

Abstract
To determine whether the PI3K/Akt signaling pathway is involved in the pathogenesis of mantle cell lymphoma (MCL), we investigated the phosphorylation status of Akt and multiple downstream targets in primary MCL cases and cell lines. Akt was phosphorylated in 12 of 12 aggressive blastoid MCL variants and in 4 of 4 MCL cell lines. In contrast, phosphorylated Akt was present in only 5 of 16 typical MCL, 3 at comparable levels to the blastoid cases, and 2 at low levels. The presence of p-Akt was accompanied by the phosphorylation of p27kip1, FRKHL-1, MDM2, Bad, mTOR, and p70S6K. Inhibition of the PI3K/Akt pathway in the MCL cell lines abrogated or reduced the phosphorylation of Akt, p27kip1, FRKHL-1, MDM2, Bad, mTOR, GSK-3β, IκB, and led to cell-cycle arrest and apoptosis. Six MCL cases (5 with activated Akt and 1 with inactive Akt) and 3 of 4 cell lines showed loss of PTEN expression. PIK3CA mutations were not detected. We conclude that constitutive activation of the PI3K/Akt pathway contributes to the pathogenesis of MCL and preferentially occurs in blastoid variants. One possible mechanism of activation is loss of PTEN expression. These data suggest that PI3K/Akt inhibitors may be effective in the treatment of Akt-activated MCL.
Introduction
Mantle cell lymphoma (MCL) is a distinct subtype of B-cell lymphoma composed of small- to medium-sized lymphoid cells, which are believed to originate from follicle mantle B cells.1 This lymphoma is characterized by the t(11;14)(q13;q32) translocation, which results in deregulated aberrant expression of cyclin D1.2,3 MCL is generally incurable, and patients have a poor prognosis with a median survival of 3 to 5 years.4,5 Approximately 20% of MCL cases show an increase in nuclear size and pleomorphism with a higher mitotic rate and are classified as blastoid variants by hematopathologists.6 These cases have a more aggressive clinical course than typical MCL, often have aneuploid or tetraploid karyotypes, and frequently possess additional genetic changes affecting critical cell-cycle control genes, including mutations and deletions of p53 and p16.6-10
Recent gene profiling studies suggest that MCL is a transcriptionally homogeneous entity and that survival can be predicted based upon a set of proliferation-related genes.11 While many individual genes involved in signaling pathways that affect cellular proliferation have been identified as overexpressed or underexpressed in MCL cases, functional evidence supporting the significance of specific gene overexpression has been quite limited.12-14
Among the overexpressed genes identified in 2 different gene expression studies of MCL are several members of the phosphatidylinositol 3-kinase (PI3K) Akt signaling pathway, including PIK3CA, Akt-1, PDK-1, and PPP1R2.13,15 This signaling pathway is involved in the transduction of extracellular stimuli that regulate fundamental cellular processes including cell-cycle progression, proliferation and cell growth, apoptosis, and survival.16,17 PI3K functions as a proximal transducer for a variety of cell-surface receptors, particularly receptor tyrosine kinases, by increasing cellular levels of the membrane lipid phospho-inositol(3,4,5)P3 (PIP3) from phospho-inositol(4,5)P2 (PIP2). The levels of PIP3 are negatively controlled by several phosphatases, including phosphatase and tensin homolog deleted on chromosome 10 (PTEN), which converts PIP3 back to PIP2. PIP3 binds protein pleckstrin homology (PH) domains facilitating protein-protein interactions.
The serine/threonine kinase Akt/protein kinase B (PKB) occupies a central position in this pathway.18 Through its PH domain, Akt binds to PIP3, facilitating the activation of Akt by phosphoinositide-dependent kinase 1 (PDK1). Activated Akt has numerous targets that are important regulators of the cell cycle, the apoptotic pathway, and the translational and transcriptional machinery. Among these targets are the proapoptotic protein Bad, the cyclin-dependent kinase inhibitor p27kip1, several forkhead family members, the mammalian target of rapamycin (mTOR), glycogen synthase kinase-3-β (GSK-3β), and the IκB kinases.19-23
Given the critical role of the PI3K/Akt pathway in cell growth and homeostasis, it is not surprising that constitutive activation of the pathway contributes to the pathogenesis of many types of cancer, including breast, lung, prostate, brain, and endometrial cancer.24 Loss of expression of negative regulators of this pathway, such as the serine phosphatase PTEN, or activating mutations of components of this pathway, such as PI3K itself, have been implicated in a wide range of human cancers. To date, activation of the PI3K/Akt pathway in lymphomas has been described only in nucleophosmin-anaplastic lymphoma kinase-positive (NPM-ALK+) anaplastic large cell lymphoma, multiple myeloma, and Hodgkin lymphoma.25-27 The identification of overexpressed elements of the PI3K/Akt pathway in global gene expression studies of MCL cell lines prompted us to perform a careful analysis of the activation status of this important pathway to determine more precisely its role in the pathogenesis of MCL.
Materials and methods
Primary cases and cell lines
Thirty-one primary MCL cases (19 typical and 12 blastoid variants) were selected from the files of the Hematopathology Section, Laboratory of Pathology, National Cancer Institute, Bethesda, MD. All cases were reviewed by an expert hematopathologist (S.P.) and classified according to the World Health Organization classification28 as either typical MCL or blastoid variant. The latter comprises cases with blastic morphology as well as cases with more heterogeneous cytology, larger cells, and prominent nucleoli. To be included in this study, all cases were required to be positive for cyclin D1 and/or t(11;14) (by immunohistochemistry and/or cytogenetics) and to have a minimum of 75% tumor cells to facilitate interpretation of the results.
Four MCL cell lines (REC-1,29 Z138C,30 NCEB-1,31 and Granta 51932) and 3 non-MCL cell lines (BJAB, Raji, SU-DHL-1) were used in this study. REC-1, Z138C, NCEB-1, BJAB, Raji, and SU-DHL-1 were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Granta 519 was grown in Dulbecco modified Eagle medium supplemented with 10% FBS and 1% penicillin/streptomycin. The MCL cells lines have been extensively characterized, and all have abnormalities of the p16, p53, or/and ataxia telangiectasia mutated, and in this respect are genetically more closely representative of blastoid MCL cases than they are of typical MCL cases.33
PI3K/Akt inhibition studies
Two PI3K inhibitors, LY294002 and wortmannin, and one Akt inhibitor designated “Akt inhibitor” (1L-6-hydroxymethyl-chiro-inositol 2-[R]-2-O-methyl-3-O-octadecylcarbonate) were purchased from Calbiochem (San Diego, CA). Cells were incubated for 48 hours with increasing concentrations of each compound in a range individually chosen for each one, based on published literature.34-36 The final concentrations used were selected by their ability to abrogate the phosphorylation of Akt without decreasing the total Akt levels and were 20 μM for LY294002, 8 nM for wortmannin, and 40 μM for Akt inhibitor. For control experiments, the MAPK p42/44 inhibitor UO126 (Calbiochem) was used at a concentration of 8 μM.
