

Abstract
The B-cell receptor (BCR) transmits life and death signals throughout B-cell development, and altered BCR signaling may be required for survival of B-lymphoma cells. We used single-cell signaling profiles to compare follicular lymphoma (FL) B cells and nonmalignant host B cells within individual patient biopsies and identified BCR-mediated signaling events specific to lymphoma B cells. Expression of CD20, Bcl-2, and BCR light chain isotype (κ or λ) distinguished FL tumor B-cell and nontumor host B-cell subsets within FL patient biopsies. BCR-mediated signaling via phosphorylation of Btk, Syk, Erk1/2, and p38 occurred more rapidly in tumor B cells from FL samples than in infiltrating nontumor B cells, achieved greater levels of per-cell signaling, and sustained this level of signaling for hours longer than nontumor B cells. The timing and magnitude of BCR-mediated signaling in nontumor B cells within an FL sample instead resembled that observed in mature B cells from the peripheral blood of healthy subjects. BCR signaling pathways that are potentiated specifically in lymphoma cells should provide new targets for therapeutic attention.
Introduction
B-cell receptor (BCR) signaling regulates several B-cell fate decisions throughout development,1-5 and continued expression of the signaling subunits of the BCR is required for survival of mature B cells.6,7 Because BCR signaling is so closely linked with survival and proliferation of B cells, it is thought that alterations in BCR signaling may support lymphomagenesis.8,9 We have previously developed an approach to identify profiles of aberrant signaling in individual malignant or normal cells.10,11 The identification of altered BCR signaling in lymphoma cells might provide new opportunities to improve cancer therapy, as targeted inhibition of signaling associated with cancer progression and maintenance has proved successful for other malignancies.12
This study of BCR signaling in lymphoma builds on extensive prior work mapping the BCR signaling events13 and our own studies of BCR signaling kinetics in primary peripheral blood B cells.14 Here we measure BCR-mediated Btk, Syk, Erk1/2, and p38 (Figure 1A) signaling kinetics within different types of B cells in follicular lymphoma (FL) tumor specimens. These 4 signaling nodes are highlighted in a model of BCR signaling activation and regulation. Btk and Syk are key proximal signal transducers required for effective BCR-mediated activation of PI3K and PLCγ2.15-17 As such, changes to BCR signaling in lymphoma might be expected to alter the activity of Btk and Syk. Downstream effectors of BCR signaling include the MAPK family proteins Erk1/2 and p38.
Figure 1.
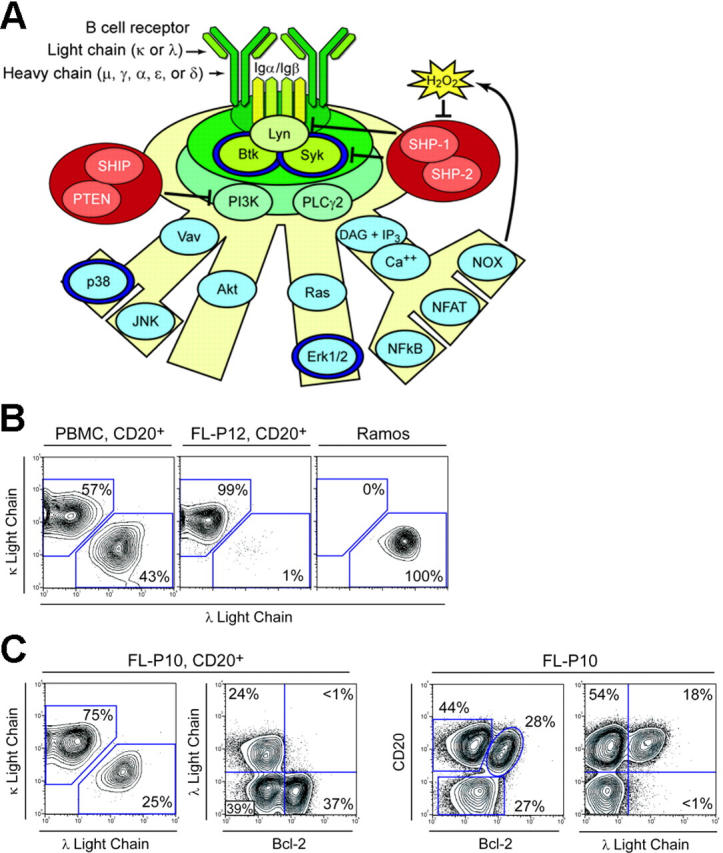
Distinguishing BCR signaling events and FL biopsy cell subsets by flow cytometry. (A) Phosphoproteins detected by flow cytometry (dark blue rings) are highlighted on a model of BCR signaling that includes regulation by protein tyrosine phosphatases. The BCR can instruct B cells to proliferate, alter innate immune signaling thresholds, induce B-cell anergy, or initiate cell death in a manner thought to depend on the strength, duration, and path of signaling. We detected BCR-mediated phosphorylation of Syk, Btk, Erk1/2, and p38 in subsets of primary human B cells. (B) Flow cytometry analysis of light chain isotype of CD20+ cells from a peripheral blood mononuclear cell (PBMC) sample (healthy donor), an FL tumor biopsy specimen from a different patient (FL-P12), and the Ramos lymphoma cell line. Ramos cells are clonal in origin and were of λ isotype. FL tumor cells in FL-P12 were κ isotype and vastly outnumbered the TIL B cells, as is commonly observed in FL tumor biopsy specimens. (C) Flow cytometry analysis of an FL biopsy specimen with an unusually large number of infiltrating nonmalignant B cells (FL-P10). Analysis of light chain isotypes present in the CD20+ subset of cells suggested that the tumor B cells were κ isotype and indicated that all B cells in the sample were exclusively κ or λ isotype. Expression of λ isotype was compared with Bcl-2 expression to identify FL tumor and nonmalignant B-cell populations. FL tumor B cells were κ isotype and overexpressed Bcl-2. Nonmalignant tumor-infiltrating host B cells did not overexpress Bcl-2 and were either κ or λ isotype. CD20 expression was also compared with Bcl-2 and with λ light chain expression in the total population of cells in the sample.
