

Abstract
Normal mouse lungs lack appreciable numbers of mast cells (MCs) or MC progenitors (MCp's), yet the appearance of mature MCs in the tracheobronchial epithelial surface is a characteristic of allergic, T-cell-dependent pulmonary inflammation. We hypothesized that pulmonary inflammation would recruit MCp's to inflamed lungs and that this recruitment would be regulated by distinct adhesion pathways. Ovalbumin-sensitized and challenged mice had a greater than 28-fold increase in the number of MCp's in the lungs. In mice lacking endothelial vascular cell adhesion molecule 1 (VCAM-1) and in wild-type mice administered blocking monoclonal antibody (mAb) to VCAM-1 but not to mucosal addressin CAM-1 (MadCAM-1), recruitment of MCp's to the inflamed lung was reduced by greater than 75%. Analysis of the integrin receptors for VCAM-1 showed that in β7 integrin-deficient mice, recruitment was reduced 73% relative to wild-type controls, and in either BALB/c or C57BL/6 mice, mAb blocking of α4, β1, or β7 integrins inhibited the recruitment of MCp's to the inflamed lung. Thus, VCAM-1 interactions with both α4β1 and α4β7 integrins are essential for the recruitment and expansion of the MCp populations in the lung during antigen-induced pulmonary inflammation. Furthermore, the MCp is currently unique among inflammatory cells in its partial dependence on α4β7 integrins for lung recruitment.
Introduction
Mast cells (MCs) develop in tissues from bone marrow-derived MC progenitors (MCp's) drawn from the intravascular compartment. The intestine has an abundant supply of MCs, and MCp's constitutively home to this organ. In contrast, the naive mouse lung has few MCp's and lacks mature MCs beyond first- and second-generation bronchi.1-3 Nonetheless, MCs accumulate in the epithelial surfaces of both large and small bronchi of mice that are sensitized systemically with ovalbumin (OVA) and challenged repetitively by aerosol inhalation to generate chronic airway inflammation.3 Furthermore, MCs are required for the development of airway hyperresponsiveness to methacholine in protocols that lack adjuvant or otherwise limit the intensity of sensitization before challenge with OVA.4-6 In humans with bronchial asthma, MCs accumulate and degranulate in both the bronchial epithelium and airway smooth muscle,7-9 accompanied by increased numbers of MCp's in the blood.10 Treatment with humanized monoclonal antibody (mAb) that blocks access of serum IgE to the high-affinity FcεRI receptor reduces the frequency of asthma exacerbations.11 These findings imply a pathophysiologic role for MCs and their expansion both in mouse models of airway inflammation and in the pathogenesis of human bronchial asthma.
Although the small airways of the mouse have scant smooth muscle, little lamina propria, and negligible bronchial circulation compared with those of the human, an increment in intraepithelial MCs and leukocyte infiltrates around bronchovascular bundles is consistently observed in inflamed respiratory mucosal surfaces in both species, leading to the suggestion that the site of extravasation is via the associated microvasculature.12,13 It is thus likely that the pathways needed for incremental MC numbers are conserved. Moreover the low levels of MCp's and scarcity of mature MCs in the naive mouse lung suggest that MCp recruitment accounts for the MC accumulation in the bronchial mucosa with the induction of allergic inflammation. To date, no studies have addressed this issue.
We previously reported that constitutive transendothelial migration of MCp's to intestine and the development of mature MCs in that organ are absent in β7 integrin null mice.2,14 The residence of these MCp's after homing to the intestine in wild-type mice was transient, as mAb blocking of α4β7 integrin depleted intestinal MCp's by half over 7 days.1 This dynamic relationship for homeostasis between bone marrow and the intestinal MCp's required interaction of α4β7 integrins on MCp's with endothelial cell mucosal addressin cellular adhesion molecule 1 (MAdCAM-1) and vascular cellular adhesion molecule 1 (VCAM-1) based on mAb blocking of MCp migration in sublethally irradiated mice reconstituted with bone marrow from wild-type donors. Nonetheless, after infection with Trichinella spiralis, β7 integrin-null mice were still able to mount a reactive mastocytosis and to clear the adult worm burden, albeit with delayed kinetics relative to wild-type control mice14 (M.F.G., Daniel S. Friend, K. F. A., unpublished data, August 2000). This observation supports the existence of an alternative inducible adhesion pathway that can mediate inflammatory recruitment of MCp independent of the α4β7 integrin.
Thus, we hypothesized that the minimal homing of MCp's to lung would be dramatically expanded by recruitment during antigen (Ag)-induced pulmonary inflammation and that the adhesion pathway(s) for this response would be different from that necessary for the abundant homing of MCp's to the intestine. Herein, we demonstrate a rapid increase in the number of MCp's in the lung with aerosolized antigen-induced inflammation. Furthermore, unlike intestinal homing, this recruitment is mediated by VCAM-1 interacting with the α4β1 integrin and the α4β7 integrin expressed by MCp's, with no role for MAdCAM-1.
Materials and methods
Animals
All mice were 6- to 16-week-old males when used. BALB/c mice were obtained from Taconic Laboratories (Germantown, NY). The β7 integrin-deficient mice (C57BL/6-Itgb7tm1Cgn)15 and their C57BL/6 controls were obtained from Jackson Laboratory (Bar Harbor, ME). VCAM-1 (Tie2Cre/LoxP)-deficient mice on a C57BL/6 background16 and their parental control strains were maintained at the Medical College of Georgia. All animal experiments were approved by the Institutional Animal Care and Use Committee and the studies were carried out in accordance with the guidelines for animal care of the National Institutes of Health and the Public Health Service.
