

Abstract
A broad spectrum of cytokines can activate the signal transducer and activator of transcription 5 (Stat5) by inducing a single tyrosine phosphorylation of the molecule. Although the process of Stat5 activation has been well studied, the mechanism by which it is inactivated is not fully understood. We demonstrate that the proteasome inhibitor MG132, but not the nuclear export inhibitor leptomycin B (LMB), stabilizes active nuclear Stat5A, whereas MG132 only partially stabilizes active cytoplasmic Stat5A. Importantly, ubiquitinated Stat5A is detected in the nucleus and the polyubiquitination of active Stat5A is K48 linked, a linkage type targeting proteins for degradation. Ubiquitination of Stat5A is recapitulated in a cell-free system, and Ubc5 is identified as the E2-conjugating enzyme for Stat5A ubiquitination. Interestingly, phosphorylation of Stat5A per se is not required for ubiquitination. Finally, C-terminal deletion analysis of Stat5A localizes the amphipathic region of amino acids 751-762 as a ubiquitination signal, possibly representing an E3 recognition motif. Taken together, these results demonstrate that the down-regulation of nuclear and cytoplasmic active Stat5A is differentially regulated. In the nucleus, ubiquitin/proteasome-mediated protein degradation is the dominant mechanism for the down-regulation of active Stat5A, whereas in the cytoplasm, protein tyrosine phasphatase is a major player in the down-regulation of active Stat5A.
Introduction
The signal transducer and activator of transcription (Stat) family of molecules play important roles in a variety of cytokine-mediated cellular responses, including cell growth, survival, differentiation, and function, by conveying signals from cytokine receptors to the nucleus and regulating cytokine-inducible gene expression.1-3 To date, 7 mammalian Stat family members have been identified, including Stat1, Stat2, Stat3, Stat4, Stat5A, Stat5B, and Stat6, and each Stat member has unique functions in cytokine signaling.3 The 2 closely related Stat5 proteins (Stat5A and Stat5B) have been of particular interest because of the broad spectrum of hematopoietic cytokines and growth factors that induce their activation.3 Gene disruptions in mice have highlighted important functions for Stat5 proteins. Stat5A deficiency disrupts prolactin-mediated functions in the lactating mammary gland,4,5 whereas Stat5B deficiency blocks growth hormone-regulated functions in the liver.5,6 Studies of Stat5A/5B nullizygous mice have illustrated key roles for Stat5A and Stat5B in prolactin-regulated ovarian function,5 repopulating potential of hematopoietic stem cells,7,8 interleukin-2 (IL-2)–induced T-cell proliferation,9 IL-3–mediated mast cell development and survival,10 and IL-5–driven differentiation of T helper 2 (Th2)–type eosinophils.11
Stat5 proteins, as other Stat members, exist normally as latent monomers in the cytoplasm, and activation of Stat5 is totally dependent upon phosphorylation of a single tyrosine residue by the Janus family of protein tyrosine kinases (Jaks).1,2 Upon cytokine binding, cytokine receptors aggregate and activate 1 or more members of the Jaks. Activated Jaks phosphorylate Stat5 proteins, which in turn dimerize, translocate to the nucleus, and activate transcription of a variety of genes.1-3 Activation of Stat5 is modulated in both magnitude and duration, such that normal function of Stat5 requires its proper activation and inactivation. Constitutive activation of Stat5 is frequently found in leukemic cells derived from BCR-ABL–induced chronic myelogenous and lymphogenous leukemia.12-15 A constitutively active Stat5A mutant is able to transform growth factor–dependent cells to growth factor independence,16 to induce a rapidly fatal myeloproliferative disease in mice,17 and to lead to an abnormal increase in numbers of T and B cells.18
Although much is known about Stat5 activation, the mechanism by which it is inactivated is not fully understood. Our recent studies have identified Shp-2, an Src homology 2 (SH2) domain–containing protein-tyrosine phosphatase, as a specific Stat5A phosphatase that dephosphorylates active Stat5A.19 Nonetheless, Shp-2 deficiency only delays dephosphorylation of Stat5A in the cytoplasm and has no effect on the down-regulation of active Stat5A in the nucleus.19 These observations suggest that additional phosphatases or alternative mechanisms to that of dephosphorylation are involved in turnover of Stat5. Previous studies have shown that proteasome inhibitors can dramatically stabilize tyrosinephosphorylated Stat5.20 However, there is no direct evidence to support a role for the proteasome in down-regulation of active Stat5.
In the current studies, we demonstrate that the mechanism involved in down-regulation of tyrosine-phosphorylated Stat5A in the nucleus differs from that in the cytoplasm, and exclusively depends on proteasome-mediated protein degradation instead of phosphatase-mediated dephosphorylation.
Materials and methods
Antibodies, cDNAs, and inhibitors
Anti–phospho-Stat5 monoclonal (30979345; ZYMED Laboratories, San Francisco, CA), anti-Stat5 monoclonal (610191; BD Biosciences, San Jose, CA), anti-Stat5A polyclonal (as previously described20), anti-Flag monoclonal (M2; Sigma-Aldrich, St Louis, MO), anti–G-6-PDH polyclonal (A9521; Sigma-Aldrich), anti-HA monoclonal (12CA5; Abcam, Cambridge, MA), anti-p53 monoclonal (Ab7; Calbiochem, San Diego, CA), anti-YY1 monoclonal (sc-7341; Santa Cruz Biotechnology, Santa Cruz, CA), anti–human Ubc5 polyclonal (A-615; Boston Biochem, Cambridge, MA), and antiubiquitin polyclonal21 antibodies were used for immunoprecipitation or Western blot analyses. The cDNAs encoding murine JAK2 and Stat5A were as previously described20 and cDNAs encoding HA-tagged wild-type ubiquitin, K48R ubiquitin, and K63R ubiquitin in mammalian expression vector pEF were kindly provided by Dr Zhijian Chen (University of Texas Southwestern Medical Center, Dallas). The proteasome inhibitor MG132 was purchased from the Peptide Institute (Louisville, KY) and the nuclear export inhibitor leptomycin B (LMB) and protease inhibitors NEM, TLCK, and TPCK were purchased from Sigma-Aldrich.
Cell culture and transfection
32D (EpoR wt) cells were cultured in RPMI-1640 containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin and supplemented with murine IL-3 (20 U/mL). Human Ubc5b was introduced into 32D (EpoR wt) cells via retrovirus-mediated gene transfer as previously described.22 Briefly, the human Ubc5b gene was cloned into a bicistronic retrovirus MSCV-IRES-GFP vector. The expression of the cloned gene and green fluorescent protein (GFP) is under murine stem cell virus (MSCV) promoter control. Conditioned media containing high-titer, amphotropic retrovirus particles were derived by cotransfection of 293T cells with the retrovirus vector expressing the cloned gene and GFP and a helper plasmid pEQPAM3 containing the required gag, pol, and env retroviral genes. This media was used to transduce 32D (EpoR wt) cells with 6 μg/mL polybrene (Sigma-Aldrich). Cells exhibiting high GFP expression were sorted and subsequently expanded as Ubc5b overexpressing cells.
