatment of cancers. Proc Natl Acad Sci U S A. 2001;98: 10572-10577. [DOI] [PubMed] [Google Scholar]
- 4.Mitsiades N, Mitsiades CS, Richardson PG, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101: 4055-4062. [DOI] [PubMed] [Google Scholar]
- 5.Catley L, Weisberg E, Tai YT, et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102: 2615-2622. [DOI] [PubMed] [Google Scholar]
- 6.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10: 3839-3852. [DOI] [PubMed] [Google Scholar]
- 7.Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci U S A. 2004;101: 540-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuino L, Bali P, Wittmann S, et al. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol Cancer Ther. 2003;2: 971-984. [PubMed] [Google Scholar]
- 9.Nimmanapalli R, Fuino L, Bali P, et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63: 5126-5135. [PubMed] [Google Scholar]
- 10.Qian DZ, Wang X, Kachhap SK, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64: 6626-6634. [DOI] [PubMed] [Google Scholar]
- 11.Arrigo AP, Tanaka K, Goldberg AL, Welch WJ. Identity of the 19S `prosome' particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature. 1988;331: 192-194. [DOI] [PubMed] [Google Scholar]
- 12.Driscoll J, Goldberg AL. The proteasome (multi-catalytic protease) is a component of the 1500-kDa proteolytic complex which degrades ubiquitin-conjugated proteins. J Biol Chem. 1990;265: 4789-4792. [PubMed] [Google Scholar]
- 13.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65: 801-847. [DOI] [PubMed] [Google Scholar]
- 14.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79: 13-21. [DOI] [PubMed] [Google Scholar]
- 15.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6: 79-87. [DOI] [PubMed] [Google Scholar]
- 16.Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays. 2000;22: 442-451. [DOI] [PubMed] [Google Scholar]
- 17.Gelman MS, Kannegaard ES, Kopito RR. A principal role for the proteasome in endoplasmic reticulum-associated degradation of misfolded intracellular cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2002;277: 11709-11714. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg AL, Akopian TN, Kisselev AF, Lee DH. Protein degradation by the proteasome and dissection of its in vivo importance with synthetic inhibitors. Mol Biol Rep. 1997;24: 69-75. [DOI] [PubMed] [Google Scholar]
- 19.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426: 895-899. [DOI] [PubMed] [Google Scholar]
- 20.Plemper RK, Wolf DH. Retrograde protein translocation: ERADication of secretory proteins in health and disease. Trends Biochem Sci. 1999;24: 266-270. [DOI] [PubMed] [Google Scholar]
- 21.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100: 9946-9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23: 7448-7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143: 1883-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10: 524-530. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol. 1999;146: 1239-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115: 727-738. [DOI] [PubMed] [Google Scholar]
- 27.Seigneurin-Berny D, Verdel A, Curtet S, et al. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21: 8035-8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417: 455-458. [DOI] [PubMed] [Google Scholar]
- 29.Guardiola AR, Yao TP. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J Biol Chem. 2002;277: 3350-3356. [DOI] [PubMed] [Google Scholar]
- 30.Blagosklonny MV, Robey R, Sackett D, et al. Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther. 2002;1: 937-941. [PubMed] [Google Scholar]
- 31.Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci U S A. 2001;98: 87-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong JC, Hong R, Schreiber SL. Structural biasing elements for in-cell histone deacetylase para-log selectivity. J Am Chem Soc. 2003;125: 5586-5587. [DOI] [PubMed] [Google Scholar]
- 33.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22: 27-55. [DOI] [PubMed] [Google Scholar]
- 34.Moalli PA, Pillay S, Weiner D, Leikin R, Rosen ST. A mechanism of resistance to glucocorticoids in multiple myeloma: transient expression of a truncated glucocorticoid receptor mRNA. Blood. 1992;79: 213-222. [PubMed] [Google Scholar]
- 35.Gomi M, Moriwaki K, Katagiri S, Kurata Y, Thompson EB. Glucocorticoid effects on myeloma cells in culture: correlation of growth inhibition with induction of glucocorticoid receptor messenger RNA. Cancer Res. 1990;50: 1873-1877. [PubMed] [Google Scholar]