OVA sensitization and challenge protocol
Groups of 2 to 3 mice received intraperitoneal injections of 10 μg OVA (A5503; Sigma-Aldrich, Grand Island, NY) adsorbed to 1 mg alum (77161; Pierce, Rockford, IL) in 200 μL of sterile Hanks balanced salt solution (HBSS) on days 0 and 7. Mice were challenged with 1% aerosolized OVA in HBSS for 30 minutes per day using a PARI nebulizer (PARI, Midlothian, VA). For most experiments, the mice were challenged on days 17 to 19 and were killed for the determination of MCp's on day 20. For time course experiments, mice were killed on day 20, approximately 20 hours after receiving the last daily challenge of the indicated number of challenges (Figure 1A).
Figure 1.

Time course for the increase in pulmonary MNCs and MCp's after OVA sensitization and challenge of BALB/c mice. (A) The protocol used for induction of pulmonary inflammation is depicted. Mice are sensitized intraperitoneally (IP Imm) with OVA and alum on days 0 and 7, and then challenged with aerosolized OVA daily, beginning on days 13, 15, 17, or 19, with assay on day 20. These mice received 7, 5, 3, or 1 daily challenges, respectively. (B) The mean number (± SEM) of lung MNCs recovered per BALB/c mouse is graphed for the various numbers of daily challenges as indicated. Values are means ± SE from 5 separate experiments, with following determinations (n) in each group: 0 (5); 1 day (3); 3 days (5); 5 days (4), 7 days (4). (C) The mean (± SE) MCp concentration (MCp's/106 MNCs) in the lungs of the same mice as in panel B. (D) The mean (± SE) total number of MCp's per lung from the same mice as in panel B. *Indicates statistical significance (P < .05) as determined by the 2-tailed Student t test.
Antibodies and the blocking of homing protocol
The mAbs anti-VCAM-1 (429, rat-IgG2a), anti-MAdCAM-1 (MECA367, rat-IgG2a), anti-αE integrin (M290, rat-IgG2a), anti-β1 integrin (9EG7, rat-IgG2a), anti-α4β7 integrin (DATK 32, ratIgG2a), and anti-β7 integrin (FIB27, rat-IgG2a) were obtained from Pharmingen (San Diego, CA). The anti-α4 integrin (PS/2, rat-IgG2b) was obtained from American Type Culture Collection (CRL-1911; Manassas, VA). All mAbs containing sodium azide were dialyzed against HBSS to eliminate the azide.
To evaluate the molecules important in pulmonary recruitment of MCp's, approximately 50 μg of isotype control or blocking mAb, in 100 μL of HBSS, was administered intraperitoneally to OVA/alum-sensitized mice. Injections were given daily starting on the first day of aerosolized OVA challenges.
Mononuclear cell (MNC) preparation and MCp assessment
Mice were killed by CO2 asphyxiation and both lungs and spleen were harvested. Lung and spleen were placed separately in 20 mL of RPMI 1640 complete (RPMI 1640 containing 100 U/mL penicillin, 100 μg/mL streptomycin, 10 μg/mL gentamicin, 2 mM l-glutamine, 0.1 mM nonessential amino acids, and 10% heat-inactivated fetal calf serum; F2442, Sigma-Aldrich, Grand Island, NY) and were processed essentially as previously described.1,2 Briefly, the lungs, perfused with 10 mL of HBSS administered via the right ventricle, were removed, finely chopped with scalpels, and transferred to 50-mL plastic tubes with 30 mL of RPMI 1640 complete plus 1 mg/mL collagenase type 4 (Worthington, Lakewood, NJ). Three enzymatic digestions were carried out for approximately 20 minutes each at 37°C. The undigested tissue clumps were collected after each time period and subjected to another enzymatic digestion, while the liberated cells were pelleted, resuspended in 44% Percoll (P1644; Sigma-Aldrich, St Louis, MO), underlayed with a 67% Percoll layer, and spun at 400g for 20 minutes at 4°C.
The MNCs were harvested from the 44/67% Percoll interfaces, and cells from the lungs of the 3 digestions were pooled and washed in RPMI 1640 complete. The number of viable cells was determined by trypan blue dye exclusion on a hemocytometer. The cells were serially diluted 2-fold in RPMI 1640 complete, and 100 μL of each dilution was added to wells of standard 96-well flat-bottomed microtiter plates (3596; Corning, Corning, NY). Typically, 24 wells were plated for each cell concentration. Lung MNCs were plated beginning at 10 000 to 30 000 cells/well. Then, each well received 100 μL of γ-irradiated (30 Gy) splenic feeder cells plus cytokines (recombinant mouse IL-3 at 20 ng/mL and recombinant mouse stem cell factor at 100 ng/mL).
The cultures were placed in humidified 37°C incubators with 5% CO2 for 12 to 14 days, and wells containing MC colonies were counted with an inverted microscope. The MC colonies were easily distinguished as large colonies of nonadherent, small-to-medium-sized cells.17,18 The MCp concentration is expressed as the number of MCp's/106 MNCs isolated from the tissue. The number of MCp's/lung is derived by multiplying the concentration of MCp's by the MNC yield per lung for individual mice or divided by the number of mice when MNCs were pooled from more than one mouse.