COS-7 cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. For transfection in COS-7 cells, various combinations of cDNAs in mammalian expression vectors were transfected into COS-7 cells using LipoFectamine (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Cells were lysed in lysis buffer (10 mM HEPES [pH 7.9], 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 3 μg/mL aprotinin, 2 μg/mL pepstain, 1 μg/mL leupeptin, 5 mM NEM, 0.25 mM TPCK, and 0.25 mM TLCK) for analysis 48 hours after transfection.
RT-PCR
Total RNA was prepared from the cells using RNAzol B (Tel-Test, Friendswood, TX). First-strand cDNA was synthesized from total RNA with the Gene Amp reverse transcriptase–polymerase chain reaction (RT-PCR) kit (QIAGEN, Valencia, CA) and subjected to semiquantitative PCR analysis. The specific primers for Stat5 target gene Cis were as follows: 5′ primer, TGGAGCAGCTGTTGATGGACCTAC; 3′ primer, AGATGATGCTTTGACAGAAGGCTATC. The β-actin gene was used as an internal RNA level control and the specific primers for β-actin were: 5′ primer, ACTCCTATGTGGGTGACGAG; 3′ primer, CAGGTCCAGACGCAGGATGGC.
Preparation of cytoplasmic and nuclear extracts
32D (EpoR wt) cells were starved overnight and pretreated with or without LMB (5 ng/mL) for 60 minutes or MG132 (40 μM) for 30 minutes, followed by stimulation with IL-3 for 15 minutes. After removal of the cytokine, the cells were continually cultured in the presence or absence of LMB or MG132 as indicated. At the indicated time points, cells (2 × 107) were collected and suspended in 100 μL of cold buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 0.2 mM EDTA, 1 mM DTT, 3 μg/mL aprotinin, 2 μg/mL pepstain, and 1 μg/mL leupeptin). After incubation on ice for 15 minutes, NP-40 was added to a final concentration of 0.5%. The mixtures were vortexed for 10 seconds and spun at 15 700g for 30 seconds. The supernatants were collected as the cytoplasmic extracts. The pellets were washed with buffer A once and resuspended in 50 μL of buffer B (20 mM HEPES [pH 7.9], 400 mM NaCl, 2 mM EDTA, 1 mM DTT, 3 μg/mL aprotinin, 2 μg/mL pepstain, and 1 μg/mL leupeptin), followed by incubation on ice for 15 minutes. The mixtures were spun at 13 000 rpm for 5 minutes and the supernatants were collected as the nuclear extracts. In ubiquitination assays, the protease inhibitors NEM, TPCK, and TLCK were added to both the cytoplasmic and nuclear extracts.
Oligodeoxynucleotide pull-down
The Stat5 optimal DNA binding sequences were determined using STAT homodimers in binding and amplification reactions with random oligodeoxynucleotides as previously described.2 The oligodeoxynucleotides containing optimal Stat5-binding sequences (sense 5′-GATCCGAATTCCAGGAATTC A-3′ and antisense 3′-GCTTAAGGTCCTTAAGTCTAG-5′) were synthesized, conjugated to biotin, and annealed to form double strands. 32D (EpoR wt) cells (2 × 107) were lysed in lysis buffer (10 mM HEPES [pH 7.9], 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 3 μg/mL aprotinin, 2 μg/mL pepstain, and 1 μg/mL leupeptin) at 4°C. The cytoplasmic or nuclear extracts were precleared with Sepharose beads. Subsequently, the extracts were incubated with 5 μL of the biotin-labeled double-stranded oligodeoxynucleotides (20 μg/mL) at 4°C for 30 minutes, followed by incubation with avidin-conjugated Sepharose beads at 4°C for 2 hours. After washing 3 times with cold lysis buffer, the proteins bound to the avidin-conjugated Sepharose beads were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis.
In vitro ubiquitination assay
Ubiquitin was purchased from Sigma-Aldrich, purified to apparent homogeneity, and radioiodinated by the chloramine-T procedure to obtain 125I-ubiquitin.23 Human erythrocyte E1 enzyme and various recombinant E2s were purified to apparent homogeneity and functionally assayed as previously described.24 Active Stat5 proteins were pulled down from nuclear extracts or total cell lysates using Stat5-binding oligodeoxynucleotides as described in “Oligodeoxynucleotide pull-down.” Subsequently, the Stat5 proteins were incubated with 20 nM of E1, 400 nM of the indicated E2s, 5 μM of ubiquitin, or 125I-ubiquitin at 37°C for 45 minutes in the reaction buffer (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 1 mM dithiothreitol, 2 mM ATP, 10 mM creatine phosphate, 40 IU/mL creatine phosphokinase). The reaction products were subjected to SDS-PAGE, followed by Western blot analyses with antiubiquitin, anti-Stat5A, or anti-Flag antibodies, or by autoradiography.
Results
Down-regulation of tyrosine-phosphorylated Stat5A in the nucleus via proteasome-mediated protein degradation
Previous studies identified Shp-2 as a Stat5A phosphatase that specifically dephosphorylates tyrosine-phosphorylated Stat5A in the cytoplasm.19,25 However, in the absence of Shp-2, down-regulation of tyrosine-phosphorylated Stat5A that had translocated into the nucleus proceeded normally.19 Thus, the mechanism by which nuclear tyrosine-phosphorylated Stat5A is inactivated is unclear. To determine the inactivation mechanism of nuclear active Stat5A, we examined down-regulation of tyrosine-phosphorylated Stat5A in 32D (EpoR wt) cells, an IL-3–dependent myeloid cell line that also expresses wild-type erythropoietin receptors (EpoR wt). 32D (EpoR wt) cells were starved and stimulated with IL-3. At different time points following removal of IL-3, cytoplasmic and nuclear extracts were prepared and levels of tyrosine-phosphorylated and total Stat5A protein in the extracts were assessed by direct Western blot analysis. In the cytoplasm, tyrosine-phosphorylated Stat5A disappeared 2 hours after removal of the cytokine, whereas the amount of total Stat5A remained constant throughout the duration of the experiment (Figure 1A). In contrast, nuclear tyrosine–phosphorylated Stat5A and total Stat5A protein levels disappeared 2 hours after cytokine removal (Figure 1A). Therefore, inactivation of cytoplasmic tyrosine-phosphorylated Stat5A is mainly through dephosphorylation, consistent with the previous findings that Shp-2 specifically dephosphorylates Stat5A in the cytoplasm,19 whereas a different mechanism is involved in the down-regulation of nuclear tyrosine-phosphorylated Stat5A.
Figure 1.

Down-regulation of Stat5A in the cytoplasm and the nucleus. (A) No effect of the nuclear export inhibitor LMB on the down-regulation of active or total Stat5A proteins in the nucleus. 32D (EpoR wt) cells were starved overnight and pretreated with or without LMB, followed by stimulation with IL-3. After removal of the cytokine, cytoplasmic and nuclear extracts were prepared at the indicated time points and subjected to Western blot analyses with anti–phospho-Stat5 (α-pStat5), anti-Stat5A (α-Stat5A), or anti–nuclear protein YY1 (α-YY1) antibodies. (B) LMB treatment resulting in nuclear accumulation of p53. As a control, cytoplasmic and nuclear extracts from Hela cells treated with LMB were subjected to Western blot analyses with anti-p53 (α-p53), anti-YY1 (α-YY1), or anti–cytoplasmic protein G-6-PDH (α–G-6-PDH) antibodies.