Immunoreagents
For immunoblotting the following antibodies were used: cyclin D1 (DCS6; BD Bioscience, Franklin Lakes, NJ), PTEN (6H2.1; Cascade Bioscience, Winchester, MA), α-tubulin (Sigma Aldrich, St Louis, MI), actin (ACTN05; LabVision, Fremont, CA), Bcl-xL (2H12; Zymed, San Francisco, CA), p-p27 (Thr157) and p27 (kip1) (R&D Systems, Minneapolis MN), p-FKHRL-1 (Ser253) and oct-1 (Santa Cruz Biotechnology, CA); p-Akt (Ser473) and Akt, p-MDM2 (Ser166), Bad, p-Bad (Ser136), p-GSK-3β (Ser9), cleaved caspase 3, p-p65 (Ser536), p65 (NF-κB), p-IκBα (Ser32), p-mTOR (Ser2448), p-p70S6 (Thr389), p-S6K (Ser235/236), anti-mouse and anti-rabbit IgG horseradish peroxidase (HRP)-linked, from Cell Signaling (Beverly, MA).
Protein extraction
Five 20-μm sections of frozen tissue embedded in Tissue-Tek (Sakura Finetek, Torrance, CA) were immersed into 5-mL sonication buffer (phosphate-buffered saline [PBS], 1 × protease inhibitor cocktail I and II, 1 × phosphatase inhibitor [Calbiochem]) and centrifuged at 200g for 5 minutes. The supernatant was discarded, and the tissue pellet was processed in the same way as the cell pellets. Pellets were dissolved in lysis buffer (PBS, 1 × cell lysis buffer, 1 × protease inhibitor cocktail I and II, 1 × phosphatase inhibitor) and incubated for 30 minutes on ice. Subsequently, the lysates were centrifuged for 15 minutes at 1600 g at 4°C, and the supernatants were used for further analysis. Nuclear and cytoplasmic lysates were prepared using a nuclear extract kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. The effectiveness of the compartmentalization was assessed by Western blot analysis using α-tubulin as a cytoplasmic marker and oct-1 as a nuclear marker. Protein concentration was determined by the bovine serum albumin protein assay reagent kit (Pierce, Rockford, IL) according to the manufacturer's protocol.
Western blot analysis and immunoprecipitation
Twenty micrograms of protein was mixed with 2 × sample buffer (375 mM Tris[tris(hydroxymethyl)aminomethane]-Cl pH 6.8, 10% SDS [sodium dodecyl sulfate], 50% glycerol, 250 mM DTT [dithiothreitol], 0.05% bromophenol-blue) and heated for 5 minutes at 95°C. Proteins were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE, Invitrogen, Carlsbad, CA) and transferred to polyvinylidene-fluoride membranes (0.45 μm, Immobilon Millipore, Bedford, MA). Nonspecific binding sites were blocked by incubation with 5% (wt/vol) nonfat dry milk in TTBS (0.1% Triton X-100, 20 mM Tris, 136 mM NaCl at pH 7.6) for 1 hour. Subsequently, membranes were incubated with primary antibody (1:1000) for 12 to 14 hours at 4°C. After incubation for 1 hour with HRP-conjugated secondary antibody (1:2000) immunoreactivity was visualized with SuperSignal West Pico Chemiluminescent kit (Pierce) and exposed to Kodak X-OMAT MR film (Kodak, Rochester, NY) for 45 seconds. Signal intensity of each band was quantified using ImageQuant software (National Institutes of Health, Bethesda, MD), and the result was normalized to total protein expression measured by α-tubulin.
Immunoprecipitation studies were performed using 250 μg total protein in 250 μL lysis buffer. Lysates were incubated overnight at 4°C with anti-bcl-xL antibody at a concentration of 2.5 μg/mL. The immune complexes were collected with 100 μL protein-G-agarose (Upstate Biotechnology, Charlottesville, VA) and analyzed by SDS-PAGE as described.
Reverse-phase protein arrays
For generation of reverse-phase protein arrays (RPPA), cells or tissues were prepared identically as for Western blotting, except that a modified lysis buffer (Pink buffer) was used.37 Lysates were pipetted into 384-well microtiter plates with 10 two-fold serial dilutions (1:20-1:29) to ensure capturing the linear range of the signal-concentration curves for low- and high-abundance proteins. Samples were spotted on nitrocellulose-coated glass slides (Grace BioLabs, Bend, OR) using a solid-pin format robotic microarrayer (Aushon BioSystems, Lexington, MA). To monitor intraslide and interslide variation, a control sample (mixture of all samples on the array) was spotted in each field. Quality control studies of processed data showed mean intra-array and inter-array coefficients of variation of 9.2% and 13.5%, respectively. Prior to use, all antibodies were screened for specificity. After incubation with a specific primary antibody, detection was carried out on an automated immunostainer (DakoCytomation, Carpinteria, CA), using the DakoCytomation catalyzed signal amplification kit (CSA-I). Total protein staining was performed with colloidal gold (Bio-Rad, Hercules, CA). The gold-stained or immunostained arrays were scanned on an Epson Perfection 4870 flatbed scanner (Epson, Long Beach, CA), and the median pixel value was calculated using P-SCAN software (Peak quantification with statistical comparative analysis; http://abs.cit.nih.gov/pscan). The data were analyzed using a dose interpolation algorithm as described previously.37 Data were adjusted to total protein expression and were displayed in a log2 scale of the dilution series. Independent 2-tailed student t test was performed for MCL with phosphorylated Akt and MCL without phosphorylated Akt, using JMP 5.0 (SAS Institute, Cary, NC).
MTT test
Cell proliferation/viability was determined by MTT (3-[4,5-dimethylthiazol-2yl]-2,5-diphenoltetrazolium bromide)-Test (Sigma Aldrich). 1 × 105 cells were plated in 96-well cell-culture plates and incubated with or without kinase inhibitors for 8, 24, or 48 hours. MTT (5 mg/mL) was added to each well, and the optical density was measured at 570 nm using a Microelisa Autoreader MR580 (Dynatech, Lebanon, PA).
Cell-cycle analysis
For cell-cycle analysis, cells were fixed in 70% ethanol for 1 hour at 4°C and then incubated with 1 mg/mL RNase A for 30 minutes at 37°C. Subsequently, cells were stained with propidium iodide (50 μg/mL PI) (Becton Dickinson, San Jose, CA) in PBS, 0.5% Tween-20, and analyzed using a Becton Dickinson flow cytometer BD FACScan (San Jose, CA) and CellQuest acquisition and analysis programs. Gating was set to exclude cell debris, cell doublets, and cell clumps.
PI3KCA mutation analysis
Genomic DNA was extracted from frozen tissue or cell pellets with known tumor cell (> 75%) content by using standard phenol-chloroform methods. After polymerase chain reaction (PCR) amplification the DNA was separated by 1.5% TAE (tris[hydroxymethyl]-aminomethane-acetate-ethylenediaminetetraacetic acid)-agarose gel electrophoresis and extracted using a QIAquick gel extraction kit (Qiagen, Valencia, CA). Purified DNA was sequenced with the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA) following the manufacturer's supplied instructions. Sequencing was performed in sense and antisense directions.
The following primers were used: exon 9 initial PCR reaction forward 5′gtcttagattggttctttcctgtc3′ and reverse 5′atggcaaagaacacaaaagg3′; exon 20 initial PCR forward 5′tggggtaaagggaatcaaaag3′ and reverse 5′cctatgcaatcggtctttgc3′ (set1) and forward 5′ttgcatacattcgaaagacc3′ and reverse 5′ggggatttttgttttgttttg3′ (set2). Sequencing primers for exon 9 were forward 5′gtcttagattggttctttcctgtc3′ and reverse 5′ttgctttttctgtaaatcatctgtg3′, for exon 20 sequencing primers were identical to PCR primers. Data were analyzed with Sequencer 4.0.2 (Applied Biosystems) and BLAST software (National Center for Biotechnology Information, Bethesda, MD).