Histology
The assessment of inflammation in mice was performed 1 day after the last of 3 daily challenges. Lung tissue was fixed in 4% paraformaldehyde overnight, washed in PBS, embedded, and sectioned as previously described.19 JB-4-embedded sections were stained for chloroacetate esterase to illustrate mast cells and neutrophils. Inflammatory infiltrates in the lung parenchyma were evaluated by counting the number of bronchovascular bundles with an inflammatory infiltrate out of a total of 20 such bundles randomly visualized in each section after Diff-Quick (Dade Behring, Newark, DE) staining. The extent of goblet cell hyperplasia in the bronchioles was assessed by a semiquantitative mucus scoring between 0 and 3. A score of 0 corresponds to absence of mucus overproduction, and a score of 3 represents blockage of the majority of bronchiolar lumena by mucus.
For illustration of the histologic changes, digital images were acquired at room temperature using a Nikon DXM 1200 digital camera (Melville, NY) fitted onto a Leica DM LB-2 light microscope through a Leica C-mount adapter (0.63 × HC f2/3′′) with ACT-1 software from the same manufacturer (Leica, Heidelberg, Germany). Images submitted were taken at 125 × magnification using a Leica HC plane 20×/0.5 numeric aperature objective lens. No further image processing was undertaken.
Statistical analysis
Data in the figures are expressed as the mean ± SEM, derived from 3 or more separate experiments with the values from pools of 2 to 3 mice or from 2 or more individual mice in each group. For the statistical calculations, each n is either the value from an individual mouse or the value from the cells pooled from 2 or 3 mice. Significance was determined using a 2-tailed Student t test. Values of P less than .05 were considered significant.
Results
Challenge with aerosolized Ag increases the numbers of lung MCp's
Under basal conditions, few MCp's2 or mature MCs3,20 are identified in the mouse lung relative to the numbers found in the bone marrow, spleen, and intestine. We evaluated the numbers of MCp's in the lung with and without the induction of pulmonary inflammation by exposure to aerosolized antigen in sensitized mice to determine whether an increase in MCp's occurred and the time frame of the response. BALB/c mice sensitized with OVA and alum on days 0 and 7, followed by daily challenges with aerosolized OVA beginning 1, 3, 5, or 7 days before the MCp assay on day 20 (Figure 1A), exhibited a dramatic increase in the number of MNCs isolated from the enzymatically dispersed lungs (Figure 1B). The MNC preparation was not contaminated with MCs, as fewer than one chloroacetate-positive MC could be detected per 5 million MNCs after 3 daily challenges. The average number of MNCs increased over the baseline value of 1.6 × 106 MNCs per mouse by 3.9-fold after 3 daily challenges (P < .05, n = 5) and progressed to 5.7-fold with 7 daily challenges (P < .05, n = 4). The concentration of MCp's isolated from the lungs also increased significantly, with a 4.8-fold increase in the mean number after 3 daily challenges (P < .05), with little further increase after 5 (6.6-fold, P < .05, n = 4) or 7 (6.8-fold) daily challenges (Figure 1C). The mean (± SE) increase in total MCp's per mouse lung relative to unchallenged controls was 2.4 ± 1.0-fold after 1 challenge, 28.1 ± 14.8-fold after 3 challenges, 42.2 ± 6.9-fold after 5 challenges, and 54.6 ± 16.1-fold after 7 challenges (Figure 1D). The increase in total lung MCp's relative to unchallenged controls was statistically significant after 3 and 5 days of challenges (P < .05).
Histologic assessment confirmed that the 3 daily aerosol antigen challenges resulted in the presence of inflammation with increased numbers of eosinophils and MNCs around the bronchovascular bundles (Figure 2). BALB/c mice showed 45% ± 15% of the blood vessels with an inflammatory infiltrate (mean ± 0.5 range of 2 mice), 35% ± 15% of the bronchioles with an inflammatory infiltrate, and a relative mucus score of 2 for both mice. C57BL/6 mice, also assessed after 3 days, exhibited an inflammatory infiltrate around 35% ± 0% of the blood vessels (mean ± 0.5 range of 2 mice), and 17% ± 2.5% of the bronchioles, and a mucus score of 1 ± 0. The magnitude of the infiltrate is more striking in the BALB/c mice than in the C57BL/6 mice, as previously demonstrated by others, after sensitization and airway antigen challenge13,21,22 (reviewed in Boyce and Austen23). No increase in lung MCs was noted in either strain after 3 challenges (data not shown)
Figure 2.