Based on the disappearance of total Stat5A proteins in the nucleus, down-regulation of nuclear active Stat5A could arise by export to the cytoplasm, protein degradation, or both. To distinguish between the 2 possibilities, we first examined the effect of LMB, a specific inhibitor of nuclear export,26-28 on the turnover of nuclear Stat5A proteins. LMB binds to the receptor for the nuclear export signal (NES), named chromosome region maintenance 1 (CRM1/exportin 1), and inhibits nuclear export.26-28 Studies have shown that LMB is able to block nuclear export of Stat proteins.29-32 Starved 32D (EpoR wt) cells were pretreated with LMB, followed by stimulation with IL-3. After removal of the cytokine, levels of tyrosine-phosphorylated Stat5A and the amount of total Stat5A proteins in cytoplasmic and nuclear extracts were assessed. The rates of disappearance for both tyrosine-phosphorylated and total Stat5A proteins in the nucleus were comparable in the presence and absence of LMB (Figure 1A, left panel). In addition, LMB had no effect on the down-regulation of cytoplasmic tyrosine-phosphorylated Stat5A or on the constant level of total cytoplasmic Stat5A proteins (Figure 1A, right panel). As a control, LMB blocked nuclear export of p53, resulting in nuclear accumulation, as reported previously33 (Figure 1B). Therefore, these data preclude export as the mechanism for turnover of Stat5A proteins in the nucleus.
Our previous studies have shown that the MG132 proteasome inhibitor stabilizes tyrosine-phosphorylated Stat5A in total cell lysates,20 suggesting that protein degradation might be involved in the down-regulation of tyrosine-phosphorylated Stat5A. Next, we examined the effect of MG132 on the down-regulation of nuclear and cytoplasmic tyrosine-phosphorylated Stat5A. Starved 32D (EpoR wt) cells were pretreated with MG132, followed by stimulation with IL-3. At different time points after cytokine removal, nuclear and cytoplasmic levels of tyrosine-phosphorylated and total Stat5A protein pools were assessed. In the nucleus, MG132 completely stabilized tyrosine-phosphorylated Stat5A as well as total Stat5A proteins (Figure 2, left blot). In contrast, in the cytoplasm, MG132 only partially stabilized tyrosine-phosphorylated Stat5A (Figure 2, right blot), consistent with the notion that Shp-2 dephosphorylates Stat5A in the cytoplasm.19 These data indicate that proteasome-mediated protein degradation might regulate the turnover of tyrosine-phosphorylated Stat5A in a subcellular compartment–specific manner.
Figure 2.

Complete stabilization of tyrosine-phosphorylated Stat5A in the nucleus by MG132. 32D (EpoR wt) cells were starved overnight and pretreated with or without MG132, followed by stimulation with IL-3. After removal of the cytokine, cytoplasmic and nuclear extracts were prepared at the indicated time points and subjected to Western blot analysis with anti–phospho-Stat5 (α-pStat5), anti-Stat5A (α-Stat5A), or anti–nuclear protein YY1 (α-YY1) antibodies.
Ubiquitination of Stat5A in the nucleus
Although proteasome inhibitors stabilize the nuclear tyrosine-phosphorylated Stat5A, it is not clear whether proteasome-mediated protein degradation is directly or indirectly involved in the down-regulation of Stat5A activation. Proteins targeted for proteasome-mediated degradation usually undergo ubiquitination. To obtain evidence that the down-regulation of Stat5A in the nucleus is directly via proteasome-mediated protein degradation, we examined the ubiquitination of Stat5A in 32D (EpoR wt) cells. Initially, we failed to detect any ubiquitinated Stat5A by immunoprecipitating Stat5A, followed by Western blot with antiubiquitin antibodies, due to the high background caused by Stat5A antibodies (data not shown). To overcome the high background, we employed biotin-labeled double-stranded oligodeoxynucleotides containing the consensus Stat5-binding sequence to pull down tyrosine-phosphorylated Stat5. Starved 32D (EpoR wt) cells were stimulated with or without IL-3 in the presence or absence of MG132. The cytoplasmic and nuclear extracts were prepared and incubated with the biotin-labeled Stat5-binding oligodeoxynucleotides and avidin-conjugated Sepharose beads. Afterward, the bound proteins were eluted from the beads and subjected to Western blot analysis with either antiubiquitin or anti-Stat5A antibodies. Ubiquitinated Stat5 proteins were detected by antiubiquitin blot in nuclear extracts derived from cells treated with IL-3 plus MG132 (Figure 3, top blot, lane 8) and confirmed by anti-Stat5A blot (Figure 3, bottom blot, lane 8). The bands, indicated as ubiquitinated-Stat5A in the top panel, matched those in the bottom panel when the 2 panels were overlaid (Figure 3). Quantitation of these bands by densitometry demonstrated that ubiquitinated Stat5A represented approximately 3% of total pulled-down Stat5A proteins from 3 independent experiments (2.6% ± 0.7%, n = 3), which is consistent with previous findings that detectable ubiquitination levels of proteins targeted for proteasome-mediated degradation, including cyclin D1, were usually very low.34 The ubiquitinated Stat5A were not detected in the nuclear extracts derived from cells treated with or without IL-3 in the absence of MG132 or without IL-3 in the presence of MG132 (Figure 3). In contrast, no ubiquitinated Stat5A proteins were detected in the cytoplasmic extracts derived from cells treated with or without IL-3 in the presence or absence of MG132 (Figure 3). Although some ubiquitinated proteins appeared in the cytoplasmic extracts derived from cells treated with IL-3 in the presence of MG132, the anti-Stat5A blot revealed that these proteins were not Stat5A (Figure 3, bottom blot, lane 4). Therefore, these results demonstrate that tyrosine-phosphorylated Stat5A in the nucleus is ubiquitinated.
Figure 3.

Detection of ubiquitinated Stat5A in the nucleus. 32D (EpoR wt) cells were starved overnight, followed by stimulation with IL-3 in the presence or absence of MG132. Cytoplasmic and nuclear extracts were prepared from these cells. Subsequently, active Stat5 was pulled down from the extracts using the biotin-labeled double-stranded Stat5-binding oligodeoxynucleotides and subjected to Western blot analyses with antiubiquitin (α-Ub) or anti-Stat5A (α-Stat5A) antibodies.
K48-linked polyubiquitination of Stat5A
The type of ubiquitin linkage determines the fate of ubiquitinated proteins. K48-linked ubiquitination generally directs proteins to the proteasome for degradation, whereas K63-linked ubiquitination usually alters protein function or protein-protein interactions.35 To determine which kind of ubiquitin linkage is involved in the ubiquitination of Stat5, we examined the ability of HA-tagged wild-type ubiquitin (WT) or ubiquitin mutants, in which K48 or K63 was substituted with R (K48R or K63R, repectively; Figure 4A), to ubiquitinate Stat5A. Stat5A and Jak2 kinase were cotransfected with WT, K48R, or K63R ubiquitin into COS-7 cells. After transfection, the cells were lysed and the cell lysates were incubated with biotin-labeled Stat5-binding oligodeoxynucleotides and avidin-conjugated Sepharose beads. The bound proteins were subjected to Western blot analysis with antibodies against HA. The tyrosine-phosphorylated Stat5A proteins by Jak2 were pulled down by the Stat5-binding oligos and ubiquitinated by WT and K63R, but not K48R, ubiquitin (Figure 4B). These data demonstrate that tyrosine-phosphorylated Stat5A can be ubiquitinated and that the polyubiquitination of Stat5A is via the K48 linkage, a type that leads to protein degradation. This observation is consistent with the ability of MG132 to stabilize nuclear Stat5A.