Signaling originating at the BCR can either promote proliferation or initiate programmed cell death, and it is not yet known whether BCR signaling in FL B cells is unaltered, amplified, or suppressed. Supporting the idea that BCR signaling might be altered in FL, evidence exists that the BCR in these malignant cells has somatic mutations that enable N-glycosylation.18,19 If BCR signaling supports the survival of lymphoma B cells, we would expect to see amplified signaling specific to FL B cells. However, BCR signaling might also induce apoptosis or cell cycle arrest in lymphoma B cells. The effectiveness of monoclonal antibody therapy directed against the idiotype of the lymphoma BCR20,21 has been directly correlated with the ability of these antibodies to induce BCR signal transduction.22 Thus, there is a need to determine whether BCR signal transduction is altered in lymphoma, to identify the direction of this alteration (potentiation or attenuation), and to dissect whether signaling changes are specific to lymphoma B cells or common to all B cells at the tumor site. The latter might occur if the lymphoma tumor microenvironment supported altered BCR signaling, in which case both normal and malignant B cells would be expected to show abnormal signaling.
Flow cytometry is particularly well suited to address these needs as it can measure multiple signaling events in individual cells while simultaneously distinguishing among cell subsets.10 Measuring several phosphoproteins by flow cytometry indicates whether signaling changes modify the per-cell level of signaling or alter the path of signaling (which phosphoproteins are activated following a common upstream signaling input). Here we measured a combination of lymphoma biomarkers and protein phosphorylation events by flow cytometry to determine whether FL cells alter BCR-mediated signaling and to identify which signaling changes were specific to lymphoma B cells. In addition, we asked how profiles of BCR signaling over time distinguished lymphomas from each other. Cell-intrinsic signaling mechanisms that distinguish lymphoma B cells from normal B cells could be explored as therapeutic targets that are specific to a patient's lymphoma cells.
Materials and methods
Isolation, storage, thawing, and equilibration of primary cells
Peripheral blood mononuclear cells (PBMCs) were isolated using density gradient separation (Ficoll-Paque Plus; Amersham Biosciences AB, Uppsala, Sweden). Tumor biopsy specimens from 7 individuals with FL were obtained with informed consent, in accordance with the Decleration of Helsinki. The study was approved by Stanford University's Administrative Panels on Human Subjects in Medical Research. Surgical biopsies of FL tumors were transferred directly from the operating room to the laboratory and used for the preparation of viable, sterile single-cell suspensions. Briefly, lymph node tissue was diced and forced through a metal sieve in a laminar flow hood into RPMI tissue culture medium. PBMCs or disaggregated FL biopsy cells were pelleted by low-speed centrifugation, resuspended in media composed of 90% fetal calf serum (HyClone, South Logan, UT) and 10% DMSO (Sigma, St Louis, MO), frozen slowly in the vapor phase of liquid nitrogen in multiple cryotubes, and stored in liquid nitrogen. For signaling analysis, an individual cryotube was thawed into 5 mL Stem Span H3000 serum-free medium (Stem Cell Technologies, Vancouver, BC), counted, pelleted, and resuspended at 6 × 106 cells/mL. Thawed PBMCs or FL tumor biopsy cells were allowed to rest at 37°C for a total of 2 hours in a 5% CO2 tissue culture incubator prior to stimulation.
Cell culture
The B-cell lymphoma cell line Ramos was from American Type Culture Collection (www.atcc.org, Manassas, VA) and was cultured in RPMI medium (Invitrogen, Carlsbad, CA), plus 10% fetal calf serum (Hy-Clone), 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM l-glutamine. Ramos cells were split at least 8 hours prior to observation of signaling such that cells achieved a density of approximately 1.0 × 106 cells/mL at the time of stimulation.
Stimulation of BCR signaling
At least 30 minutes before stimulation, 300 μL media containing 2 × 106 PBMCs, FL tumor biopsy, or Ramos cells were aliquoted into flow cytometry tubes (Falcon 2052; BD-Biosciences, San Jose, CA) and allowed to continue resting at 37°C in the 5% CO2 tissue culture incubator. Cells were then stimulated as previously performed with PBMCs14 by addition of 6 μL of a 0.5-mg/mL solution of the indicated F(ab′)2 to tubes, achieving a final concentration of 10 μg/mL of each F(ab′)2. Cross-linking of B-cell receptors was achieved using goat polyclonal α-IgM and α-IgG F(ab′)2 fragments (Biosource International, Camarillo, CA). When used, H2O2 was at 3.3-mM final concentration and was added as 2 μL of a 500-mM stock solution immediately prior to BCR cross-linking. Time-course studies of signaling kinetics were performed by addition of stimulus to tubes individually in reverse time points with simultaneous fixation of all samples in unison at “time zero.” Immediately after addition of stimulus, cells were returned to the 37°C incubator to allow signal transduction for the indicated time. To stop signaling, cells were fixed by addition of 15 μL of 32% paraformaldehyde (PFA; Electron Microscopy Services, Fort Washington, PA) to each 300-μL tube of cells for a final concentration of approximately 1.6%. To determine basal levels of phosphorylation, unstimulated cells were maintained in parallel with stimulated cells and fixed at time zero (labeled as “0 minute” in figures). Cells were fixed for 5 minutes at room temperature, pelleted, permeabilized by resuspension in 2 mL ice cold methanol for 10 minutes, and stored at 4°C for fewer than 3 days before being stained for flow cytometry. FL tumor B cells were identified as IgM (FL-P08, 09, 10) or IgG (FL-P07, 11) isotype Ig heavy chain based on the heavy chain isotype of the Bcl-2–expressing cells.