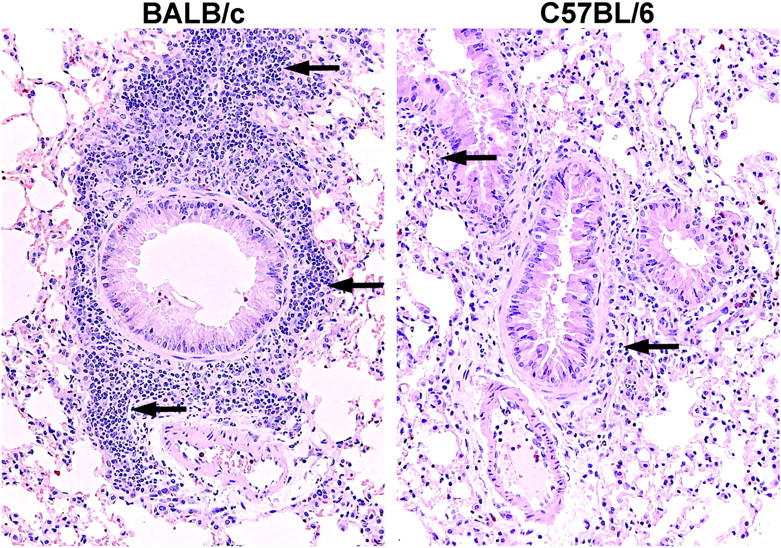
Histology of BALB/c and C57BL/6 mouse lung after OVA sensitization and 3 days of aerosolized OVA challenge. Both strains show an inflammatory response around the bronchovascular bundles after 3 OVA challenges (arrows), the routine time for assessment of lung MCp's. The lung from the BALB/c mouse (left panel) shows much more leukocyte infiltration than does the lung from the C57BL/6 mouse (right panel).
The increase in lung MCp's required active sensitization and challenge with the same antigen. Mice sensitized with alum alone or with an irrelevant protein (goat Ig) adsorbed to alum and challenged with OVA showed no increase in pulmonary MNCs or MCp's (data not shown). Given the dramatic increase in lung MCp's after only 3 daily challenges, we used this time point in the subsequent experiments in which we sought to evaluate the molecules on the surface of the MCp's and the endothelium that were involved in this recruitment.
VCAM-1 deficiency and mAb blockade of VCAM-1 but not MAdCAM-1 reduces the recruitment of MCp's to the inflamed lung
In our previous studies, α4β7 integrin interacted with both VCAM-1 and MAdCAM-1 in the basal homing of MCp's to the intestine.1 For evaluation of the role of VCAM-1 on recruitment of MCp's to inflamed lung, endothelial VCAM-1-deficient mice were compared with wild-type C57BL/6 mice.16 After 3 challenges on days 17 to 19 and analysis on day 20, sensitized C57BL/6 mice exhibited a 2-fold increase in the mean number of lung MNCs recovered in 3 separate experiments (Figure 3A). There was a 7-fold increase in the mean MCp concentration in the lung (Figure 3B) and a 13.6-fold increase in the mean number of total MCp's per lung (Figure 3C). In mice lacking endothelial VCAM-1, there was a similar 2-fold increase in the number of MNCs, indicating that this aspect of the inflammatory response had not been suppressed by the lack of endothelial VCAM-1. However, there was no increment in the concentration of lung MCp's and only a 2-fold increase in the total number of lung MCp's. These findings represent an 85% ± 3% reduction in MCp concentration (mean ± SE, n = 3) and an 87% ± 7% reduction in total MCp's in the lungs of challenged VCAM-1-deficient mice relative to the challenged C57BL/6 mice. This indicates a role for VCAM-1 in inflammatory recruitment that is not evident for basal homing of MCp's to the lung.
Figure 3.

Effect of endothelial VCAM-1 deficiency on MCp recruitment to the inflamed lung. (A) Number of lung MNCs recovered per mouse from OVA-sensitized C57BL/6 (▪) and VCAM-1-deficient (□) mice with and without aerosolized OVA challenges. Values are the mean ± SEM from 3 separate experiments with MNCs pooled from 2 to 3 mice in each experiment. (B) Concentration of MCp's (MCp's/106 MNCs) in the lungs of the same mice. (C) Total number of MCp's per lung in the same mice. *Indicates statistical significance (P < .05) as determined by the 1-tailed Student t test.
We further evaluated the role of VCAM-1 and any role for MAdCAM-1 using blocking mAbs directed against these molecules individually. Sensitized C57BL/6 mice received the mAbs just before each challenge. Relative to nonchallenged controls, the number of MNCs/mouse lung increased 2- to 3-fold in mice receiving HBSS or MAdCAM-1 Ab (Figure 4A). Mice receiving anti-VCAM-1 showed a reduction in the number of MNCs recovered that was not statistically significant (41% ± 3.5%, mean ± SE, n = 4). The administration of anti-MAdCAM-1 (Figure 4B-C) did not reduce pulmonary recruitment of MCp's whereas mice receiving anti-VCAM-1 had significant reductions in MCp concentration (58% ± 12% reduction, mean ± SE, P < .05, n = 4; Figure 4B) and in total MCp's/mouse lung (75% ± 6.6% reduction, P < .01; Figure 4C).
Figure 4.

Effect of mAb to VCAM-1 and MAdCAM-1 on MCp recruitment to the inflamed lung. (A) Number of lung MNCs recovered per mouse from sensitized C57BL/6 mice not challenged with aerosolized OVA (No Challenge) or that had received injections of HBSS or the indicated mAb and then challenged with aerosolized OVA. Values are the mean ± SEM from 3 experiments with 4 determinations in each group. (B) Concentration of MCp's (MCp's/106 MNCs) in the lungs of the same mice. (C) Total number of MCp's per lung in the same mice. *P < .05, **P < .01, as determined by the 2-tailed Student t test.