Figure 4.
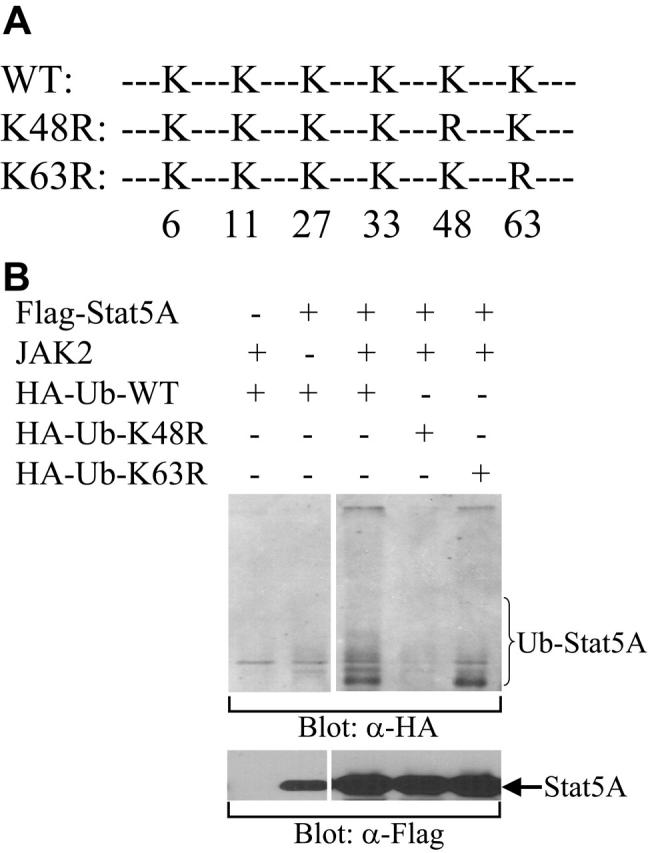
K48-linked polyubiquitination of Stat5A. (A) A schematic diagram of HA-tagged wild-type (WT) and mutant forms of ubiquitin, in which K to R substitution were introduced at position 48 (K48R) or 63 (K63R). (B) Ubiquitination of Stat5A by wild-type and K63R mutant, but not K48R mutant, ubiquitins. HA-tagged wild-type (HA-Ub-WT), K48R (HA-Ub-K48R), or K63R (HA-Ub-K63R) ubiquitin was cotransfected with Stat5A into COS-7 cells in the presence or absence of JAK2 kinase. Following transfection, Stat5 proteins were pulled down from cell lysates using biotin-labeled double-stranded Stat5-binding oligodeoxynucleotides and avidin-conjugated Sepharose beads. Bound proteins were subjected to Western blot analysis with anti-HA (α-HA) antibody.
Ubiquitination of Stat5A in a cell-free system and identification of Ubc5 as the conjugating enzyme (E2) for Stat5A ubiquitination
To further confirm the ubiquitination of Stat5A and to identify the enzyme(s) involved in Stat5A ubiquitination, we examined the ubiquitination of Stat5A in a cell-free system. Ubiquitination requires the sequential action of 3 enzymes, including an activating enzyme (E1) that activates ubiquitin, a conjugating enzyme (E2) that transiently carries the activated ubiquitin, and a ligase (E3) that transfers the activated ubiquitin from the E2 to the substrate.36 There is a single E1, a limited number of E2s, and a larger number of E3s in the entire ubiquitination system.37-39 Each E2 serves several E3s, and each E3 specifically recognizes a set of substrates.37-39 We detected ubiquitination of tyrosine-phosphorylated Stat5A in the nucleus of 32D (EpoR wt) cells. We believed that nuclear active Stat5A should form a complex with its E3 ligase. Therefore, we pulled down the tyrosine-phosphorylated Stat5A from the nuclear extracts of IL-3–activated 32D (EpoR wt) cells with the biotin-labeled Stat5-binding oligoes and avidin-conjugated Sepharose beads. After extensive washing, the beads were incubated with human E1 and a panel of different, recombinant E2 isoforms as well as I125-labeled ubiquitin. After reaction, the mixture was subjected to SDS-PAGE and autoradiography. Among the 6 different E2s we examined, Ubc5b, but not the other 5 E2s or the absence of E2, was able to promote ubiquitination of the nuclear Stat5 complex (Figure 5A).
Figure 5.

Ubiquitination of Stat5A in a cell-free system and identification of Ubc5 as the E2 for Stat5A ubiquitination. (A) Ubiquitination of Stat5A by Ubc5 and 125I-ubiquitin in a cell-free system. Active Stat5 proteins were pulled down from 32D (EpoR wt) cell nuclear extracts using biotin-labeled double-stranded Stat5-binding oligodeoxynucleotides and avidin-conjugated Sepharose beads. The beads were incubated with human E1, an E2 as indicated, and 125I-ubiquitin. Subsequently, the reaction products were subjected to SDS-PAGE, followed by autoradiography. (B) Ubiquitination of Stat5A by Ubc5 and nonradioactive ubiquitin in a cell-free system. The pulled down Stat5 proteins from panel A were incubated with human E1, an E2 as indicated, and nonradioactive ubiquitin. Subsequently, the reaction products were subjected to SDS-PAGE, followed by Western blot analyses with antiubiquitin (α-Ub) or anti-Stat5A (α-Stat5A) antibodies.
However, there was a possibility that Stat5 associated proteins, but not Stat5 itself, was ubiquitinated by the Ubc5-containing cell-free ubiquitination system. To exclude this possibility, we performed the same cell-free ubiquitination assay on the nuclear Stat5 complex with nonradioactive ubiquitins. Subsequently, the reaction mixture was subjected to SDS-PAGE, followed by Western blot analysis with either antiubiquitin or anti-Stat5A antibodies. Antiubiquitin antibodies detected the high-molecular-weight proteins, possibly polyubiquitinated Stat5, in the reaction mixture containing Ubc5b or Ubc5c, another member of Ubc5 E2 family, but not other types of E2s or no E2 (Figure 5B, top blot). Importantly, anti-Stat5A antibodies also detected the low-molecular-weight, less-ubiquitinated Stat5A in the reaction mixture containing Ubc5b or Ubc5c, but not other types of E2s or no E2 (Figure 5B, bottom blot). The apparent difference in relative distribution of Stat5-ubiquitin conjugate bands when detected by 125I-ubiquitin (Figure 5A) or antiubiquitin antibody (Figure 5B, top blot) versus anti-Stat5 antibody (Figure 5B, bottom blot) is a consequence of the stoichiometry of the ubiquitin adducts and their sensitivity of detection, a common observation in protein ubiquitination that has been described previously.21,40 When multiubiquitinated adducts are detected by either 125I-ubiquitin or antiubiquitin antibody, the sensitivity of detection increases with the addition of each ubiquitin moiety, tending to skew detection toward hyperubiquitinated species. In contrast, when ubiquitinated proteins are detected by monitoring the target protein, the sensitivity of detection directly reflects the stoichiometric abundance of target proteins that are ubiquitinated to various degrees, with hypoubiquitinated proteins being more abundant than hyperubiquitinated proteins. Similar differences in distribution were noted early in the ubiquitination field for 125I-ubiquitin-lysozyme versus ubiquitin-125I-lysozyme adducts.40 Taken together, these data demonstrate that nuclear tyrosine–phosphorylated Stat5A can be ubiquitinated in a cell-free system and that Ubc5 is the conjugating E2 for Stat5A ubiquitination.