Intracellular phosphospecific flow cytometry
PFA-fixed, methanol-permeabilized cells were rehydrated by addition of 2 mL phosphate-buffered saline (PBS), resuspension by vortexing, and then centrifugation. The cell pellet was washed once with 2 mL PBS + 1% BSA (Sigma), resuspended in 50 μL PBS + 1% BSA, and then split evenly into 2 new fluorescence-activated cell sorting (FACS) tubes for staining. An antibody mix (50 μL) containing directly conjugated phosphospecific antibodies was added to each tube of cells and staining proceeded for 20 minutes at room temperature. Antibodies conjugated to Alexa (Ax) dye (Molecular Probes, Eugene, OR) or other fluorophores (all from BD Pharmingen, San Diego, CA) included phospho-Btk(Y551)/Itk(Y511)–Ax488, phospho-p38(T180/Y182)–Ax488, phospho-Erk1/2(T202/Y204)–PE, phospho-Syk(Y352)/Zap70(Y319)–Ax647, and CD20-cytoplasmic-PercPCy5.5. Antibodies were applied in panels (p38/Erk/CD20/Syk and Btk/CD20) for data shown in Figures 2, 3, 4. For Figures 1 and 5, 6, 7, λ-FITC F(ab)2, κ-FITC or κ-PE F(ab)2, and Bcl-2-PE were combined with different dye conjugates of the same phosphospecific antibodies in other panels (λ/κ/CD20, Syk/Bcl-2/CD20/Erk1/2, λ/Bcl-2/CD20/Btk, λ/Bcl-2/CD20/Syk, λ/Bcl-2/CD20/Erk1/2, and λ/Bcl-2/CD20/p38). Between 50 000 and 800 000 ungated events were collected for each sample on a benchtop FACSCalibur dual-laser cytometer (Becton Dickinson, Franklin Lakes, NJ).
Figure 2.

Activation of BCR signaling varies among primary FL samples and contrasts with that of normal B cells and a lymphoma cell line. (A) Flow cytometry analysis of signaling in PBMCs from a healthy blood donor, Ramos cells, and 5 FL patient samples (FL-P07–11) stimulated by BCR cross-linking (α-μ/γ) for various times in a short time course (1, 2, 4, 8, or 16 minutes) or left unstimulated (0 minute). BCR-mediated signaling in the CD20+ B-cell subset was compared by coloring heat map squares relative to the unstimulated PBMC sample. (B) As in panel A, with the addition of H2O2 just prior to BCR cross-linking. (C) Histogram data underlying the heat maps in panels A and B are shown for 2 samples, the lymphoma cell line Ramos and FL-P08.
Figure 3.

Rapid activation of BCR-mediated signaling in the presence of H2O2 is a common feature of FL sample B cells and differs from normal PBMC B cells. Differences in initiation of BCR-mediated signaling were compared in 5 tumor biopsy samples from individuals with FL and 5 PBMC samples from healthy donors. Samples were stimulated for 4 minutes by a combination of BCR cross-linking and H2O2 (α-μ/γ + H2O2) or left unstimulated. The average MFI of each phosphoprotein in CD20+ B cells has been graphed. Error bars indicate standard deviation.
Figure 4.

Altered BCR signaling kinetics distinguish individual FL samples. BCR-mediated phosphorylation of Btk, Syk, Erk1/2, and p38 was observed at numerous times over a 2-hour time course in 3 representative FL samples (FL-P07, FL-P10, FL-P11). Cells were stimulated by BCR cross-linking and H2O2 (α-μ/γ + H2O2) or left unstimulated (0 minute), and the MFI of CD20+ B cells was graphed for each phosphoprotein. The area under the curve has been shaded to indicate the sum of signaling induction over time in B cells from each FL sample. As a reference, the same plot of the average MFI and standard deviation observed in 3 samples of normal PBMCs (PBMC Average) is shown on each graph.
Figure 5.

Tumor cell–specific BCR-mediated Btk, Syk, Erk1/2, and p38 signaling. Flow cytometry contour plots of FL patient biopsy cells (FL-P09) stimulated by BCR cross-linking plus H2O2 (α-μ/γ + H2O2) for 4 or 30 minutes or left unstimulated (0 minute). BCR-mediated phosphorylation of Btk, Syk, Erk1/2, and p38 was compared in FL B cells (CD20+ Bcl-2hi λ–, dark arrow) and nonmalignant B cells (CD20hi Bcl-2lo, light arrow).
Figure 6.

Tumor cell–specific BCR-mediated Syk signaling. (A) Flow cytometry contour plots of FL patient biopsy cells (FL-P10) stimulated by BCR cross-linking alone (α-μ/γ) or by a combination of BCR cross-linking and H2O2 (α-μ/γ+ H2O2) for various times (4, 16, 30, 60, 90 minutes) or left unstimulated (0 minute). BCR-mediated phosphorylation of Syk was compared in CD20+ Bcl-2+ FL B cells and CD20+ Bcl-2– nonmalignant B cells (refer to Figure 1). Dark arrows indicate greater Syk signaling in Bcl-2+ FL tumor B cells than in nonmalignant host B cells. Light arrows indicate λ isotype B cells that failed to activate Syk in response to BCR cross-linking. (B) Flow cytometry contour plots of FL patient biopsy cells (FL-P10) stimulated by a combination of BCR cross-linking and H2O2 (α-μ/γ + H2O2) for various times (4, 16, 30, 60, or 90 minutes) or left unstimulated (0 minute). BCR-mediated phosphorylation of Syk was measured in CD20+ κ isotype B cells and compared with that in λ isotype nonmalignant B cells (all λ– cells are κ+). Dark arrows indicate sustained Syk signaling in Bcl-2+ FL tumor B cells. Light arrows indicate where Syk signaling in λ isotype B cells differed from κ isotype B cells.