The β7 integrin deficiency and mAb blockade of α4, β1, β7, but not αE, integrins reduce the recruitment of MCp's to the inflamed lung
VCAM-1 can interact with either the α4β7 or the α4β1 integrin heterodimers.24-26 We examined MCp recruitment in β7 integrin-deficient mice and then used antibody blocking to address specifically which integrins were critical to the response. The numbers of MNCs recovered from wild-type and β7 integrin-deficient mice after the induction of allergic inflammation were similar (Figure 5A), and the cellular infiltrates involved similar numbers of inflamed blood vessels in both strains (Figure 6), 35% ± 0% versus 38% ± 18% in C57BL/6 and β7 integrin-deficient mice, respectively (mean ± 0.5 range, n = 2), similar numbers of inflamed bronchioles, 18% ± 3% and 23% ± 8%, respectively, and similar increments in mucous production, 1 ± 0 and 1 ± 0, respectively. The baseline concentrations and total number of MCp's in the lungs of wild-type and β7 integrin-deficient strains were similar and consistent with our previous enumeration of lung MCp's in naive wild-type and β7 integrin-deficient mice.2 After challenge with aerosolized Ag, and relative to the levels in wild-type mice, the recruited MCp concentration in the β7 integrin-deficient mice was reduced by 64% ± 11.7% (Figure 5B; mean ± SE, n = 7, P < .05) and total lung MCp's by 73% ± 8.6% (Figure 5C; P < .05). Sensitized, unchallenged wild-type and β7 integrin-deficient mice had 91 ± 7 and 70 ± 6 ng/mL of anti-OVA IgE, respectively (mean ± 0.5 range, n = 2), and the mice challenged with aerosolized OVA had 123 ± 3 and 120 ± 7 ng/mL, respectively, indicating that the absence of β7 did not impair sensitization and amplification of the Th2 response.
Figure 5.

Effect of β7 integrin deficiency on MCp recruitment to the inflamed lung. (A) Number of lung MNCs recovered per mouse from OVA-sensitized C57BL/6 (▪) and β7 integrin-deficient (□) mice with and without aerosolized OVA challenges. Values are the mean ± SEM from 6 separate experiments with 7 determinations in each group. (B) Concentration of MCp's (MCp's/106 MNCs) in the lungs of the same mice. (C) Total number of MCp's per mouse in the lungs of the same mice. *P < .05, **P < .01, as determined by the 2-tailed Student t test.
Figure 6.
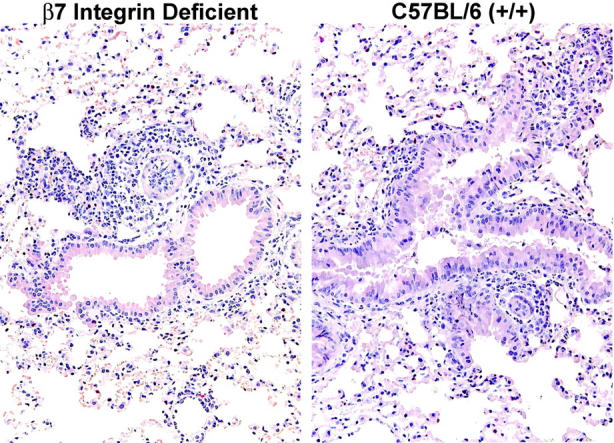
Histology of β7 integrin-deficient and C57BL/6 mouse lung after OVA sensitization and 3 days of aerosolized OVA challenge. The lung from the β7 integrin-deficient mouse (left panel) shows leukocyte infiltration around the bronchovascular bundles, similar to that observed in lung from the wild-type C57BL/6 mouse treated in parallel (right panel).
To distinguish the contribution of α4β1, α4β7, and αEβ7 integrins to the recruitment of MCp's after the induction of pulmonary inflammation, we used mAb directed to the individual components of the heterodimers α4, αE, β1, and β7. In C57BL/6 mice, no inhibition of MNCs was noted (Figure 7A). The lung MCp concentration (per 106 MNCs; Figure 7B) and the total lung MCp's (Figure 7C) were significantly inhibited by mAb to α4, β1, β7, or α4β7 integrin. Thus, in the C57BL/6 strain, the recruitment of lung MCp's was inhibited by 83% ± 4.5% with mAb to α4 integrin (P < .05, n = 3), by 60% ± 2.8% with mAb to β1 integrin (P < .05, n = 5), by 67% ± 1.2% with mAb to β7 integrin (P < .05, n = 3), and by 43% ± 7% (P < .05, n = 3) with a mAb to a combinatorial epitope of the α4β7 integrin heterodimer. The inhibition of MCp recruitment to lung by anti-β7 integrin mAb was very similar to that observed in the β7 integrin null strain, 67% versus 73%, respectively.
Figure 7.

Effect of mAb to α4, β1, β7, or α4B7 integrins on MCp recruitment to the inflamed lung in C57BL/6 mice. (A) Number of lung MNCs per mouse recovered from OVA-sensitized BALB/c mice not challenged (No Chall) or from mice that had received injections of HBSS or the indicated mAb and then challenged with aerosolized OVA. Values are the mean ± SEM from 6 separate experiments with following determinations in each group: HBSS (9); anti-α4 (3); anti-β1 (5); anti-β7 (3); anti-α4β7 (3). (B) Concentration of MCp's (MCp's/106 MNCs) in the lungs of the same mice. (C) Total number of MCp's per mouse in the lungs of the same mice. *Indicates statistical significance (P < .05) as determined by the 2-tailed Student t test.