Last, we examined whether overexpression of Ubc5 could affect activation of Stat5A and subsequent expression of its target genes. Although 32D (EpoR wt) cells had easily detectable levels of endogenous Ubc5, introduction of human Ubc5b further elevated levels of Ubc5 in these cells (Figure 6A). Nonetheless, overexpression of Ubc5b failed to affect IL-3–induced Stat5A activation (Figure 6B), down-regulation of Stat5A upon removal of IL-3 (Figure 6C), or induction of Stat5 target gene Cis (Figure 6D). Our findings that elevation of Ubc5 proteins over endogenous levels has no detectable effect on proteasome-mediated degradation of Stat5A suggest either that endogenous levels of Ubc5 proteins already saturate a rate-limiting amount of E2 or that E2 is not the rate-limiting factor for proteasome-mediated degradation of Stat5A.
Figure 6.
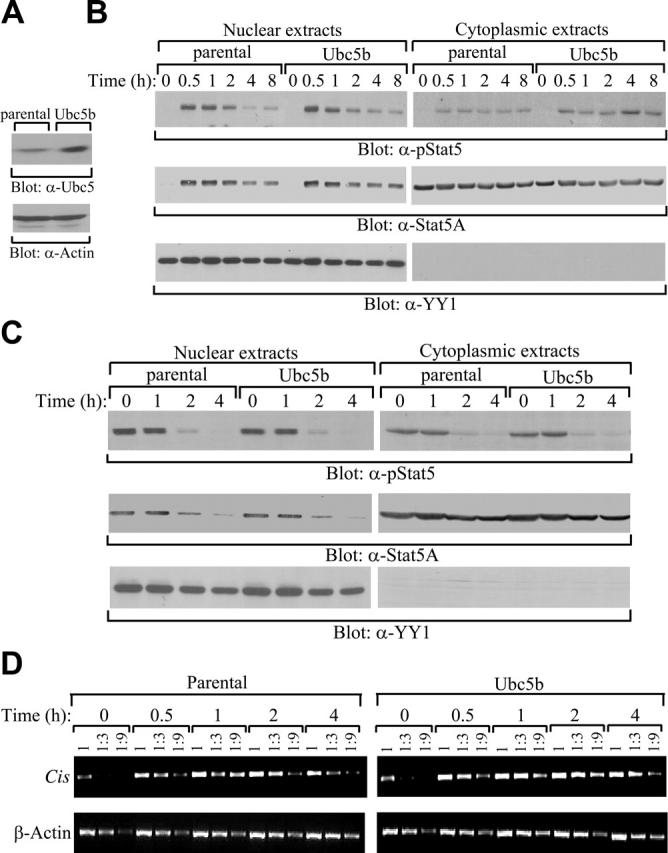
No detectable effect of overexpression of Ubc5 on activation of Stat5A and expression of Stat5 target gene. (A) Overexpression of Ubc5b in 32D (EpoR wt) cells. Cell lysates from parental 32D (EpoR wt) cells (parental) and 32D (EpoR wt) cells overexpressing human Ubc5b (Ubc5b) were subjected to direct Western blot analyses with anti-Ubc5b antibodies. (B) No effect of Ubc5b overexpression on activation of Stat5A. Parental 32D (EpoR wt) cells or 32D (EpoR wt) cells overexpressing human Ubc5b were starved overnight, followed by stimulation with IL-3. In the continuous presence of IL-3, cytoplasmic and nuclear extracts were prepared at the indicated time points and subjected to Western blot analyses with anti–phospho-Stat5 (α-pStat5), anti-Stat5A (α-Stat5A), or anti–nuclear protein YY1 (α-YY1) antibodies. (C) No effect of Ubc5b overexpression on down-regulation of Stat5A in the cytoplasm or the nucleus. Parental 32D (EpoR wt) cells or 32D (EpoR wt) cells overexpressing human Ubc5b were starved overnight, followed by stimulation with IL-3. After removal of the cytokine, cytoplasmic and nuclear extracts were prepared at the indicated time points and subjected to Western blot analyses with anti–phospho-Stat5 (α-pStat5), anti-Stat5A (α-Stat5A), or anti–nuclear protein YY1 (α-YY1) antibodies. (D) No effect of Ubc5b overexpression on expression of Stat5 target gene. Parental 32D (EpoR wt) cells or 32D (EpoR wt) cells overexpressing human Ubc5b were starved overnight, followed by stimulation with IL-3. At the indicated time points, total RNA and subsequent cDNA were prepared. Serial dilutions of the cDNA were subjected to PCR using primers designed to detect the Stat5 target gene, Cis. The β-actin gene was used as an internal RNA level control.
Phosphorylation of Stat5A per se is not required for its ubiquitination and identification of a potential amphipathic α-helical region within the Stat5 C-terminal domain as the Stat5A ubiquitination signal
Phosphorylation of the critical tyrosine residue 694 of Stat5A by Jaks is a prerequisite for Stat5A proteins to dimerize, translocate to the nucleus, and bind to DNA containing Stat5-binding sequences.1-3 Since ubiquitination of Stat5A mainly occurs in nucleus (Figure 3) and nonphosphorylated Stat5A monomers cannot enter the nucleus, it is reasonable to hypothesize that phosphorylation of Stat5A is required for its ubiquitination. Nonetheless, we further examined whether phosphorylation by itself is required for ubiquitination. Immunoprecipitation of nonphosphorylated Stat5A by anti-Stat5A antibodies, followed by in vitro ubiquitination assay and Western blot with antiubiquitin antibodies, generated high background, which prevented us from detection of ubiquitination of Stat5A. The biotin-labeled double-stranded Stat5-binding oligodeoxynucleotides could reduce the high background and enabled us to detect ubiquitination of tyrosine-phosphorylated Stat5A (Figure 3), but could not pull down nonphosphorylated Stat5. To overcome these obstacles, we examined ubiquitination of Stat5AY-F694, a Stat5A mutant with the Tyr-694 mutated to Phe. Flag-tagged Stat5AY-F694 (Stat5AY-F694FL) was coexpressed with Jak2 kinase in the presence or absence of wild-type Stat5A in COS-7 cells. As expected, Stat5AY-F694FL could not be tyrosine-phosphorylated by Jak2 with or without coexpression of wild-type Stat5A (Figure 7A, top blot). However, in the presence, but not absence, of wild-type Stat5A, Stat5AY-F694FL could form heterodimer with tyrosine-phosphorylated wild-type Stat5A and be pulled down by the biotin-labeled double-stranded Stat5-binding oligodeoxynucleotides (Figure 7A, bottom blot). As a control, Flag-tagged wild-type Stat5A (Stat5AFL) could be tyrosine-phosphorylated by Jak2 and pulled down by the Stat5-binding oligodeoxynucleotides (Figure 7A). Surprisingly, Stat5AY-F694FL pulled down by the Stat5-binding oligodeoxynucleotides was able to be ubiquitinated in vitro, similar to tyrosine-phosphorylated Stat5AFL (Figure 7B). These results demonstrate that phosphorylation is not required for ubiquitionation per se, although it is essential for Stat5 dimerization, nucleus translocation, and DNA binding.