Figure 7.

Tumor cell–specific BCR-mediated signaling is common in FL. (A) BCR-mediated signaling at 4 and 30 minutes following BCR cross-linking in the presence of H2O2 (α-μ/γ+ H2O2) was measured in subsets of the CD20+ B cells from 4 FL patient samples (FL-P07, FL-P08, FL-P09, FL-P11). FL B cells (CD20+ Bcl-2hi nontumor light chain–, dark arrow) and tumor-infiltrating nonmalignant B cells (CD20+ Bcl-2lo nontumor light chain+, light arrow) were distinguished and their signaling was compared by coloring heat map squares relative to the fold induction of phosphorylation relative to the unstimulated sample (0 minute). (B) Flow cytometry analysis of FL patient biopsy cells (FL-P12) stimulated by a combination of BCR cross-linking and H2O2 (α-μ/γ+ H2O2) for various times (4, 30, 60, or 90 minutes) or left unstimulated (0 minute). BCR-mediated phosphorylation of Erk1/2 and p38 was measured in nonmalignant B cells (CD20+ λ isotype, light arrow) and compared with that in FL B cells (CD20+ Bcl-2+ λ–, dark arrow).
Results
Individual cell detection of tumor and nontumor cell populations within lymphoma biopsies
We first asked whether B-cell receptor isotype and hallmark molecular features of FL tumor cells could be used as flow cytometry biomarkers10 to distinguish malignant B cells from infiltrating host B and T cells. In normal PBMCs, the κ/λ ratio was 1.3:1 (Figure 1B). However, clonal expansion of FL B cells skews the proportion of BCR isotypes in FL tumor samples to favor the lymphoma B cells (Figure 1B). As an example of absolute clonality, the λ isotype lymphoma B-cell line Ramos is shown (Figure 1B). The nontumor light chain isotype can thus be used as a marker for the host nontumor B cells.
We next sought to add another biomarker that would further distinguish the B-cell populations in FL patient samples. In the vast majority of FL cases,23,24 the hallmark translocation t(14;18) juxtaposes the BCL2 gene and the immunoglobulin heavy chain promoter. The resulting deregulation of the BCL2 gene enables overexpression of Bcl-2 protein in lymphoma cells.25 To test our ability to discriminate host and lymphoma B-cell populations, we examined an FL sample with an uncommonly large number of tumor-infiltrating nonmalignant cells present in the biopsy. A subpopulation of 28% of the total cells (38% of the B cells) that overexpressed Bcl-2 was detected by flow cytometry in this FL sample (Figure 1C). The κ/λ ratio in the CD20+ subset of cells was 3:1, suggesting that the tumor B-cell isotype was κ (Figure 1C). By comparing λ, the nontumor light chain isotype, against Bcl-2, it was seen that Bcl-2 overexpression was restricted to the κ isotype FL tumor cells (Figure 1C). Notably, the nontumor host B cells infiltrating this sample retained a more normal κ/λ ratio of 1.6:1 (39%:24%). FL patient specimens are more typically composed almost entirely of FL B cells, as was the case with FL-P12 (Figure 1B).
These 3 FL biomarkers were used for the remainder of the study. CD20 expression was used to identify the B-cell subset, the overexpression of Bcl-2 served as a marker for FL tumor B cells, and the nontumor light chain isotype was used to positively identify a subset of nontumor B cells.
BCR signaling effectors are not constitutively phosphorylated in FL
We first assessed the resting phosphorylation level of BCR signaling effectors Btk, Syk, Erk1/2, and p38 in FL B cells from several patient samples, PBMC B cells, and Ramos B cells (Figure 2A, 0 minute column; Figure S1, available on the Blood website; see the Supplemental Figures link at the top of the online article). The median fluorescence intensity (MFI) of samples was compared by coloring heat map squares or histogram peaks according to the log10 fold difference between the sample and B cells from normal PBMCs. In this color scale, black indicates no difference, lighter yellow indicates increased phosphoprotein MFI, and blue indicates decreased phosphoprotein MFI, relative to the control. In order to demonstrate the relationship between the heat map data and the primary flow cytometry data, stimulation of Btk, Syk, Erk1/2, and p38 phosphorylation in 2 samples is shown with both heat maps (Figure 2A-B) and histograms (Figure 2C) for Ramos and FL-P08.
Per-cell levels of basal Btk, Syk, and p38 phosphorylation were virtually identical in resting PBMC and FL samples (Figure 2B). The average level of basal Erk1/2 phosphorylation in FL was also comparable with normal PBMCs. Notably, Erk1/2 phosphorylation varied more among FL samples than it did in samples of normal PBMC B cells (Figure 2B). Ramos cells are shown as an example of high basal (constitutive) phosphorylation (Figure 2). This high basal phosphorylation in Ramos contrasted with primary FL B cells. Because resting levels of BCR signaling effectors were comparable in FL and PMBC B cells, we next asked whether these phosphoproteins were present and able to respond to BCR stimulation.
Altered BCR-mediated signaling in FL
In order to examine the initiation of BCR-mediated signaling, the same samples were stimulated by cross-linking BCR heavy chains (α-μ/γ), and phosphorylation was analyzed during the initiation of signaling in the first 16 minutes (Figure 2A, 1 minute-16 minutes). The color scale indicates fold difference in phosphorylation of CD20+ B cells stimulated by BCR cross-linking over the level in resting PBMCs.