In BALB/c mice receiving intraperitoneal injections of mAb immediately before challenge with aerosolized OVA, no significant reduction of MNC yields was found (Figure 8A). In contrast, both the concentration of MCp's (Figure 8B) and the total number of pulmonary MCp's/mouse (Figure 8C) were consistently decreased in mice treated with anti-α4, anti-β1, or anti-β7 relative to mice given HBSS or the isotype-matched anti-αE. Mice treated with anti-α4 demonstrated significant reductions in both MCp concentration (73% ± 7.4%; mean ± SE, n = 3) and in total MCp's (65% ± 21%) relative to HBSS-treated mice (P < .05 for both measures). Mice treated with anti-β1 had a 57% ± 15% reduction in MCp concentration (mean ± SE, n = 3) and a significant 62% ± 17% reduction in total MCp's (P < .05). Mice treated with anti-β7 had a significant 42% ± 10% reduction in MCp concentration (mean ± SE, P < .05, n = 8) and a 26% ± 23% reduction in total lung MCp's. Mice given anti-αE and mice deficient in αE integrin (data not shown) showed no reduction in MCp recruitment.
Figure 8.

Effect of mAb to α4, αE, β1, or β7 integrins on MCp recruitment to the inflamed lung of BALB/c mice. (A) Number of lung MNCs per mouse recovered from OVA-sensitized BALB/c mice not challenged (No Chall) or from mice that had been given injections of HBSS (HBSS) or the indicated mAb and then challenged with aerosolized OVA. Values are the mean ± SEM from 4 separate experiments with following determinations in each group: HBSS (6); anti-αE (3); anti-α4 (3); anti-β1 (5); anti-β7 (8). (B) Concentration of MCp's (MCp's/106 MNCs) in the lungs of the same mice. (C) Total number of MCp's per mouse in the lungs of the same mice. *Indicates statistical significance (P < .05) as determined by the 2-tailed Student t test.
Discussion
The lung of the naive mouse has sparse MCp's1,2 and few submucosal or intraepithelial MCs in the bronchi, yet there is a distinct increase in intraepithelial MCs with sensitization and airway challenge with Ag.3,20 We therefore sought to determine whether this increment reflected de novo recruitment of blood-born MCp's via transendothelial migration to the lung. With systemic sensitization and various intervals for daily airway challenge before assay, we found that a small accumulation of pulmonary MCp's occurred after even a single challenge of a systemically sensitized mouse, with a large increase (> 28-fold) after 3 or more challenges (Figure 1). At day 3, there was a substantial cellular infiltration of the bronchovascular bundles and mucus in the airways, with the changes being greater in the BALB/c mice than in the C57BL/6 mice (Figure 2). The recovery of MNCs also was 2- to 4-fold greater in BALB/c than in C57BL/6 mice. The magnitude and rate of the increase in MCp's from baseline to day 3 suggests that the major contribution is from influx. The MCp's were identified by limiting dilution and clonal expansion from isolated pulmonary MNCs purified by density gradient centrifugation, and these preparations did not contain detectable mature MCs. Moreover, histologic analysis confirmed that the acute experimental conditions selected did not generate a reactive mastocytosis in the epithelium or submucosa, as was observed with chronic antigen exposure.3 Also, the efficacy of blocking antibody to the adhesion molecules involved in transendothelial migration of MCp's indicates that recruitment rather than proliferation accounts for the expanded lineage reservoir.
Our earlier study on the mechanism of basal MCp homing to the intestine suggested that ongoing transendothelial influx was the major mechanism maintaining the baseline reservoir in this organ.1,2 MCp's were absent in the small intestine of β7 integrin-deficient mice, but normal numbers were detected in the stomach and colon of the same animals. Thus, mucosal tissues use strikingly distinct and specific mechanisms to regulate MCp homing. Based on studies with blocking Ab, homing of MCp's to the intestine used the interaction of α4β7 integrin with either MAdCAM-1 or VCAM-1 with no role for α4β1 integrin.1,2 In determining which of these molecules might be important in the inflammatory recruitment of MCp's to the lung, only VCAM-1 was expected to contribute, since its low-level constitutive expression is upregulated during an inflammatory response and this results in more eosinophils and lymphocytes being recruited to the lung.27-30 As anticipated, VCAM-1-deficient mice recruited minimal MCp's with pulmonary inflammation, the total influx per lung being reduced by approximately 90% compared with that in the wild-type mice (Figure 3) and contrasting with the modest decrement in total numbers of MNCs in these mice. The recruitment response of total MCp's per lung, but not total MNCs, was also significantly inhibited in wild-type mice given anti-VCAM-1 before inhalation challenge (Figure 4). Thus, VCAM-1-dependent adhesion pathways are critical for MCp's relative to other MNCs recruited during experimental pulmonary inflammation. MAdCAM-1 is neither constitutively nor inducibly expressed in this tissue28,31 and the administration of mAb to MAdCAM-1 in sensitized and inhalation-challenged mice did not decrease recruitment of MCp's to the lung (Figure 4).