Figure 7.

Ubiquitination of nonphosphorylated Stat5A in vitro and identification of the region between amino acid residues 751 and 762 within the Stat5A C-terminal domain as the Stat5A ubiquitination signal. (A) Nonphosphorylated Stat5AY-F694 forming a heterodimer with tyrosine-phosphorylated wild-type Stat5A. Flag-tagged wild-type Stat5A (Stat5AFL) or Flag-tagged mutant Stat5A with Tyr-694 mutated to Phe (Stat5AY-F694FL) was coexpressed with Jak2 in the presence or absence of wild-type Stat5A in COS-7 cells. Cell lysates were immunoprecipitated with anti-Flag antibodies. Precipitated proteins were subjected to Western blot analyses with anti–phospho-Stat5 (α-pStat5) (top blot) or anti-Flag (α-Flag) (middle blot) antibodies. Cell lysates were incubated with the biotin-labeled double-stranded Stat5-binding oligodeoxynucleotides and avidin-conjugated Sepharose beads. Bound proteins were subsequently subjected to Western blot analyses with anti-Flag antibodies (bottom blot). (B) Ubiquitination of nonphosphorylated Stat5AY-F694 in vitro. Stat5AFL or Stat5AY-F694FL was coexpressed with wild-type Stat5A and Jak2 kinase in COS-7 cells. Cell lysates were incubated with the biotin-labeled double-stranded Stat5-binding oligodeoxynucleotides and avidin-conjugated Sepharose beads. Bound proteins were subjected to cell-free ubiquitination in vitro and ubiquitinated forms (*) were identified as higher-molecular-weight species detected by Western blot analyses with anti-Flag antibodies. (C) A schematic diagram of N-terminal Flag-tagged full-length (Flag-Stat5A) and carboxyl-truncated Stat5A at amino acid 773 (Flag-Stat5AΔ773), 762 (Flag-Stat5AΔ762), 751 (Flag-Stat5AΔ751), or 740 (Flag-Stat5AΔ740). (D) The region between amino acid 751 and 762 within the Stat5A C-terminal domain in the control of Stat5A ubiquitination. 32D (EpoR wt) cells expressing N-terminal Flag-tagged wild-type (WT) or carboxyl-truncated Stat5A as shown in panel A (Δ773, Δ762, Δ751, or Δ740) were stimulated with IL-3. The wild-type or carboxyl-truncated Stat5A proteins were pulled down from cell lysates using biotin-labeled double-stranded Stat5-binding oligodeoxynucleotides and avidin-conjugated Sepharose beads. Following cell-free ubiquitination in vitro, N-terminal Flag-tagged wild-type or carboxyl-truncated Stat5A proteins were subjected to SDS-PAGE and ubiquitinated forms (*) were identified as higher-molecular-weight species detected by Western blot analyses with anti-Flag antibodies.
E3 ligase determines the substrate specificity through binding to specific ubiquitination signals and its interaction with the target protein is essential for target ubiquitination. Finally, we sought to define the ubiquitination signal of Stat5A that directs its ubiquitination. Previous studies have identified an amphipathic α-helical region between amino acids 751-762 within the Stat5A C-terminal domain that is involved in its down-regulation.20 Thus, we constructed a series of carboxyl-truncated Stat5A genes harboring an N-terminal Flag tag (Figure 7C) and established IL-3–dependent 32D cell lines that stably expressed each of these Stat5A mutants separately. The starved cell lines were stimulated with IL-3. After removing the cytokine, the nuclear active wild-type Stat5A or carboxyl-truncated Stat5A proteins were isolated with the biotin-labeled Stat5-binding oligodeoxynucleotides and avidin-conjugated Sepharose beads. Following the in vitro cell-free ubiquitination assay with exogenous E1 and the E2, Ubc5b, the reaction mixtures were subjected to Western blot analysis with anti-Flag antibodies. The tyrosine-phosphorylated Stat5A proteins with deletions to amino acids 773 or 762 were able to be ubiquitinated in the cell-free ubiquitination system comparable with the tyrosine-phosphorylated full-length wild-type Stat5A protein (Figure 7D). However, the tyrosine-phosphorylated Stat5A proteins with deletions to amino acid 751 or 740 failed to be ubiquitinated (Figure 7D). These results demonstrate that the predicted amphipathic α-helical region between residues 751-76241,42 with no lysine residue directs the ubiquitination of Stat5A. Therefore, these findings provide a mechanism for how this region controls the stability of tyrosine-phosphorylated Stat5A, a phenomenon observed in previous studies.20
Discussion
In the present studies, we have discovered different mechanisms for down-regulation of active Stat5A in the nucleus versus the cytoplasm. In the nucleus, proteasome-mediated protein degradation is the dominant mechanism, evidenced by disappearance of nuclear Stat5A protein with time after cytokine withdrawal, stabilization of nuclear active Stat5A by proteasome inhibitor MG132 but not nuclear export inhibitor LMB, and detection of ubiquitinated nuclear active Stat5A. In addition, polyubiquitination of tyrosine-phosphorylated Stat5A is via a K48 linkage, a type targeting proteins for degradation, and can be recapitulated in a cell-free system. Moreover, we have identified Ubc5 as the E2 for Stat5A ubiquitination and the potential amphipathic α-helical region between amino acid residues 751 and 762 within the Stat5A C-terminal domain as a ubiquitination signal that controls Stat5A ubiquitination.
Although previous studies have shown that proteasome inhibitors can dramatically stabilize tyrosine-phosphorylated Stat5A, they have failed to demonstrate either a significant reduction of Stat5A protein levels or ubiquitination of Stat5A.20 Until the present studies, there have been no direct evidences to support protein degradation as the mechanism for the down-regulation of active Stat5A. One of the reasons might be that previous studies analyze the down-regulation of total active Stat5A in the whole-cell lysates, which include both nuclear and cytoplasmic Stat5A.20 In fact, the down-regulation of cytoplasmic active Stat5A is mainly via Shp-2–mediated dephosphorylation.19 Thus, the inclusion of cytoplasmic Stat5A in the analysis might have hindered detection of degradation and ubiquitination of nuclear tyrosine–phosphorylated Stat5A. In addition, the high background caused by the immunoprecitation antibodies in Western blot analysis and the presence of large amounts of nonphosphorylated Stat5A in the immunoprecitates complicates detection of ubiquitinated nuclear Stat5A. In the current studies, important factors that may have contributed to our successful detection of ubiquitinated Stat5A include the separate analyses of nuclear and cytoplasmic active Stat5A and the use of biotin-labeled Stat5-binding oligodeoxynucleotides, which specifically pull down tyrosine-phosphorylated Stat5. These important factors may have facilitated discovery of the ubiquitin/proteasome-mediated down-regulation of nuclear active Stat5A.