Surprisingly, signaling via Btk, Syk, and p38 phosphorylation was often absent in FL B cells stimulated by BCR cross-linking (Figure 2A). Stimulation of Ramos cells is shown in order to demonstrate the effectiveness of BCR cross-linking (Figure 2). BCR cross-linking did not induce Btk phosphorylation in FL sample B cells and only infrequently stimulated Syk phosphorylation (Figure 2A). Levels of BCR-mediated Erk1/2 phosphorylation were independent of Btk, Syk, and p38 phosphorylation and were observed to vary widely among cases of FL (Figure 2A). The widespread variation in Erk1/2 signaling kinetics among FL biopsy samples from different individuals contrasted with consistent signaling kinetics we observed in PBMC samples from different healthy blood donors.14 BCR-mediated phosphorylation of p38 was significantly attenuated in all 5 FL samples, compared with normal PBMC B cells (Figure 2A).
Both Erk1/2 and p38 phosphorylation are considered to be downstream signaling events that depend on upstream Syk activity (Figure 1A). However, phosphorylation of Syk was not related to Erk1/2 or p38 phosphorylation in primary FL sample B cells. In a particularly striking example (FL-P09), BCR cross-linking was followed by lower than normal levels of both Syk and Btk phosphorylation in parallel with greater than normal induction of Erk1/2 phosphorylation and no induction of p38 phosphorylation (Figure 2A). Per-cell analysis comparing Erk1/2 and Syk phosphorylation indicated that the MFI of Erk1/2 phosphorylation in FL B cells increased 5- to 10-fold, independent of an increase in Syk phosphorylation (Figure S2).
H2O2 enables rapid activation of BCR-mediated signaling in FL
In normal B cells, BCR signaling is controlled by BCR-mediated protein tyrosine phosphatases (PTPs) that dephosphorylate Btk, Syk, Erk1/2, and p38 by 8 to 16 minutes following stimulation.14 The lack of BCR-mediated signaling in FL B cells suggested that increased basal phosphatase activity might be present in FL cells. H2O2, the primary redox species generated by the BCR,26 regulates the magnitude and duration of BCR signaling and reversibly inhibits BCR-mediated PTP activity.14 In order to study the role of PTPs in FL, BCR-mediated signaling was initiated immediately after the addition of 3.3 mM H2O2 (α-μ/γ+ H2O2) in FL, PBMCs, and Ramos cells. Addition of 3.3 mM H2O2 alone did not lead to significant signaling in primary B cells but activated BCR signaling pathway phosphoproteins in Ramos cells (Figure S3).
BCR cross-linking in the presence of H2O2 led to extremely rapid and robust phosphorylation of Btk, Syk, Erk1/2, and p38 (Figure 2B). Following BCR cross-linking in the presence of H2O2, levels of protein phosphorylation usually accumulated rapidly (Figure 2B). We had previously observed that normal B cells from peripheral blood required 30 to 60 minutes to reach maximum Erk1/2 phosphorylation following BCR cross-linking in the presence of H2O2.14 In only 4 minutes, BCR-mediated Erk1/2 phosphorylation in many FL samples reached this maximum observed in normal B cells (Figure 2C: FL-P08, FL-P09, FL-P10, FL-P11). Erk1/2 phosphorylation went on to exceed the normal maximum MFI in fewer than 16 minutes. p38 signaling was initiated more slowly than Erk1/2 signaling and followed a different pattern of activation. Cells with high Erk1/2 phosphorylation did not necessarily have higher p38 phosphorylation (Figure 2A-B).
These results of basal and BCR-mediated phosphorylation in the 5 cases of FL and in PBMC B cells are summarized in Figure 3. Individual variation was observed in basal phosphorylation, but no significant difference in average basal phosphorylation was observed between FL and PBMCs. Typically, BCR-mediated phosphorylation in B cells from normal PBMCs was only slightly increased above basal levels at 4 minutes following BCR cross-linking and H2O2. In contrast, phosphorylation of Btk, Syk, Erk1/2, and p38 were, on average, significantly higher in FL B cells than in PBMC B cells at 4 minutes following BCR stimulation (Figure 3).
BCR signaling kinetics distinguish individual cases of FL
We sought to integrate FL signaling measurements over time and obtain a kinetic profile of BCR stimulation in FL that could be compared with that of normal B cells. We measured the phosphorylation of Btk, Syk, Erk1/2, and p38 at 10 time points over a 2-hour period following BCR cross-linking in the presence of H2O2 (Figure 4). The MFI for each phosphoprotein was measured in CD20+ B cells from 3 FL patient samples (Figure 4: FL-P07, FL-P10, FL-P11). BCR signaling kinetics measured in comparable experiments using PBMCs14 are displayed for comparison with FL B cells.
BCR signaling strength could be amplified either by extending the duration of time a phosphoprotein spends at the normal maximum level of activity or by temporarily increasing the activity above the maximum level. In an example of the first (FL-P07), Erk1/2 phosphorylation was observed to plateau at the maximum observed in normal PBMCs within the first 4 minutes following BCR stimulation plus H2O2 (Figure 4). This rapid activation of signaling distinguished all FL samples from normal PBMCs. In addition, in some FL samples (eg, FL-P10 and FL-P11) Erk1/2 phosphorylation was sustained at greater than the normal PBMC maximum for approximately 90 minutes. The integral of Erk1/2 phosphorylation over time was significantly greater in each FL sample than in normal PBMCs (Figure 4). Differences in BCR-mediated signaling between FL-P07 and FL-P11 observed in the presence of H2O2 are notable because these 2 samples exhibited similar suppression of BCR-mediated signaling phenotypes following BCR cross-linking alone (Figure 2B). The signal from all of the CD20+ B cells in each sample is shown here (Figure 4), but it was known that FL tumor samples had significant populations of nontumor B cells. In subsequent experiments, we asked whether these alterations to BCR signaling were specific to the FL tumor B cells.