As both α4β1 and α4β7 integrins mediate binding to VCAM-1, we evaluated which of these integrins was involved in recruitment of MCp's to inflamed lung. Although the β7 integrin-deficient mice did not differ from wild-type C57BL/6 mice in their low baseline levels of lung MCp's, the incremental recruitment of MCp's was strikingly attenuated in the null strain (Figure 5). These β7 integrin-deficient mice had no decrement in any of the other parameters of sensitization and inflammation monitored (Figure 6). The blocking of MCp recruitment by mAbs in wild-type C57BL/6 mice showed that specificity for α4, β1, or β7 integrins markedly suppressed total lung MCp recruitment, with inhibition by the anti-β7 integrin being comparable to that observed in the β7 integrin-deficient mice (Figure 7). In wild-type BALB/c mice, blocking of MCp recruitment was significant for total lung MCp's when the mAbs were directed to α4 and β1 integrins, with some effect for β7 integrin and none for αE (Figure 8). When expressed as inhibition of recruited MCp's per 106 MNCs, the effects of mAbs to α4 and β7 integrins were significant, with a substantial effect with mAb to β1 integrins and none for mAb to αE integrin. The α4β1 integrin has been clearly implicated in recruitment of other leukocytes to sites of allergic inflammation,27,32 but we know of no other leukocyte in which α4β7 plays such a prominent role in trafficking to the lung. The fact that mAb to heterodimeric α4β7 integrin failed to similarly suppress the elicitation of MCp's in lung (Figure 7) may be due to differences in epitopes blocked by binding of the various mAbs. Importantly, blockade of the αE integrin subunit, the only other partner for β7, had no effect at all, further supporting the involvement of the α4β7 dimer.
Just as integrin-dependent transendothelial migration regulates basal MCp homing to the intestine, we now find that integrin-dependent transendothelial migration is the basis for inflammation-induced expansion of MCp's in the lung, a tissue that normally contains very few cells of this lineage in laboratory strains of mice. This recruitment of MCp's likely reflects the critical dependence on the induction of VCAM-1, as the constitutive MAdCAM-1 is not expressed by the pulmonary endothelium. The induction of VCAM-1 was sufficiently rapid to permit recruitment of MCp's within 72 hours of antigen challenge, during the initiation of Th2 cell-type pulmonary inflammation. Subsequent amplification of the inflammatory response by mucosal Th2 cells may permit proliferation and maturation of interepithelial MCs. Notably, whereas α4β1 integrin played no role in intestinal homing of MCp's via VCAM-1 and MAdCAM-1 for which α4β7 integrin was critical, α4β1 is a critical MCp integrin for VCAM-1-based recruitment to lung. However, the MCp is currently unique in also using α4β7 integrin for this migration.
Acknowledgments
Dr Abonia's current address is Cincinnati Children's Hospital Medical Center, 3333 Burnet Ave, ML7028, Cincinnati, OH 45229-3039.
Prepublished online as Blood First Edition Paper, May 2, 2006; DOI 10.1182/blood-2005-12-012781.
Supported by grants AI 057991, HL 036110, AI 031599, AI 052353, AI 048802, and AI 047379 from the National Institutes of Health and the P. E. Lindahl Fund from The Royal Swedish Academy of Sciences.
J.P.A., K.F.A., and M.F.G. designed the experiments; J.P.A., J.H., T.J., T.S., Y.X., and M.F.G. performed the experiments and analyzed the data; P. K. and R.A.F. provided vital reagents; and J.P.A., J.A.B., K.F.A., and M.F.G. wrote the manuscript.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Abonia JP, Austen KF, Rollins BJ, et al. Constitutive homing of mast cell progenitors to the intestine depends on autologous expression of the chemokine receptor CXCR2. Blood. 2005;105: 4308-4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurish MF, Tao H, Abonia JP, et al. Intestinal mast cell progenitors require CD49dβ7(α4β7 integrin) for tissue-specific homing. J Exp Med. 2001;194: 1243-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ikeda RK, Miller M, Nayar J, et al. Accumulation of peribronchial mast cells in a mouse model of ovalbumin allergen induced chronic airway inflammation: modulation by immunostimulatory DNA sequences. J Immunol. 2003;171: 4860-4867. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi T, Miura T, Haba T, et al. An essential role of mast cells in the development of airway hyperresponsiveness in a murine asthma model. J Immunol. 2000;164: 3855-3861. [DOI] [PubMed] [Google Scholar]
- 5.Williams CM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med. 2000;192: 455-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taube C, Wei X, Swasey CH, et al. Mast cells, FcepsilonRI, and IL-13 are required for development of airway hyperresponsiveness after aerosolized allergen exposure in the absence of adjuvant. J Immunol. 2004;172: 6398-6406. [DOI] [PubMed] [Google Scholar]
- 7.Wenzel SE, Fowler AA III, Schwartz LB. Activation of pulmonary mast cells by bronchoalveolar allergen challenge: in vivo release of histamine and tryptase in atopic subjects with and without asthma. Am Rev Respir Dis. 1988;137: 1002-1008. [DOI] [PubMed] [Google Scholar]
- 8.Laitinen LA, Laitinen A, Haahtela T. Airway mucosal inflammation even in patients with newly diagnosed asthma. Am Rev Respir Dis. 1993;147: 697-704. [DOI] [PubMed] [Google Scholar]