- 36.Chen Q, Van der Sluis PC, Boulware D, Hazlehurst LA, Dalton WS. The FA/BRCApathway is involved in melphalan-induced DNAinterstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood. 2005;106: 698-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellamy WT, Dalton WS, Gleason MC, Grogan TM, Trent JM. Development and characterization of a melphalan-resistant human multiple myeloma cell line. Cancer Res. 1991;51: 995-1002. [PubMed] [Google Scholar]
- 38.Ross DD, Yang W, Abruzzo LV, et al. Atypical multidrug resistance: breast cancer resistance protein messenger RNA expression in mitoxantrone-selected cell lines. J Natl Cancer Inst. 1999;91: 429-433. [DOI] [PubMed] [Google Scholar]
- 39.Dalton WS, Durie BG, Alberts DS, Gerlach JH, Cress AE. Characterization of a new drug-resistant human myeloma cell line that expresses P-glycoprotein. Cancer Res. 1986;46: 5125-5130. [PubMed] [Google Scholar]
- 40.Bellamy W, Dalton WS, Meltzer P, Dorr RT. Role of glutathione and its associated enzymes in multidrug-resistant human myeloma cells. Biochem Pharmacol. 1989;38: 787-793. [DOI] [PubMed] [Google Scholar]
- 41.Bellamy W, Dorr RT, Dalton WS, Alberts DS. Direct relation of DNA lesions in multidrug-resistant human myeloma cells to intracellular doxorubicin concentration. Cancer Res. 1988;48: 6360-6364. [PubMed] [Google Scholar]
- 42.Bellamy W, Dalton WS, Kailey JM, et al. Verapamil reversal of doxorubicin resistance in multidrug-resistant human myeloma cells and association with drug accumulation and DNA damage. Cancer Res. 1988;48: 6365-6370. [PubMed] [Google Scholar]
- 43.Chauhan D, Uchiyama H, Urashima M, Yamamoto K, Anderson KC. Regulation of interleukin 6 in multiple myeloma and bone marrow stromal cells. Stem Cells. 1995;13(suppl 2): 35-39. [PubMed] [Google Scholar]
- 44.Chauhan D, Uchiyama H, Akbarali Y, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87: 1104-1112. [PubMed] [Google Scholar]
- 45.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93: 1658-1667. [PMC free article] [PubMed] [Google Scholar]
- 46.Damiano JS, Dalton WS. Integrin-mediated drug resistance in multiple myeloma. Leuk Lymphoma. 2000;38: 71-81. [DOI] [PubMed] [Google Scholar]
- 47.Piperno G, LeDizet M, Chang XJ. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. 1987;104: 289-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piperno G, Fuller MT. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. J Cell Biol. 1985;101: 2085-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100: 4389-4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97: 10014-10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blagosklonny MV, Robey R, Sackett DL, et al. Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther. 2002;1: 937-941. [PubMed] [Google Scholar]
- 52.Chai F, Evdokiou A, Young GP, Zalewski PD. Involvement of p21(Waf1/Cip1) and its cleavage by DEVD-caspase during apoptosis of colorectal cancer cells induced by butyrate. Carcinogenesis. 2000;21: 7-14. [DOI] [PubMed] [Google Scholar]
- 53.Catley L, Tai YT, Shringarpure R, et al. Proteasomal degradation of topoisomerase I is preceded by c-Jun NH2-terminal kinase activation, Fas up-regulation, and poly(ADP-ribose) polymerase cleavage in SN38-mediated cytotoxicity against multiple myeloma. Cancer Res. 2004;64: 8746-8753. [DOI] [PubMed] [Google Scholar]
- 54.Mitsiades N, Mitsiades CS, Richardson PG, et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101: 2377-2380. [DOI] [PubMed] [Google Scholar]
- 55.Cusack JC Jr, Liu R, Houston M, et al. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic nuclear factor-kappaB inhibition. Cancer Res. 2001;61: 3535-3540. [PubMed] [Google Scholar]
- 56.Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003;102: 3765-3774. [DOI] [PubMed] [Google Scholar]
- 57.Hideshima T, Bradner JE, Wong J, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005;102: 8567-8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381: 571-579. [DOI] [PubMed] [Google Scholar]
- 59.Mitsiades C, Mitsiades N, McMullan CJ, et al. Antimyeloma activity of heat shock protein-90 inhibiton. Blood. 2006;107: 1092-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bali P, Pranpat M, Bradner JE, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90. J Biol Chem. 2005;280: 26729-26734. [DOI] [PubMed] [Google Scholar]