Results
The PI3K/Akt pathway is preferentially activated in the blastoid variant of MCL and in MCL cell lines
To determine the activation status of the PI3K/Akt pathway in MCL, we initially studied the phosphorylation of Akt in 19 typical MCL and 12 blastoid variants, as well as in 4 MCL cell lines: REC-1, Z138C, NCEB-1, and Granta 519. Western blot analysis revealed high levels of p-Akt in all 12 blastoid variants of MCL and in the 4 cell lines (Figure 1A). In contrast, p-Akt was detected in only 5 of 19 typical MCL cases, with high levels in 3 (cases 14, 16, 18) and lower levels in 2 (cases 24 and 27).
Figure 1.

Akt is preferentially activated in the blastoid variant of MCL and in MCL cell lines and is associated with phosphorylation of several downstream targets. (A) Western blot analysis for activated p-Akt (Ser473) in MCL cases and cell lines (Granta 519 [G], NCEB-1 [N], REC-1 [R], Z138C [Z]). High levels of p-Akt were detected in 12 of 12 blastoid MCLs (cases 1-12) and in the 4 cell lines, whereas only 5 of 19 typical MCLs displayed p-Akt—3 at high levels (cases 14, 16, and 18) and 2 at low levels (cases 24 and 27). Actin is shown as loading control. (B) Western blot analyses of downstream Akt targets shown for representative MCL cases, the Granta 519 cell line, and a follicular hyperplasia control (FHP). Blastoid variants (cases 1-4) and the Granta 519 cell line show high levels of p-Akt and multiple phosphorylated downstream targets (p-FRKHL-1, p-MDM2, p-p27kip1, p-Bad), compared with typical MCL (cases 22-24). The low activation of Akt in case 24 results in modest phosphorylation of the downstream targets. Note that GSK-3β, a classic Akt target, is phosphorylated in both p-Akt+ and p-Akt-cases. The negative control follicular hyperplasia (FHP) shows no activation of the Akt pathway. Cyclin D1 is strongly positive in all MCL cases. Actin shows equal loading of protein for each lane.
Regardless of MCL subtype, the presence of p-Akt was accompanied by the phosphorylation of multiple classic Akt targets, including the cell-cycle and apoptosis regulatory proteins p27kip1, FRKHL-1, MDM-2, and Bad. Cases with activated Akt had corresponding levels of the phosphorylated form of each of these target proteins, while cases without detectable p-Akt totally lacked the phosphorylated target (Figure 1B). Interestingly, Akt target GSK-3β was phosphorylated in both Akt-activated and Akt-inactive cases. This result suggests that other cellular kinases participate in the phosphorylation (inactivation) of GSK-3β in MCL.
To assess additional targets of Akt activation and to quantify the levels of the phosphorylated proteins, RPPA was used.38 This technology allows one to quantitate proteins of interest using nanograms of protein extract rather than the microgram quantities required for Western blot analysis, which enabled us to study several additional targets on our limited clinical material, including mTOR, p70S6K, and S6K. The RPPA data confirmed the results of the Western analysis of the initial targets and also showed strong correlations between p-Akt and the latter 3 targets (P values < .013 for all targets) (Table 1). GSK-3β showed a weaker, but still significant, correlation (P = .042). This result is consistent with the notion that while GSK-3β can be phosphorylated by cellular kinases other than Akt in MCL, activation of Akt still can influence the total levels of phosphorylated GSK-3β.
Table 1.
Relative expression levels of various downstream targets in Akt-active and Akt-inactivate MCL cases from RPPA assay
| Akt target | MCL with p-Akt, mean (σ)* | MCL without p-Akt, mean (σ)* | P† |
|---|---|---|---|
| P-MDM2 (Ser166) | 3.31 (0.29) | 1.17 (0.17) | < .001 |
| P-Bad (Ser136) | 5.0 (0.35) | 1.37 (0.37) | < .001 |
| P-FKHRL-1 (Ser253) | 4.4 (0.27) | 1.90 (0.32) | < .001 |
| P-GSK-3β (Ser9) | 4.57 (0.36) | 3.28 (0.36) | .042 |
| P-mTOR (Ser2448) | 5.03 (0.35) | 2.22 (0.56) | .001 |
| P-p70S6K (Thr389) | 2.61 (0.38) | 1.0 (0.10) | .013 |
| P-S6K (Ser235/236) | 4.09 (0.21) | 1.88 (0.38) | < .001 |
The mean expression levels for each target were calculated from 17 typical MCL and 12 blastoid variants
Mean expression levels (log2) with standard deviations calculated, as described in “Reverse-phase protein assays”
Independent 2-tailed Student t test performed for MCL cases with phosphorylated Akt versus MCL without phosphorylated Akt
Constitutive activation of Akt in MCL cell lines
Since Akt can be activated as a result of growth factors present in serum,39,40 we wished to determine whether phosphorylation of Akt in MCL was serum independent. The 4 MCL cell lines were grown in serum-free medium for 48 hours, harvested, and Western analysis for p-Akt was performed. No significant decrease in p-Akt protein levels was observed under these conditions in any of the 4 MCL cell lines (Figure 2), confirming the constitutive activation of Akt. In contrast, the presence of p-Akt in the Burkitt lymphoma cell line BJAB was strictly serum dependent.
Figure 2.

Akt is constitutively activated in MCL cell lines. Serum-depletion experiments were performed with all 4 MCL cell lines. Cells were grown for 48 hours with (FBS +) or without serum (FBS -). Western blot analysis with p-Akt under serum starvation shows no significant decrease of p-Akt in MCL cell lines (shown are Granta 519, NCEB-1, and Z138C). In contrast, p-Akt levels in the Burkitt cell line BJAB are dramatically decreased under serum-starvation conditions. Total Akt levels are shown as loading control.
Activation of Akt and downstream targets are dependent upon PI3K activity in MCL cell lines
Although the phosphorylation sites of the downstream targets studied are well-established Akt targets, it was possible that these proteins could have been activated through alternative kinases. We therefore studied the effect of 2 PI3K inhibitors, LY294002 and wortmannin, and one Akt inhibitor on the phosphorylation of Akt and downstream targets. Three different inhibitors were used to minimize the possibility of “off-target” effects.
Cells from the 4 MCL cell lines (REC-1, Granta 519, NCEB-1, and Z138C) were incubated with predetermined concentrations of the 3 inhibitors (as described in “Materials and methods”), and Western analysis for p-Akt and downstream targets were analyzed after 8, 24, and 48 hours. All 4 cell lines showed complete inhibition of the phosphorylation of Akt by 8 hours with each of the inhibitors, followed by the loss of phosphorylated Bad, p27kip1, FRKHL-1, and MDM-2 within 24 hours and a marked decrease in the levels of phosphorylated mTOR and its pathway targets p70S6K and S6K (Figure 3). In contrast, treatment of the cells with a MAPK p44/42 inhibitor under identical conditions had no effect on the phosphorylation status of Akt and selected targets (data not shown). These results indicate that activation of the PI3K/Akt pathway is necessary for phosphorylation of Akt and the downstream target proteins.
Figure 3.