Altered BCR signaling specific to FL tumor B cells
We next examined the tumor-infiltrating nontumor B cells from a typical FL specimen (FL-P09) where host TIL B cells comprised only 1.4% of the cells (Figure 5). Phosphorylation of Btk, Syk, Erk1/2, and p38 in FL tumor B cells (Bcl-2 overexpressing, κ isotype/λ–, CD20+) was compared with that in nontumor B cells (CD20hi, Bcl-2lo, mixture of κ and λ). In malignant B cells from FL-P09, significantly greater than normal responses of Btk, Syk, Erk1/2, and p38 were observed following BCR cross-linking in the presence of H2O2. In contrast, signaling in the nonmalignant B cells infiltrating this FL sample was lower than normal, indicating that the altered signaling responses were specific to the FL tumor B cells.
In another sample (FL-P10), 62% of the B cells in the tumor biopsy were nonmalignant host B cells (Figure 1). BCR-mediated Syk signaling kinetics were examined with and without H2O2 in tumor and nontumor B-cell subsets (Figure 6). Following BCR cross-linking alone, Syk phosphorylation was observed only in the FL tumor B cells that overexpressed Bcl-2 (Figure 6A, dark arrows). Following BCR cross-linking in the presence of H2O2, Syk was rapidly phosphorylated in both FL and nonmalignant B cells. However, FL tumor B cells had a higher per-cell level of Syk phosphorylation than nonmalignant B cells and sustained this level for at least 90 minutes (Figure 6A). In contrast, nonmalignant B cells displayed signaling kinetics equivalent to those of normal PBMC B cells when stimulated by BCR cross-linking in the presence of H2O2. With H2O2 present, Syk phosphorylation in λ isotype B cells was compared with the level in κ isotype cells from the same stimulated sample or with resting cells (Figure 6B). Syk phosphorylation in λ isotype nontumor B cells followed kinetics comparable with normal PBMC B cells, and declined to basal levels by 60 minutes (Figure 6B), unlike κ isotype B cells from the same sample or FL tumor B cells that overexpressed Bcl-2 (Figure 6B). In summary, altered BCR-mediated Syk signaling was specific to FL tumor B cells.
In order to determine how commonly signaling in FL tumor B cells differed from the tumor-infiltrating host B cells, we distinguished between tumor and nontumor B-cell signaling in 4 FL samples following BCR cross-linking in the presence of H2O2 (Figure 7). Malignant FL B cells were distinguished by Bcl-2 expression and clonal BCR light chain isotype (FL), whereas nontumor B cells were identified by nontumor BCR light chain isotype and low Bcl-2 expression (TIL B). Consistent with prior results, we observed that FL and TIL B cells had similar resting phosphoprotein levels (0 minute), but following stimulation by BCR cross-linking in the presence of H2O2 the FL tumor B cells displayed greater than normal signaling of Btk, Syk, Erk1/2, and p38, while the nonmalignant TIL B cells displayed normal or suppressed signaling responses (Figure 7A). Consistent with previous results, a lack of signaling in response to BCR cross-linking alone was observed for these FL samples (Figure S4). Most commonly, Erk1/2 phosphorylation following BCR cross-linking in the presence of H2O2 was significantly greater in FL B cells than in host TIL B cells (Figure 7A).
In order to highlight the flow cytometry data underlying the heat map, histogram data for Erk1/2 and p38 are shown for an additional FL sample (FL-P12) that was representative of the commonly observed difference between FL and TIL B cells within a sample (Figure 7B). In malignant B cells from FL-P12 overexpressing Bcl-2, Erk1/2, and p38, phosphorylation following BCR cross-linking in the presence of H2O2 was greater than normal (Figure 7B, FL). In contrast, nonmalignant λ isotype B cells displayed Erk1/2 and p38 phosphorylation that was more typical of normal PBMC B cells (Figure 7B, TIL B).
Discussion
By following the phosphorylation initiated by BCR signaling over time, it was possible to identify differences in BCR signaling kinetics between lymphoma and nonmalignant B-cell populations from healthy individuals and even within FL tumors. Distinguishing the signaling of tumor and nonmalignant B cells (Figures 5, 6) indicated that the BCR signaling network of lymphoma cells differed from normal B cells in the speed, duration, and per-cell magnitude of the signal sent. In comparison with normal PBMC B cells from healthy blood donors, individual lymphoma B cells activated BCR network signaling more quickly and stayed at the maximum level of signaling for a longer period of time. We identified time points and states under which signaling nodes could be compared between different FL tumors. BCR cross-linking in the presence of H2O2 will be particularly useful, since large differences in BCR signaling kinetics among individuals become apparent (Figure 4). The level of signaling achieved at 4 minutes and 30 minutes proved useful in distinguishing among FL samples (Figures 4 and 7).