- 9.Brightling CE, Bradding P, Symon FA, et al. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346: 1699-1705. [DOI] [PubMed] [Google Scholar]
- 10.Mwamtemi HH, Koike K, Kinoshita T, et al. An increase in circulating mast cell colony-forming cells in asthma. J Immunol. 2001;166: 4672-4677. [DOI] [PubMed] [Google Scholar]
- 11.Busse W, Corren J, Lanier BQ, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108: 184-190. [DOI] [PubMed] [Google Scholar]
- 12.Pabst R, Tschernig T. Perivascular capillaries in the lung: an important but neglected vascular bed in immune reactions? J Allergy Clin Immunol. 2002;110: 209-214. [DOI] [PubMed] [Google Scholar]
- 13.Singh B, Shinagawa K, Taube C, Gelfand EW, Pabst R. Strain-specific differences in perivascular inflammation in lungs in two murine models of allergic airway inflammation. Clin Exp Immunol. 2005;141: 223-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Artis D, Humphreys NE, Potten CS, et al. Beta7 integrin-deficient mice: delayed leukocyte recruitment and attenuated protective immunity in the small intestine during enteric helminth infection. Eur J Immunol. 2000;30: 1656-1664. [DOI] [PubMed] [Google Scholar]
- 15.Wagner N, Lohler J, Kunkel EJ, et al. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature. 1996;382: 366-370. [DOI] [PubMed] [Google Scholar]
- 16.Koni PA, Joshi SK, Temann UA, et al. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193: 741-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crapper RM, Schrader JW. Frequency of mast cell precursors in normal tissues determined by an in vitro assay: antigen induces parallel increases in the frequency of P cell precursors and mast cells. J Immunol. 1983;131: 923-928. [PubMed] [Google Scholar]
- 18.Guy-Grand D, Dy M, Luffau G, Vassalli P. Gut mucosal mast cells: origin, traffic, and differentiation. J Exp Med. 1984;160: 12-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friend DS, Ghildyal N, Austen KF, et al. Mast cells that reside at different locations in the jejunum of mice infected with Trichinella spiralis exhibit sequential changes in their granule ultrastructure and chymase phenotype. J Cell Biol. 1996;135: 279-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humbles AA, Lu B, Friend DS, et al. The murine CCR3 receptor regulates both the role of eosinophils and mast cells in allergen-induced airway inflammation and hyperresponsiveness. Proc Natl Acad Sci U S A. 2002;99: 1479-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corry DB, Grunig G, Hadeiba H, et al. Requirements for allergen-induced airway hyperreactivity in T and B cell-deficient mice. Mol Med. 1998;4: 344-355. [PMC free article] [PubMed] [Google Scholar]
- 22.McIntire JJ, Umetsu SE, Akbari O, et al. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol. 2001;2: 1109-1116. [DOI] [PubMed] [Google Scholar]
- 23.Boyce JA, Austen KF. No audible wheezing: nuggets and conundrums from mouse asthma models. J Exp Med. 2005;201: 1869-1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruegg C, Postigo AA, Sikorski EE, et al. Role of integrin alpha 4 beta 7/alpha 4 beta P in lymphocyte adherence to fibronectin and VCAM-1 and in homotypic cell clustering. J Cell Biol. 1992;117: 179-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berlin C, Berg EL, Briskin MJ, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74: 185-195. [DOI] [PubMed] [Google Scholar]
- 26.Andrew DP, Berlin C, Honda S, et al. Distinct but overlapping epitopes are involved in alpha 4 beta 7-mediated adhesion to vascular cell adhesion molecule-1, mucosal addressin-1, fibronectin, and lymphocyte aggregation. J Immunol. 1994; 153: 3847-3861. [PubMed] [Google Scholar]
- 27.Nakajima H, Sano H, Nishimura T, Yoshida S, Iwamoto I. Role of vascular cell adhesion molecule 1/very late activation antigen 4 and intercellular adhesion molecule 1/lymphocyte function-associated antigen 1 interactions in antigen-induced eosinophil and T cell recruitment into the tissue. J Exp Med. 1994;179: 1145-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu B, Wagner N, Pham LN, et al. Lymphocyte homing to bronchus-associated lymphoid tissue (BALT) is mediated by L-selectin/PNAd, alpha4beta1 integrin/VCAM-1, and LFA-1 adhesion pathways. J Exp Med. 2003;197: 1255-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chin JE, Hatfield CA, Winterrowd GE, et al. Airway recruitment of leukocytes in mice is dependent on alpha4-integrins and vascular cell adhesion molecule-1. Am J Physiol. 1997;272: L219-L229. [DOI] [PubMed] [Google Scholar]
- 30.Wolber FM, Curtis JL, Milik AM, et al. Lymphocyte recruitment and the kinetics of adhesion receptor expression during the pulmonary immune response to particulate antigen. Am J Pathol. 1997; 151: 1715-1727. [PMC free article] [PubMed] [Google Scholar]
- 31.Briskin M, Winsor-Hines D, Shyjan A, et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am J Pathol. 1997; 151: 97-110. [PMC free article] [PubMed] [Google Scholar]
- 32.Richards IM, Kolbasa KP, Hatfield CA, et al. Role of very late activation antigen-4 in the antigen-induced accumulation of eosinophils and lymphocytes in the lungs and airway lumen of sensitized brown Norway rats. Am J Respir Cell Mol Biol. 1996;15: 172-183. [DOI] [PubMed] [Google Scholar]