Activation of Akt and downstream targets are dependent upon PI3K activity. PI3K/Akt pathway inhibition studies were performed with all 4 MCL cell lines using 3 different inhibitors: LY294002, wortmannin, and Akt inhibitor (Calbiochem). Comparable results were obtained for each line, and results with Granta 519 using LY294002 or Akt inhibitor are shown in representative experiments. Akt inactivation occurs after 8 hours (8 h), followed by the abrogation of phosphorylation of Bad, FRKHL-1, MDM2, and p27kip1 after 24 hours (24 h). Translational control proteins mTOR, p70S6K, and S6K show a time-dependent decrease in phospho-protein levels. S6K and total Akt levels are shown as loading control.
Inhibition of the PI3K/Akt pathway decreases activation of the NF-κB pathway
Constitutive activation of NF-κB has been reported in MCL cell lines, and treatment with proteasome inhibitors induces apoptosis, presumably by preventing the degradation of p-IκB.41,42 NF-κB is classically activated upon stimulation with proinflammatory cytokines such as tumor necrosis factor-α (TNF-α),43 however, PI3K/Akt also has been shown to activate the NF-κB pathway under certain circumstances.44,45 To assess the presence of cross talk between the PI3K/Akt pathway and the NF-κB pathway in MCL, we treated 2 of the cell lines (Granta 519 and Z138C) with the PI3K inhibitors and assessed several parameters of NF-κB activation. The use of LY294002, wortmannin, or Akt inhibitor equally decreased the activation status of the NF-κB pathway. Western blot analysis using an antibody that recognizes the serine-32 phosphorylated form of IκBα revealed a significant decrease in IκBα levels within 24 hours, and a loss of p65 phosphorylation (Figure 4A,C). This was accompanied by the accumulation of cytoplasmic p65 and a gradual decrease in nuclear p65, as shown in cytoplasmic and nuclear extracts at 4 time points (Figure 4B,D).
Figure 4.

Inhibition of the PI3K/Akt pathway decreases activation of the NF-κB pathway. PI3K/Akt pathway inhibition studies were performed with the Granta 519 and Z138C cell lines using 3 different inhibitors: LY294002, Akt inhibitor, and wortmannin. Comparable results were obtained for both lines, and results with Granta 519 using LY294002 or Akt inhibitor are shown as representative experiments. (A,C) Western blot analysis reveals Akt inactivation by 8 hours, and a time-dependent decrease in phosphorylated IκBα and p65. Total Akt and α-tubulin are shown as loading controls. (B,D) Western blot analysis of separated nuclear and cytoplasmic fractions reveals a gradual accumulation of the NF-κB p65 subunit in the cytoplasm following addition of LY294002 or Akt inhibitor and a corresponding reduction in nuclear NF-κB. α-tubulin is shown as loading control for cytoplasmic protein; oct-1 as loading control for nuclear protein.
Inhibition of the PI3K/Akt pathway induces apoptosis and suggests a Bad-mediated mechanism
Akt phosphorylation in response to PI3K activation has been shown to mediate antiapoptotic signaling. Indeed, after incubation with LY294002, wortmannin, or Akt inhibitor, Granta 519 and Z138C MCL cell lines underwent apoptosis. The percentage of apoptotic cells increased significantly over 2 days of treatment, with about 38% apoptotic cells observed after 48 hours of LY294002 treatment, and 28% observed after 48 hours of Akt-inhibitor treatment, as detected by flow cytometry (Figure 5A,C). This finding was accompanied by the appearance of the cleaved caspase-3 after 24 and 48 hours (Figure 5B,D).
Figure 5.

Inhibition of the PI3K/Akt pathway induces cell-cycle arrest and apoptosis. PI3K/Akt pathway inhibition studies were performed with 2 MCL cell lines (Granta 519 and Z138C) and a non-Akt-activated control cell line (RAJI) using 3 different inhibitors: LY294002, Akt inhibitor, and wortmannin. Comparable results were obtained for each MCL cell line, and results with Granta 519 using LY294002 and Akt inhibitor are shown as representative experiments in comparison to RAJI. (A,C) Cell-cycle analysis by flow cytometry indicates G1-S phase arrest in the MCL cell line Granta 519 at 24 hours and an increase in apoptotic cells after 48 hours of treatment. In contrast, the non-Akt-activated control cell line RAJI shows no significant alterations under treatment. (B,D) Western blot analysis shows a corresponding activation of pro-caspase-3 as assessed by appearance of the cleaved caspase-3, the loss of phosphorylated Bad, and the subsequent accumulation of Bad-bcl-xL complexes after 24 hours (Western blot preceded by immunoprecipitation [IP] with bcl-xL). Bcl-xL is shown as a loading control for the IP; α-tubulin for the standard Western.
Activated Akt has been shown to phosphorylate and inhibit the proapoptotic protein Bad. Therefore, we wondered if caspase activation might occur through a Bad-mediated pathway. The addition of the PI3K/Akt inhibitors resulted in decreased phosphorylation of Bad, as shown in Figure 5B and 5D. Furthermore, immunoprecipitation of treated cell lysates with anti-bcl-xL antibody showed a temporally related increase in Bad-bcl-xL complexes after 8, 24, and 48 hours (Figure 5B,D), consistent with inactivation of the antiapoptotic protein bcl-xL.
Inhibition of the PI3K/Akt pathway induces G1 arrest, inhibits proliferation, and leads to up-regulation of p27kip1 and loss of cyclin D1 expression
Inhibition of the PI3K/Akt pathway has been shown to induce G1 arrest in a variety of cancers through up-regulation of the cell-cycle inhibitor p27kip1.26,46 Treatment of Granta 519 and Z138C with LY294002, wortmannin, or Akt inhibitor resulted in a reduction of cell proliferation/viability by 50% after 48 hours as assessed using the MTT assay (Figure 6A,C). Cell-cycle analysis revealed that 90% of the viable cells had accumulated in the G1 fraction by 24 hours. G1 arrest was associated with a rapid decrease in phosphorylated p27kip1, which targets the protein for degradation, followed by an increase in total p27kip1 levels (Figure 6B,D). Accompanying the increase in total p27kip1 levels was the abrogation of phosphorylation of the forkhead transcription factor FRKHL-1, which is known to activate transcription of p27kip1.47,48 In addition to the up-regulation of p27kip1, we observed a dramatic down-regulation of cyclin D1 after 24 hours of inhibition treatment. Cyclin D1 nuclear export and degradation is regulated by phosphorylation by GSK-3β, which is inactivated when phosphorylated at serine 9.49 In proliferating cells GSK-3β was highly phosphorylated (inactive), and treatment with inhibitors resulted in dephosphorylation, and presumably activation, of GSK-3β as shown in Western analysis with the phosphor-site-specific antibody. Cdk4 and cyclin E protein levels remained stable during the time course.
Figure 6.