A key measurement in evaluation of signaling kinetics is the starting level of phosphorylation, also known as the basal state or level of constitutive signaling. Constitutive signaling that results in increased basal phosphorylation is commonly observed in proliferative disorders of immune system cells.10,11 In FL, the average basal phosphorylation of Btk, Syk, Erk1/2, and p38 proteins did not differ between B cells from FL tumors and from healthy peripheral blood (Figure 3). This lack of constitutive increases in signaling in primary FL tumors contrasted with constitutive signaling observed in the Ramos cell line (Figure 2). However, the variance of basal Erk1/2 phosphorylation was greater than normal in FL patient samples. This variability in Erk1/2 phosphorylation is significant because greater than normal variance in signaling indicates a parameter that might be useful in stratifying FL cases according to signaling.10,11
Although phosphoprotein levels in FL B cells started at approximately the same point as in normal PBMC B cells, the response to BCR-mediated stimulation best distinguished FL B cells from normal PBMCs and nonmalignant B cells. A surprising result was that phosphorylation of Btk, Syk, and p38 following BCR cross-linking alone was lower in B cells from FL samples than in normal cells (Figure 2C). We used Ramos and several other B-cell lines as controls to optimize conditions to study BCR signaling. Unlike Btk, Syk, and p38, phosphorylation of Erk1/2 following BCR cross-linking was increased in some FL sample B cells and decreased in others. Potentiated Erk1/2 phosphorylation following BCR cross-linking in the FL-P09 sample was independent of Syk and Btk phosphorylation (Figure 2 and Figure S2A), suggesting that a novel signaling route between the BCR and Erk1/2 can be activated in FL B cells. This alternative connection to Erk1/2 may not be regulated by the same factors that control Syk and Btk activity. Such differences in the response of phosphoproteins to stimulation are not apparent in resting cells. These results further highlight the importance of measuring both signaling activity following stimulation and basal differences in protein phosphorylation in signaling profiles.10
The attenuation of BCR-mediated signaling we observed in FL (Figure 2A) could be explained by a loss or mutation of upstream signaling molecules. Alternatively, this attenuation of BCR-mediated signaling could also be the result of increased negative regulation. Restoration of signaling in FL B cells following inactivation of negative regulation would distinguish between these possibilities by indicating the signaling pathways were intact but suppressed. The ability of BCR cross-linking in the presence of H2O2 to activate robust signaling via all 4 phosphoproteins (Figure 2C) indicates that the attenuation of some BCR signaling events in FL (Figure 2B) is due to an increase in negative regulation, rather than a permanent loss or mutation of an upstream activator.
After PTPs were inhibited with H2O2, significant differences were observed among patient samples. Signaling kinetics in FL-P11 and FL-P07 were similar following BCR cross-linking (Figure 2B), but phosphatase inhibition by H2O2 additionally distinguished greater than the normal maximum Btk and p38 signaling in FL-P11 that was not seen in FL-P07 (Figure 2B). In another sample (FL-P10), Syk phosphorylation following BCR cross-linking was totally suppressed in the nonmalignant B cells, but was active in the FL B cells (Figure 6). When H2O2 was present during BCR stimulation, nonmalignant λ isotype B cells from FL-P10 could be stimulated, and Syk signaling was comparable with that in normal PBMC B cells (Figure 6). In the tumor B cells from FL-P10, Syk phosphorylation following BCR cross-linking in the presence of H2O2 was sustained much longer than in the nontumor B cells (Figure 6). Taken together, these results suggest that the PTP activity that regulates Syk signaling was heightened in B cells within this FL sample. Tumor-cell–intrinsic changes to BCR-mediated signaling enabled phosphorylation of Syk in B cells from FL tumors following BCR cross-linking alone. Such tumor cell–specific signaling changes may promote lymphomagenesis or sustain FL tumor B cells.
A model consistent with our results would be that BCR signaling can occur more easily in FL cells and is commonly suppressed by a PTP feedback mechanism. If BCR cross-linking did not completely overcome this PTP regulation, then BCR-mediated signaling would appear suppressed, as in Figures 2A and 7. Furthermore, addition of an inhibitor of PTPs, such as H2O2, would be expected to inactivate PTP feedback regulation of BCR signaling in lymphoma cells, reverse this phenotype, and enable greater than normal signaling, as we frequently observed. This model is consistent with the detection of common BCR mutations in FL.18,19 Such mutations might promote close associations of BCR subunits, alter the interaction of BCR subunits with coreceptors on the surface of the tumor B cells, or enable additional tumor-stromal interactions. Differences in the expression of the BCR or coreceptors that positively (CD19) and negatively (CD22) regulate BCR signaling might also account for altered signaling that we observed in FL B cells (Figure 7). Although we observed no difference in per-cell expression of BCR heavy chain among FL samples, the per-cell level of CD19 and CD22 varied widely among FL samples (data not shown).
This is the first study to date that compares signaling kinetics in primary tumor and nonmalignant cells of the same lineage within the same patient biopsy specimen. A key advantage of using tumor-infiltrating host cells for comparison is that any differences in signaling between tumor and nonmalignant cells are completely specific to the cells (cell intrinsic) and are not caused by differences in sample handling or in tissue origin. BCR signaling in nonmalignant B cells within FL samples could be altered by the tumor microenvironment, and in one case we observed that BCR cross-linking that activated FL B cells was insufficient to activate infiltrating host B cells. A significant remaining challenge is to map differences in BCR signaling kinetics between populations of normal B cells (eg, bone marrow developmental subsets, B cells at different stages of maturity,14 B cells in different organ compartments) for comparison with signaling profiles observed in B-cell proliferative disorders. FL B cells might retain aspects of the normal BCR signaling phenotype of germinal center B cells, or it may be that FL B cells have acquired an independent phenotype. The infiltrating nonmalignant B cells, whose BCR signaling was often like that of normal peripheral blood B cells, suggest that the FL B cells acquired signaling phenotypes unlike that of the host's normal B-cell population. Signaling differences between normal B cells and other B-cell malignancies, such as diffuse large B-cell lymphoma, can also be interrogated, as in FL. Signaling changes specific to tumor B cells could drive the progression of tumor cells or support their continued proliferation and survival, and such B-cell–specific signaling mechanisms might be the targets of new therapies.
Supplementary Material
Acknowledgments
We thank R. Ihrie for review of this paper and W. K. Weng for many useful discussions.
Prepublished online as Blood First Edition Paper, July 11, 2006; DOI 10.1182/blood-2006-02-003921.