The ubiquitin-proteasome system exists in both the cytoplasm and the nucleus.43 Precedence for nuclear proteasome–mediated down-regulation of several other transcription factors, including Smad2, MyoD, and the dioxin receptor, has been established.44-46 These transcription factors are ubiquitinated and degraded in the nucleus. Interestingly, another group of proteins, including cyclin D1,47 p53,33 p27kip1,48 IκBα,49 and activated Smad3,50 are ubiquitinated in the nucleus but are exported to the cytoplasm for degradation by the ubiquitin-proteasome system. The lack of an effect of the nuclear export inhibitor LMB on the turnover of the nuclear active Stat5A clearly demonstrates that Stat5A is ubiquitinated and degraded in the nucleus without cytoplasmic export, thereby adding Stat5A to the list of transcription factors that undergo proteasome-mediated degradation in the nucleus. However, the partial stabilization of cytoplasmic active Stat5A by MG132 suggests that some portion of the cytoplasmic active Stat5A is also down-regulated via proteasome-mediated protein degradation, although it is difficult to detect ubiquitinated-Stat5A in cytoplasm.
The E3 ligases determine the substrate specificity for target proteins via an E3 recognition motif, a trans-acting element on target proteins specifically interacting with its cognate E3 ligase.37,39 The finding that tyrosine-phosphorylated Stat5A proteins with deletion to amino acid 762, but not to amino acid 751, were able to be ubiquitinated demonstrates that the small region between amino acids 751 and 762 is involved in the ubiquitination of Stat5A. Noteworthy, the entire carboxy terminal domain of Stat5A, from amino acids 701 to 794, has no lysine residues, the canonical site for ubiquitination. Thus, this small region cannot be ubiquitinated per se, but likely interacts in trans with a putative Stat5A-specific E3 ligase and functions as an E3 recognition motif to direct Stat5A ubiquitination. It is well known that E3 ligase recognition motif can be distant from sites of ubiquitination.37,39 The exact ubiquitination site(s) of Stat5A has yet to be determined; however, there are a number of lysine residues distantly upstream of the small region.
The region between residues 751 and 762 of Stat5A, which is predicted to adopt an amphipathic α-helical structure characteristic of a transcriptional activation domain, has previously been shown to indeed function as a transcriptional activation domain.20 The ability of a single region to direct both transcription activation and proteasome-mediated degradation has been described for several transcription factors, including c-Myc, p53, c-Fos, c-Jun, and VP16.51,52 Nevertheless, the functional relationship between ubiquitination and transcription activation differs for different transcription factors. In the case of VP16, ubiquitination of its transcription activation domain signals both transcription activation and proteasome-mediated degradation.53 In contrast, the sequence that directs ubiquitination of c-Jun simply overlaps with the transcription activation sequence within this transcription factor.54 The exact correlation between the Stat5A transcription activity and proteasome-dependent degradation and whether the 2 functions require the same interactions remain to be evaluated. It is possible that this small region recruits a transcriptional complex that contains the Stat5A-specific E3 ligase, initiating ubiquitination of the tyrosine-phosphorylated Stat5A proteins. It is equally possible that this region independently recruits 2 protein complexes, 1 that mediates transcriptional activation, and 1 that mediates ubiquitin-dependent degradation. Purification of the proteins that associate with this small region of Stat5A will likely distinguish these 2 possibilities and would also help to identify the Stat5A E3 ligase.
Previous studies have shown that diverse mechanisms are involved in the turnover of different tyrosine-phosphorylated Stat molecules.20 Proteasome inhibitors have no effect on the down-regulation of the tyrosine-phosphorylated forms of Stat1, Stat2, or Stat3, whereas they clearly stabilizes the tyrosine-phosphorylated forms of Stat4, Stat5, and Stat6.20 The discovery in the present studies of ubiquitin-mediated protein degradation of the nuclear tyrosine–phosphorylated Stat5A, as well as the recent finding that phosphatase TC-PTP is able to dephosphorylate active Stat1 but not Stat5 in the nucleus,55 support the notion that protein phosphatases are responsible for the down-regulation of Stat1, Stat2, and Stat3, whereas proteasome-dependent protein degradation is involved in the turnover of Stat4, Stat5, and Stat6. Importantly, recent studies have identified a nuclear E3 ligase, SLIM, for Stat4.56 Interestingly, the same studies have shown that SLIM might also function as an E3 ligase for Stat1.56 Although dephosphorylation could be a major way to down-regulate tyrosine-phosphorylated Stat1 molecules, it is possible that proteasome-mediated proteolysis might also be involved in the down-regulation. Consequently, although we believe that tyrosine-phosphorylated Stat5A is down-regulated in the nucleus mainly by proteasome-mediated degradation, the possibility that some dephosphorylation of Stat5A may occur in the nucleus cannot be excluded. Nonetheless, identification of the E3 ligase for Stat5A would greatly facilitate studies of Stat5 down-regulation. In addition, the proteasome inhibitor–induced active Stat5A stabilization can be transferred onto Stat1 simply by replacing its carboxyl-terminus with that of Stat5A,20 indicating that the carboxyl-domains of different Stats might determine the mechanism by which active Stats are down-regulated. Previous studies have identified a 38–amino acid region of the carboxyl domain of Stat6 that determines the stability of its tyrosine-phosphorylated forms.57 This region does not contain a predicted amphipathic α-helical region and does not have sequence homology with the carboxyl domain of Stat5A. Therefore, it does not appear that a shared domain, by sequence or predicted structure, can be associated with a comparable functional property. Correspondingly, the carboxyl domains of Stat proteins, including Stat4, Stat5, and Stat6, are so diverse that no region with sequence or predicted structural similarity has been identified. Nonetheless, the physiologic significance of using different ways to down-regulate Stat proteins has yet to be determined.
Acknowledgments
We thank Debra K. Newman for critical review of this manuscript and for helpful discussion. We gratefully acknowledge the technical support of Shoua Yang and Guoping Fu.
Prepublished online as Blood First Edition Paper, March 28, 2006; DOI 10.1182/blood-2005-12-4777.