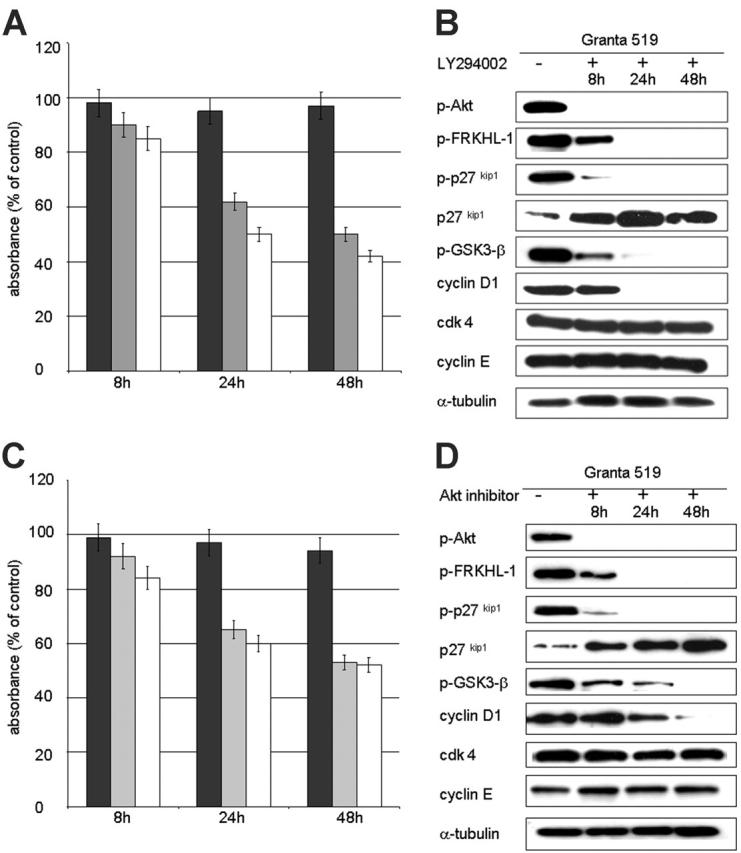
Inhibition of the PI3K/Akt pathway leads to up-regulation of p27kip1 and loss of cyclin D1 expression. PI3K/Akt pathway inhibition studies were performed with the 4 MCL cell lines and the non-Akt-activated control cell line RAJI using 3 different inhibitors: LY294002, wortmannin, and Akt inhibitor (Calbiochem). Comparable results were obtained for each MCL cell line, and results with Granta 519 using LY294002 or Akt inhibitor are shown as representative experiments. (A,C) Proliferation/viability as assessed by MTT test. Results are averages of 3 individual experiments and are displayed as percent absorbance of control cells (untreated) with Granta 519 (□), Z138C ( ), and RAJI (▪). A significant reduction in absorbance to 50% of control values after 48 hours is seen in the MCL cell lines, whereas the non-Akt-activated control RAJI shows no significant reduction. (B,D) Western blot analysis with Granta 519 demonstrates Akt inactivation at 8 hours, followed by abrogation of phosphorylation of FRKHL-1, p27kip1, and GSK-3β by 24 hours. The cell-cycle inhibitor p27kip1 shows a gradual increase in expression levels over time, and cyclin D1 is dramatically down-regulated. In contrast, cdk4 and cyclin E remain constant. α-tubulin expression is shown as loading control.
), and RAJI (▪). A significant reduction in absorbance to 50% of control values after 48 hours is seen in the MCL cell lines, whereas the non-Akt-activated control RAJI shows no significant reduction. (B,D) Western blot analysis with Granta 519 demonstrates Akt inactivation at 8 hours, followed by abrogation of phosphorylation of FRKHL-1, p27kip1, and GSK-3β by 24 hours. The cell-cycle inhibitor p27kip1 shows a gradual increase in expression levels over time, and cyclin D1 is dramatically down-regulated. In contrast, cdk4 and cyclin E remain constant. α-tubulin expression is shown as loading control.
Loss of PTEN, but not PIK3CA, mutations potentially contribute to the activation of Akt in MCL
To further study the mechanism underlying the activation of Akt in MCL, we analyzed PTEN protein expression levels and screened for somatic mutations within the helical and kinase domains (exons 9 and 20) of the PIK3CA gene. Loss of PTEN expression generally occurs as a result of mutation, deletion, or promoter methylation and has been shown to be a common abnormality in cancer and results in the activation of the PI3K/Akt pathway.50 Of the 4 MCL cell lines, 3 showed no expression of PTEN, and 6 of the 31 MCL cases showed markedly decreased expression (Figure 7). Of the 6 cases with decreased PTEN expression, 4 were blastoid variants with high Akt activation (cases 8, 10, 11, 12), 1 was a typical MCL with low Akt activation (case 24), and 1 case was a typical MCL without detectable Akt activation (case 25). These data raise the possibility that loss of PTEN may account for Akt activation in a subset of the Akt-activated MCL cases.
Figure 7.

Loss of PTEN contributes to the activation of Akt in a subset of cases. Western blot analysis for PTEN was performed for all MCL cases and cell lines. SUDHL-1 (S) served as positive control. PTEN expression was decreased in 5 of 17 (29%) cases with activated Akt, including 4 high-expressing blastoid MCL (cases 8, 10, 11, 12) and 1 low-expressing typical MCL (case 24). In contrast, 1 of 14 cases without detectable Akt activation (case 25) showed decreased expression of PTEN. Actin is shown as loading control.
Activating mutations of PI3K is a recently described mechanism by which pathway activation may occur.51 We therefore analyzed all 31 MCL cases and the 4 cell lines for somatic mutations of PIK3CA. Sequence analysis was performed to assess the 2 hotspot regions (exon 9 and exon 20), where more than 80% of the reported mutations reside. However, none of the MCL cases or cell lines were found to have activating somatic mutations (data not shown).
Discussion
The PI3K/Akt pathway is a major signaling pathway that plays a critical regulatory role in vital cellular processes controlling cell growth and cell death. It is therefore not surprising that it has been found to be activated in a high percentage of human malignancies, including colorectal, gastric, prostate, thyroid, endometrial, lung, brain, and breast cancer.51-53 There have been only a limited number of studies of lymphoid neoplasms, where the pathway has been reported to be activated in Hodgkin lymphoma, some non-Hodgkin lymphomas, and multiple myeloma.25-27,54 In this study, we demonstrate that the PI3K/Akt pathway is constitutively activated in a subset of MCL. This subset includes all of the aggressive blastoid MCL variants and a small subset of typical MCL cases. Akt and multiple downstream targets of the PI3K/Akt pathway were found phosphorylated at high levels in all 12 cases of blastoid MCL and in 4 MCL cell lines, whereas p-Akt was detected in only 5 of 19 typical cases, 3 at levels comparable to that of the blastoid cases, and 2 at lower levels. Because we did not have sufficient follow-up information on these latter 5 cases, it is not clear whether they constitute an aggressive subgroup within the typical MCL group. Our data are consistent with recent gene expression profiling and proteomic studies that have identified several overexpressed genes in the PI3K/Akt pathway in cases of MCL and MCL lymphoma cell lines.13,15
We found strong associations between the presence of activated Akt and the phosphorylation of multiple targets of the PI3K/Akt pathway in both the MCL cases and cell lines. The strongest correlations were found with MDM2 (Ser166), Bad (Ser136), FKHRL-1 (Ser253), and p27kip1 (Thr157). Phosphorylation of MDM2 by Akt promotes its nuclear localization where it antagonizes p53 activity.55 Phosphorylation of Bad and FKHRL-1 promotes their cytoplasmic retention and results in the inactivation of the proapoptotic activity of Bad20 and in decreased transcriptional activation of proapoptotic genes activated by FKHRL-1.47,56 FKHRL-1 not only functions as a proapoptotic transcription factor, but it also plays an important role in inhibiting cell-cycle progression and cell proliferation through transcriptional repression of cyclin D157 and transcriptional activation of p27kip1.47,48 Phosphorylation of p27kip1 by Akt at Thr 157 leads to its cytoplasmic retention and proteasomal degradation, further reducing the levels of this critical negative regulator of G1/S progression.21,58
Strong correlations between p-Akt and the presence of phosphorylated mTOR (Ser2448) and targets p70S6K (Thr389) and S6K (Ser235/236) also were identified, although these were somewhat weaker than the previously mentioned targets. While the PI3K/Akt pathway is a major activator of mTOR through its ability to inactivate the inhibitory tuberin-hamartin complex,59 mTOR and its targets also can be activated by Akt-independent nutrient sensing pathways as well.60,61 Interestingly, GSK-3β, a well-studied Akt target involved in the control of cyclin D1 levels, was expressed in both Akt-activated and Akt-inactive MCL and showed only a weak correlation with the presence of p-Akt in the RPPA assay, suggesting that other cellular kinases also are capable of phosphorylating GSK-3β in MCL. Nonetheless, the overall phosphorylation patterns in the MCL cases suggest that activated Akt affects multiple targets.