G.P.N. and R.L. contributed equally to this work.
Supported by the National Institutes of Health (CA 34233 and CA 33399 to R.L.), the National Heart, Lung, and Blood Institute (N01-HV-28183I to G.P.N.), and a Specialized Center of Research grant from the Leukemia and Lymphoma Society (R.L. and G.P.N). J.M.I. is a Leukemia and Lymphoma Society Fellow. R.L is a Clinical Research Professor of the American Cancer Society.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Fleming HE, Paige CJ. Pre-B cell receptor signaling mediates selective response to IL-7 at the pro-B to pre-B cell transition via an ERK/MAP kinase-dependent pathway. Immunity. 2001;15: 521-531. [DOI] [PubMed] [Google Scholar]
- 2.Reth M, Wienands J. Initiation and processing of signals from the B cell antigen receptor. Annu Rev Immunol. 1997;15: 453-479. [DOI] [PubMed] [Google Scholar]
- 3.Allman D, Srivastava B, Lindsley RC. Alternative routes to maturity: branch points and pathways for generating follicular and marginal zone B cells. Immunol Rev. 2004;197: 147-160. [DOI] [PubMed] [Google Scholar]
- 4.Hartley SB, Cooke MP, Fulcher DA, et al. Elimination of self-reactive B lymphocytes proceeds in two stages: arrested development and cell death. Cell. 1993;72: 325-335. [DOI] [PubMed] [Google Scholar]
- 5.Casellas R, Shih TA, Kleinewietfeld M, et al. Contribution of receptor editing to the antibody repertoire. Science. 2001;291: 1541-1544. [DOI] [PubMed] [Google Scholar]
- 6.Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004;117: 787-800. [DOI] [PubMed] [Google Scholar]
- 7.Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90: 1073-1083. [DOI] [PubMed] [Google Scholar]
- 8.Petlickovski A, Laurenti L, Li X, et al. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood. 2005;105: 4820-4827. [DOI] [PubMed] [Google Scholar]
- 9.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5: 251-262. [DOI] [PubMed] [Google Scholar]
- 10.Irish JM, Kotecha N, Nolan GP. Mapping normal and cancer cell signalling networks: towards single-cell proteomics. Nat Rev Cancer. 2006;6: 146-155. [DOI] [PubMed] [Google Scholar]
- 11.Irish JM, Hovland R, Krutzik PO, et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118: 217-228. [DOI] [PubMed] [Google Scholar]
- 12.Klein S, McCormick F, Levitzki A. Killing time for cancer cells. Nat Rev Cancer. 2005;5: 573-580. [DOI] [PubMed] [Google Scholar]
- 13.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2: 945-956. [DOI] [PubMed] [Google Scholar]
- 14.Irish JM, Czerwinski DK, Nolan GP, Levy R. Kinetics of B cell receptor signalling in human B cell subsets mapped by phosphospecific flow cytometry. J Immunol. 2006;177: 1581-1589. [DOI] [PubMed] [Google Scholar]
- 15.Kurosaki T, Johnson SA, Pao L, Sada K, Yamamura H, Cambier JC. Role of the Syk autophosphorylation site and SH2 domains in B cell antigen receptor signaling. J Exp Med. 1995;182: 1815-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takata M, Kurosaki T. A role for Bruton's tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med. 1996;184: 31-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beitz LO, Fruman DA, Kurosaki T, Cantley LC, Scharenberg AM. SYK is upstream of phosphoinositide 3-kinase in B cell receptor signaling. J Biol Chem. 1999;274: 32662-32666. [DOI] [PubMed] [Google Scholar]
- 18.McCann KJ, Johnson PW, Stevenson FK, Ottensmeier CH. Universal N-glycosylation sites introduced into the B-cell receptor of follicular lymphoma by somatic mutation: a second tumorigenic event? Leukemia. 2006;20: 530-534. [DOI] [PubMed] [Google Scholar]
- 19.Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood. 2002;99: 2562-2568. [DOI] [PubMed] [Google Scholar]
- 20.Miller RA, Maloney DG, Warnke R, Levy R. Treatment of B-cell lymphoma with monoclonal anti-idio-type antibody. N Engl J Med. 1982;306: 517-522. [DOI] [PubMed] [Google Scholar]
- 21.Davis TA, Maloney DG, Czerwinski DK, Liles TM, Levy R. Anti-idiotype antibodies can induce long-term complete remissions in non-Hodgkin's lymphoma without eradicating the malignant clone. Blood. 1998;92: 1184-1190. [PubMed] [Google Scholar]
- 22.Vuist WM, Levy R, Maloney DG. Lymphoma regression induced by monoclonal anti-idiotypic antibodies correlates with their ability to induce Ig signal transduction and is not prevented by tumor expression of high levels of bcl-2 protein. Blood. 1994;83: 899-906. [PubMed] [Google Scholar]
- 23.Zelenetz AD, Chu G, Galili N, et al. Enhanced detection of the t(14;18) translocation in malignant lymphoma using pulsed-field gel electrophoresis. Blood. 1991;78: 1552-1560. [PubMed] [Google Scholar]
- 24.Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47: 19-28. [DOI] [PubMed] [Google Scholar]
- 25.Ngan BY, Chen-Levy Z, Weiss LM, Warnke RA, Cleary ML. Expression in non-Hodgkin's lymphoma of the bcl-2 protein associated with the t(14;18) chromosomal translocation. N Engl J Med. 1988;318: 1638-1644. [DOI] [PubMed] [Google Scholar]
- 26.Singh DK, Kumar D, Siddiqui Z, Basu SK, Kumar V, Rao KV. The strength of receptor signaling is centrally controlled through a cooperative loop between Ca2+ and an oxidant signal. Cell. 2005;121: 281-293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.