Supported by grants from the National Institute of Health (RO1 AI52327 to R.W.; R01 GM34009 to A.L.H.; and R01 HL073284 to D.W.) and by the American Cancer Society (RSG CCG-106204 to D.W.).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Darnell JE Jr. STATs and gene regulation. Science. 1997;277: 1630-1635. [DOI] [PubMed] [Google Scholar]
- 2.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84: 331-334. [DOI] [PubMed] [Google Scholar]
- 3.Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13: 211-217. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11: 179-186. [DOI] [PubMed] [Google Scholar]
- 5.Teglund S, McKay C, Schuetz E, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93: 841-850. [DOI] [PubMed] [Google Scholar]
- 6.Udy GB, Towers RP, Snell RG, et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A. 1997;94: 7239-7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradley HL, Hawley TS, Bunting KD. Cell intrinsic defects in cytokine responsiveness of STAT5-deficient hematopoietic stem cells. Blood. 2002;100: 3983-3989. [DOI] [PubMed] [Google Scholar]
- 8.Bunting KD, Bradley HL, Hawley TS, Moriggl R, Sorrentino BP, Ihle JN. Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5. Blood. 2002;99: 479-487. [DOI] [PubMed] [Google Scholar]
- 9.Moriggl R, Topham DJ, Teglund S, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10: 249-259. [DOI] [PubMed] [Google Scholar]
- 10.Shelburne CP, McCoy ME, Piekorz R, et al. Stat5 expression is critical for mast cell development and survival. Blood. 2003;102: 1290-1297. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Chen L, Huang Z, et al. Cutting edge: IL-5 primes Th2 cytokine-producing capacity in eosinophils through a STAT5-dependent mechanism. J Immunol. 2004;173: 2918-2922. [DOI] [PubMed] [Google Scholar]
- 12.Danial NN, Pernis A, Rothman PB. Jak-STAT signaling induced by the v-abl oncogene. Science. 1995;269: 1875-1877. [DOI] [PubMed] [Google Scholar]
- 13.Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13: 247-254. [PubMed] [Google Scholar]
- 14.Weber-Nordt RM, Egen C, Wehinger J, et al. Constitutive activation of STAT proteins in primary lymphoid and myeloid leukemia cells and in Epstein-Barr virus (EBV)-related lymphoma cell lines. Blood. 1996;88: 809-816. [PubMed] [Google Scholar]
- 15.Takemoto S, Mulloy JC, Cereseto A, et al. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc Natl Acad Sci U S A. 1997;94: 13897-13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onishi M, Nosaka T, Misawa K, et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998;18: 3871-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwaller J, Parganas E, Wang D, et al. Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol Cell. 2000;6: 693-704. [DOI] [PubMed] [Google Scholar]
- 18.Burchill MA, Goetz CA, Prlic M, et al. Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: development of CD4+ CD25+ regulatory T cells versus CD8+ memory T cells. J Immunol. 2003;171: 5853-5864. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Wen R, Yang S, et al. Identification of Shp-2 as a Stat5A phosphatase. J Biol Chem. 2003;278: 16520-16527. [DOI] [PubMed] [Google Scholar]
- 20.Wang D, Moriggl R, Stravopodis D, et al. A small amphipathic alpha-helical region is required for transcriptional activities and proteasome-dependent turnover of the tyrosine-phosphorylated Stat5. EMBO J. 2000;19: 392-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haas AL, Bright PM. The immunochemical detection and quantitation of intracellular ubiquitin-protein conjugates. J Biol Chem. 1985;260: 12464-12473. [PubMed] [Google Scholar]
- 22.Wen R, Chen Y, Schuman J, et al. An important role of phospholipase Cgamma1 in pre-B-cell development and allelic exclusion. EMBO J. 2004;23: 4007-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haas AL, Rose IA. Hemin inhibits ATP-dependent ubiquitin-dependent proteolysis: role of hemin in regulating ubiquitin conjugate degradation. Proc Natl Acad Sci U S A. 1981;78: 6845-6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haas AL, Bright PM. The resolution and characterization of putative ubiquitin carrier protein isozymes from rabbit reticulocytes. J Biol Chem. 1988;263: 13258-13267. [PubMed] [Google Scholar]
- 25.Yu CL, Jin YJ, Burakoff SJ. Cytosolic tyrosine dephosphorylation of STAT5: potential role of SHP-2 in STAT5 regulation. J Biol Chem. 2000;275: 599-604. [DOI] [PubMed] [Google Scholar]
- 26.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90: 1051-1060. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda M, Asano S, Nakamura T, et al. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390: 308-311. [DOI] [PubMed] [Google Scholar]
- 28.Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278: 141-144. [DOI] [PubMed] [Google Scholar]
- 29.Begitt A, Meyer T, van Rossum M, Vinkemeier U. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiledcoil domain. Proc Natl Acad Sci U S A. 2000;97: 10418-10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharya S, Schindler C. Regulation of Stat3 nuclear export. J Clin Invest. 2003;111: 553-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banninger G, Reich NC. STAT2 nuclear trafficking. J Biol Chem. 2004;279: 39199-39206. [DOI] [PubMed] [Google Scholar]
- 32.Zeng R, Aoki Y, Yoshida M, Arai K, Watanabe S. Stat5B shuttles between cytoplasm and nucleus in a cytokine-dependent and -independent manner. J Immunol. 2002;168: 4567-4575. [DOI] [PubMed] [Google Scholar]
- 33.Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18: 7288-7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11: 957-972. [DOI] [PubMed] [Google Scholar]
- 35.Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2: 169-178. [DOI] [PubMed] [Google Scholar]
- 36.Hershko A, Heller H, Elias S, Ciechanover A. Components of ubiquitin-protein ligase system: resolution, affinity purification, and role in protein breakdown. J Biol Chem. 1983;258: 8206-8214. [PubMed] [Google Scholar]
- 37.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70: 503-533. [DOI] [PubMed] [Google Scholar]
- 38.Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30: 405-439. [DOI] [PubMed] [Google Scholar]
- 39.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67: 425-479. [DOI] [PubMed] [Google Scholar]
- 40.Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A. 1980;77: 1783-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garnier J, Osguthorpe DJ, Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol. 1978;120: 97-120. [DOI] [PubMed] [Google Scholar]
- 42.Moriggl R, Gouilleux-Gruart V, Jahne R, et al. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol. 1996;16: 5691-5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rivett AJ. Intracellular distribution of proteasomes. Curr Opin Immunol. 1998;10: 110-114. [DOI] [PubMed] [Google Scholar]
- 44.Lo RS, Massague J. Ubiquitin-dependent degradation of TGF-beta-activated smad2. Nat Cell Biol. 1999;1: 472-478. [DOI] [PubMed] [Google Scholar]
- 45.Roberts BJ, Whitelaw ML. Degradation of the basic helix-loop-helix/Per-ARNT-Sim homology domain dioxin receptor via the ubiquitin/proteasome pathway. J Biol Chem. 1999;274: 36351-36356. [DOI] [PubMed] [Google Scholar]
- 46.Floyd ZE, Trausch-Azar JS, Reinstein E, Ciechanover A, Schwartz AL. The nuclear ubiquitin-proteasome system degrades MyoD. J Biol Chem. 2001;276: 22468-22475. [DOI] [PubMed] [Google Scholar]
- 47.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12: 3499-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398: 160-165. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez MS, Thompson J, Hay RT, Dargemont C. Nuclear retention of IkappaBalpha protects it from signal-induced degradation and inhibits nuclear factor kappaB transcriptional activation. J Biol Chem. 1999;274: 9108-9115. [DOI] [PubMed] [Google Scholar]
- 50.Fukuchi M, Imamura T, Chiba T, et al. Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of ROC1 and associated proteins. Mol Biol Cell. 2001;12: 1431-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas D, Tyers M. Transcriptional regulation: kamikaze activators. Curr Biol. 2000;10: R341-R343. [DOI] [PubMed] [Google Scholar]
- 52.Salghetti SE, Muratani M, Wijnen H, Futcher B, Tansey WP. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc Natl Acad Sci U S A. 2000;97: 3118-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salghetti SE, Caudy AA, Chenoweth JG, Tansey WP. Regulation of transcriptional activation domain function by ubiquitin. Science. 2001;293: 1651-1653. [DOI] [PubMed] [Google Scholar]
- 54.Musti AM, Treier M, Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275: 400-402. [DOI] [PubMed] [Google Scholar]
- 55.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22: 5662-5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanaka T, Soriano MA, Grusby MJ. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity. 2005;22: 729-736. [DOI] [PubMed] [Google Scholar]
- 57.Moriggl R, Berchtold S, Friedrich K, et al. Comparison of the transactivation domains of Stat5 and Stat6 in lymphoid cells and mammary epithelial cells. Mol Cell Biol. 1997;17: 3663-3678. [DOI] [PMC free article] [PubMed] [Google Scholar]