Inhibition studies in the cell lines provided additional insights into the activity of this pathway in MCL and further showed that the effects of blocking the pathway in the cell lines are catastrophic, resulting in a block in proliferation, G1/S cell-cycle arrest, and apoptosis. Not only did inhibition of the pathway result in abrogation of the phosphorylation of FKHRL-1 Bad, p27kip1, and MDM2, it also resulted in the complete reduction of cyclin D1 levels within 24 hours of treatment, with no effect on cyclin E or CDK4 levels. This was of particular interest to us because the primary oncogenic event in MCL is the deregulated transcription of cyclin D1 message caused by the t(11;14) translocation. To our knowledge, modulation of cyclin D1 levels as a result of pharmacologic manipulation of the PI3K/Akt pathway has not been reported previously in MCL and suggests a potential point of intervention in this disease.
The dramatic loss of cyclin D1 following Akt inhibition potentially could occur through the reactivation of the GSK-3β kinase, which targets cyclin D1 for degradation,62 and/or the inactivation of mTOR and its direct targets, which regulate the translation of cyclin D1 message. Inhibition of mTOR with rapamycin has been shown previously to result in a decrease of cyclin D1 protein levels in several solid cancer models,63,64 and it is possible that the inactivation of mTOR may play a major role in decreasing cyclin D1 in MCL as well. However, a recent study of 2 MCL cell lines failed to demonstrate any reduction in cyclin D1 levels, although rapamycin treatment was effective in inducing cell-cycle arrest and apoptosis.65 Thus, at this point it is unclear what the relative contributions of GSK-3β and mTOR are in reducing the cyclin D1 levels following their inhibition through the PI3K/Akt pathway.
As discussed, in the MCL cell lines, GSK-3β phosphorylation was completely abrogated by inhibition of the PI3K/Akt pathway. This is consistent with a large body of data indicating that Akt and/or other downstream kinases play a major role in regulating the activity of GSK-3β.66 However, unlike most of the Akt targets studied that showed no evidence of phosphorylation in the absence of activated Akt, phosphorylated GSK-3β was present in all primary MCL studied, although cases with activated Akt, as a group, had higher levels. These data suggest that in vivo, there are alternative mechanisms responsible for the phosphorylation of GSK-3β that are not activated in the cell culture environment. Therefore, it will be important to assess the response of GSK-3β and cyclin D1 in MCL cases to PI3K/Akt inhibitors in vivo.
MCL cells underwent apoptosis following treatment with the PI3K/Akt inhibitors. The increase in apoptotic fraction seen by flow cytometry over time was accompanied by the loss of Bad phosphorylation, the association of nonphosphorylated Bad with bcl-xL, and the activation of pro-caspase 3. The initiation of apoptosis following inhibition of the PI3K pathway has been linked previously to inactivation of antiapoptotic proteins such as bcl-xL, through complex formation with dephosphorylated Bad,67,68 and our data are consistent with this mechanism.
Studies of the MCL cell lines also showed that Akt can activate the NF-κB pathway. Following treatment with inhibitors, we observed an inactivation of the NF-κB pathway as assessed by loss of phosphorylation of IκBα and the subsequent gradual loss of nuclear p65 accompanied by the accumulation of cytoplasmic p65. These data indicate that the PI3K/Akt pathway is capable of modulating the NF-κB pathway under the cell-culture conditions of this study. In vivo, the environment is much more complex, and it is likely that Akt is but one of several modulators of the NF-κB pathway. Cross talk between the PI3K and NF-κB pathway has been reported previously in a number of well-studied systems.44,69
A variety of oncogenic events have been linked to activation of the PI3K/Akt pathway.70 These include: (1) activation of membrane growth receptors through mutation or gene amplification, (2) overexpression of the growth factors themselves, (3) amplification of the Akt gene, (4) activation of intracellular mediators through mutation or gene amplification, and (5) loss of negative regulators of Akt activity, such as PTEN. While we have not yet undertaken a comprehensive investigation of all of these, we have initiated studies into several possible mechanisms. We found that Akt remained activated in the MCL cell lines in the absence of serum, suggesting that the activity of the pathway is not due to exogenous growth factor stimulation of receptors. We also examined 2 of the more common mechanisms of Akt activation, PTEN loss and PI3K mutation, and found a possible role for PTEN. Loss of PTEN expression has been reported in a variety of cancers, including lung, breast, prostate, colon, endometrial, and glioblastoma,71-74 and has been shown to occur through mutation, deletion, and epigenetic mechanisms. There are relatively few studies exploring PTEN abnormalities in lymphomas.75-78 Several cases of MCL included in one of these studies were reported to lack genetic alterations; however, PTEN expression was not evaluated.78 In the current study, we found loss of PTEN expression in 5 of the 17 cases with activated Akt, while only 1 of 14 cases without p-Akt expression showed loss of expression. This finding suggests that PTEN inactivation may be more common than initially thought in MCL and may contribute to constitutive activation of the PI3K/Akt pathway in a percentage of these lymphomas. Further investigation of this gene, particularly in blastoid MCL cases and other lymphomas with activated Akt, is warranted.
Mutations of PIK3CA have been reported recently in a variety of cancers.51,79-82 These mutations result in the constitutive activation of PI3K and the consequent phosphorylation and activation of Akt.83 In our cases with activated Akt, we were not able to identify activating mutations within either of the 2 hotspot regions (exon 9 and exon 20) of the gene. It should be noted that enhanced PI3K activity also has been attributed to gene amplification,84 and we have not ruled out this possibility. There are clearly many other potential causes for Akt activation in the MCL cases, including gene amplification of Akt itself, activation/mutation of rat sarcoma virus (RAS) oncogene or other signaling transducers, and activation/mutation of a wide range of cellular receptors, and these will need to be more fully investigated.
In summary, we have found constitutive activation of the PI3K/Akt signaling pathway in a subset of MCL that includes all of the aggressive blastoid variants. This signaling results in the activation of a number of well-known target genes and pathways, including the NF-κB pathway, a second major signaling pathway postulated to be activated independently in MCL. These constitutively activated signaling cascades lead to enhanced survival and cell proliferation, and inhibition of apoptosis. Our data demonstrate the central role of this pathway in MCL survival and suggest that specific PI3K or Akt kinase inhibitors may prove useful in the treatment of Akt-activated MCL.
Acknowledgments
The authors thank John Austin and Peter Honkanen of Aushon Biosystems for expert technical assistance and Lynn Young for assisting in p-scan analysis.
Supported by the Intramural Research Program of the NCI, NIH.
Prepublished online as Blood First Edition Paper, April 27, 2006; DOI 10.1182/blood-2006-04-015586.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Raffeld M, Jaffe ES. bcl-1, t(11;14), and mantle cell derived neoplasms. Blood. 1991;78: 259-263. [PubMed] [Google Scholar]
- 2.Meeker TC, Sellers W, Harvey R, et al. Cloning of the t(11;14)(q13;q32) translocation breakpoints from two human leukemia cell lines. Leukemia. 1991;5: 733-737. [PubMed] [Google Scholar]
- 3.Tsujimoto Y, Jaffe E, Cossman J, Gorham J, Nowell PC, Croce CM. Clustering of breakpoints on chromosome 11 in human B-cell neoplasms with the t(11;14) chromosome translocation. Nature. 1985;315: 340-343. [DOI] [PubMed] [Google Scholar]
- 4.Bosch F, Lopez-Guillermo A, Campo E, et al. Mantle cell lymphoma: presenting features, response to therapy, and prognostic factors. Cancer. 1998;82: 567-575. [DOI] [PubMed] [Google Scholar]
- 5.Argatoff LH, Connors JM, Klasa RJ, Horsman DE, Gascoyne RD. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood. 1997;89: 2067-2078. [PubMed] [Google Scholar]
- 6.Bernard M, Gressin R, Lefrere F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia. 2001;15: 1785-1791. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez L, Fest T, Cazorla M, et al. p53 gene mutations and protein overexpression are associated with aggressive variants of mantle cell lymphomas. Blood. 1996;87: 3351-3359. [PubMed] [Google Scholar]
- 8.Louie DC, Offit K, Jaslow R, et al. p53 overexpression as a marker of poor prognosis in mantle cell lymphomas with t(11;14)(q13;q32). Blood. 1995;86: 2892-2899. [PubMed] [Google Scholar]
- 9.Pinyol M, Cobo F, Bea S, et al. p16(INK4a) gene inactivation by deletions, mutations, and hypermethylation is associated with transformed and aggressive variants of non-Hodgkin's lymphomas. Blood. 1998;91: 2977-2984. [PubMed] [Google Scholar]
- 10.Ott G, Kalla J, Ott MM, et al. Blastoid variants of mantle cell lymphoma: frequent bcl-1 rearrangements at the major translocation cluster region and tetraploid chromosome clones. Blood. 1997; 89: 1421-1429. [PubMed] [Google Scholar]
- 11.Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3: 185-197. [DOI] [PubMed] [Google Scholar]
- 12.De Vos S, Krug U, Hofmann WK, et al. Cell cycle alterations in the blastoid variant of mantle cell lymphoma (MCL-BV) as detected by gene expression profiling of mantle cell lymphoma (MCL) and MCL-BV. Diagn Mol Pathol. 2003;12: 35-43. [DOI] [PubMed] [Google Scholar]
- 13.Rizzatti EG, Falcao RP, Panepucci RA, et al. Gene expression profiling of mantle cell lymphoma cells reveals aberrant expression of genes from the PI3K-AKT, WNT and TGFbeta signalling pathways. Br J Haematol. 2005;130: 516-526. [DOI] [PubMed] [Google Scholar]
- 14.Hofmann WK, de Vos S, Tsukasaki K, et al. Altered apoptosis pathways in mantle cell lymphoma detected by oligonucleotide microarray. Blood. 2001;98: 787-794. [DOI] [PubMed] [Google Scholar]
- 15.Ghobrial IM, McCormick DJ, Kaufmann SH, et al. Proteomic analysis of mantle-cell lymphoma by protein microarray. Blood. 2005;105: 3722-3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296: 1655-1657. [DOI] [PubMed] [Google Scholar]
- 17.Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003; 17: 590-603. [DOI] [PubMed] [Google Scholar]
- 18.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9: 59-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278: 687-689. [DOI] [PubMed] [Google Scholar]
- 20.Gajewski TF, Thompson CB. Apoptosis meets signal transduction: elimination of a BAD influence. Cell. 1996;87: 589-592. [DOI] [PubMed] [Google Scholar]
- 21.Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8: 1153-1160. [DOI] [PubMed] [Google Scholar]
- 22.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378: 785-789. [DOI] [PubMed] [Google Scholar]
- 23.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8: 179-183. [DOI] [PubMed] [Google Scholar]
- 24.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4: 257-262. [DOI] [PubMed] [Google Scholar]
- 25.Dutton A, Reynolds GM, Dawson CW, Young LS, Murray PG. Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of Hodgkin's lymphoma cells through a mechanism involving Akt kinase and mTOR. J Pathol. 2005; 205: 498-506. [DOI] [PubMed] [Google Scholar]
- 26.Rassidakis GZ, Feretzaki M, Atwell C, et al. Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood. 2005;105: 827-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pene F, Claessens YE, Muller O, et al. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene. 2002; 21: 6587-6597. [DOI] [PubMed] [Google Scholar]
- 28.The Non-Hodgkin's Lymphoma Classification Project: a clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. Blood. 1997;89: 3909-3918. [PubMed] [Google Scholar]
- 29.de Leeuw RJ, Davies JJ, Rosenwald A, et al. Comprehensive whole genome array CGH profiling of mantle cell lymphoma model genomes. Hum Mol Genet. 2004;13: 1827-1837. [DOI] [PubMed] [Google Scholar]
- 30.Medeiros LJ, Estrov Z, Rassidakis GZ. Z-138 cell line was derived from a patient with blastoid variant mantle cell lymphoma. Leuk Res. 2006;30: 497-501. [DOI] [PubMed] [Google Scholar]
- 31.Saltman DL, Cachia PG, Dewar AE, et al. Characterization of a new non-Hodgkin's lymphoma cell line (NCEB-1) with a chromosomal (11:14) translocation [t(11:14)(q13;q32)]. Blood. 1988;72: 2026-2030. [PubMed] [Google Scholar]
- 32.Jadayel DM, Lukas J, Nacheva E, et al. Potential role for concurrent abnormalities of the cyclin D1, p16CDKN2 and p15CDKN2B genes in certain B cell non-Hodgkin's lymphomas: functional studies in a cell line (Granta 519). Leukemia. 1997; 11: 64-72. [DOI] [PubMed] [Google Scholar]
- 33.Camps J, Salaverria I, Garcia MJ, et al. Genomic imbalances and patterns of karyotypic variability in mantle-cell lymphoma cell lines. Leuk Res. 2006;30: 923-934. [DOI] [PubMed] [Google Scholar]
- 34.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994;269: 5241-5248. [PubMed] [Google Scholar]
- 35.Arcaro A, Wymann MP. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem J. 1993;296: 297-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Y, Qiao L, Wang S, et al. 3-(Hydroxymethyl)-bearing phosphatidylinositol ether lipid analogues and carbonate surrogates block PI3-K, Akt, and cancer cell growth. J Med Chem. 2000;43: 3045-3051. [DOI] [PubMed] [Google Scholar]
- 37.Nishizuka S, Charboneau L, Young L, et al. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci U S A. 2003;100: 14229-14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paweletz CP, Charboneau L, Bichsel VE, et al. Reverse phase protein microarrays which capture disease progression show activation of prosurvival pathways at the cancer invasion front. Oncogene. 2001;20: 1981-1989. [DOI] [PubMed] [Google Scholar]
- 39.Ananthanarayanan B, Ni Q, Zhang J. Signal propagation from membrane messengers to nuclear effectors revealed by reporters of phosphoinositide dynamics and Akt activity. Proc Natl Acad Sci U S A. 2005;102: 15081-15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wlodarski P, Kasprzycka M, Liu X, et al. Activation of mammalian target of rapamycin in transformed B lymphocytes is nutrient dependent but independent of Akt, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase, insulin growth factor-I, and serum. Cancer Res. 2005;65: 7800-7808. [DOI] [PubMed] [Google Scholar]
- 41.Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol. 2003;171: 88-95. [DOI] [PubMed] [Google Scholar]
- 42.Perez-Galan P, Roue G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006;107: 257-264. [DOI] [PubMed] [Google Scholar]
- 43.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284: 309-313. [DOI] [PubMed] [Google Scholar]
- 44.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401: 86-90. [DOI] [PubMed] [Google Scholar]
- 45.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401: 82-85. [DOI] [PubMed] [Google Scholar]
- 46.Collado M, Medema RH, Garcia-Cao I, et al. Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27Kip1. J Biol Chem. 2000;275: 21960-21968. [DOI] [PubMed] [Google Scholar]
- 47.Stahl M, Dijkers PF, Kops GJ, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168: 5024-5031. [DOI] [PubMed] [Google Scholar]
- 48.Dijkers PF, Medema RH, Pals C, et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol Cell Biol. 2000;20: 9138-9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14: 3102-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parsons R. Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol. 2004;15: 171-176. [DOI] [PubMed] [Google Scholar]
- 51.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3: 1221-1224. [DOI] [PubMed] [Google Scholar]
- 52.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24: 7465-7474. [DOI] [PubMed] [Google Scholar]
- 53.Balsara BR, Pei J, Mitsuuchi Y, et al. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis. 2004;25: 2053-2059. [DOI] [PubMed] [Google Scholar]
- 54.Fillmore GC, Wang Q, Carey MJ, Kim CH, Elenitoba-Johnson KS, Lim MS. Expression of Akt (protein kinase B) and its isoforms in malignant lymphomas. Leuk Lymphoma. 2005;46: 1765-1773. [DOI] [PubMed] [Google Scholar]
- 55.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98: 11598-11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suhara T, Kim HS, Kirshenbaum LA, Walsh K. Suppression of Akt signaling induces Fas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol Cell Biol. 2002;22: 680-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt M, Fernandez de Mattos S, van der Horst A, et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22: 7842-7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Viglietto G, Motti ML, Bruni P, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8: 1136-1144. [DOI] [PubMed] [Google Scholar]
- 59.Manning BD, Cantley LC. United at last: the tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem Soc Trans. 2003;31: 573-578. [DOI] [PubMed] [Google Scholar]
- 60.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115: 577-590. [DOI] [PubMed] [Google Scholar]
- 61.Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6: 91-99. [DOI] [PubMed] [Google Scholar]
- 62.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12: 3499-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grewe M, Gansauge F, Schmid RM, Adler G, Seufferlein T. Regulation of cell growth and cyclin D1 expression by the constitutively active FRAP-p70s6K pathway in human pancreatic cancer cells. Cancer Res. 1999;59: 3581-3587. [PubMed] [Google Scholar]
- 64.Mita MM, Mita A, Rowinsky EK. The molecular target of rapamycin (mTOR) as a therapeutic target against cancer. Cancer Biol Ther. 2003;2: S169-S177. [PubMed] [Google Scholar]
- 65.Hipp S, Ringshausen I, Oelsner M, Bogner C, Peschel C, Decker T. Inhibition of the mammalian target of rapamycin and the induction of cell cycle arrest in mantle cell lymphoma cells. Haematologica. 2005;90: 1433-1434. [PubMed] [Google Scholar]
- 66.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2: 339-345. [PubMed] [Google Scholar]
- 67.Tan Y, Demeter MR, Ruan H, Comb MJ. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem. 2000; 275: 25865-25869. [DOI] [PubMed] [Google Scholar]
- 68.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13: 1899-1911. [DOI] [PubMed] [Google Scholar]
- 69.Pianetti S, Arsura M, Romieu-Mourez R, Coffey RJ, Sonenshein GE. Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene. 2001;20: 1287-1299. [DOI] [PubMed] [Google Scholar]
- 70.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94: 29-86. [DOI] [PubMed] [Google Scholar]
- 71.Zhou XP, Marsh DJ, Morrison CD, et al. Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am J Hum Genet. 2003;73: 1191-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pesche S, Latil A, Muzeau F, et al. PTEN/MMAC1/TEP1 involvement in primary prostate cancers. Oncogene. 1998;16: 2879-2883. [DOI] [PubMed] [Google Scholar]
- 73.Zhou XP, Li YJ, Hoang-Xuan K, et al. Mutational analysis of the PTEN gene in gliomas: molecular and pathological correlations. Int J Cancer. 1999; 84: 150-154. [DOI] [PubMed] [Google Scholar]
- 74.Tolkacheva T, Chan AM. Inhibition of H-Ras transformation by the PTEN/MMAC1/TEP1 tumor suppressor gene. Oncogene. 2000;19: 680-689. [DOI] [PubMed] [Google Scholar]
- 75.Gronbaek K, Zeuthen J, Guldberg P, Ralfkiaer E, Hou-Jensen K. Alterations of the MMAC1/PTEN gene in lymphoid malignancies. Blood. 1998;91: 4388-4390. [PubMed] [Google Scholar]
- 76.Nakahara Y, Nagai H, Kinoshita T, et al. Mutational analysis of the PTEN/MMAC1 gene in non-Hodgkin's lymphoma. Leukemia. 1998;12: 1277-1280. [DOI] [PubMed] [Google Scholar]
- 77.Sakai A, Thieblemont C, Wellmann A, Jaffe ES, Raffeld M. PTEN gene alterations in lymphoid neoplasms. Blood. 1998;92: 3410-3415. [PubMed] [Google Scholar]
- 78.Butler MP, Wang SI, Chaganti RS, Parsons R, Dalla-Favera R. Analysis of PTEN mutations and deletions in B-cell non-Hodgkin's lymphomas. Genes Chromosomes Cancer. 1999;24: 322-327. [PubMed] [Google Scholar]
- 79.Lee S, Choi EJ, Jin C, Kim DH. Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol. 2005;97: 26-34. [DOI] [PubMed] [Google Scholar]
- 80.Bachman KE, Argani P, Samuels Y, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3: 772-775. [DOI] [PubMed] [Google Scholar]
- 81.Bertelsen BI, Steine SJ, Sandvei R, Molven A, Laerum OD. Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: frequent PIK3CA amplification and AKT phosphorylation. Int J Cancer. 2006;118: 1877-1883. [DOI] [PubMed] [Google Scholar]
- 82.Wu G, Mambo E, Guo Z, et al. Uncommon mutation, but common amplifications, of the PIK3CA gene in thyroid tumors. J Clin Endocrinol Metab. 2005;90: 4688-4693. [DOI] [PubMed] [Google Scholar]
- 83.Samuels Y, Diaz LA Jr, Schmidt-Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7: 561-573. [DOI] [PubMed] [Google Scholar]
- 84.Shayesteh L, Lu Y, Kuo WL, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21: 99-102. [DOI] [PubMed] [Google Scholar]