

Abstract
Deregulation of signal transduction pathways (STPs) may promote leukemogenesis by conferring cell proliferation and survival advantages in acute myelogenous leukemia (AML). Several agents targeting STPs are under development; however, redundancy and cross-talk between STPs could activate multiple downstream effectors and this could negate the effect of single-target inhibition. The frequency of concurrent activation of multiple STPs in AML and the prognostic relevance of STP activation in AML are unknown. STP protein expression (PKCα, ERK2, pERK2, AKT, and pAKT) was measured by Western blot in samples from 188 patients with newly diagnosed, untreated AML. In univariate and multivariate analysis high levels of PKCα, ERK, pERK, and pAKT, but not AKT, were adverse factors for survival as was the combination variable PKCα-ERK2&pERK2-pAKT. Survival progressively decreased as the number of activated pathways increased. Patients were more likely to have none or all 3 pathways activated than was predicted based on the frequency of individual pathway activation, strongly suggesting that cross-activation occurred. Simultaneous activation of multiple STPs is common in AML and has a progressively worse adverse effect on prognosis. It is thus likely that only combinations of agents that target the multiply activated STPs will be beneficial for patients with AML.
Introduction
Proliferation, differentiation, and apoptosis of normal and leukemic hematopoietic stem cells are partially regulated by external signals received from chemokines and cytokines and by interactions with the local microenvironment. The response to these signals is, in turn, transmitted from the cell surface to the nucleus through the extensive series of signal transduction pathways (STPs), including the JAK/STAT, RAS/Raf/MEK/ERK, and PI3K/AKT pathways.1 There is extensive cross-talk and cross-activation among these pathways, so that the activation of one pathway often leads to the activation of others (Figure 1). The disruption of normal signaling through these pathways, occurring as a result of either the mutation of pathway components or alterations in the internal and external (from chemokines, cytokines, or stroma) signals received, is thought to contribute to leukemogenesis by perturbing the rates of proliferation, differentiation, and apoptosis.1-3
Figure 1.
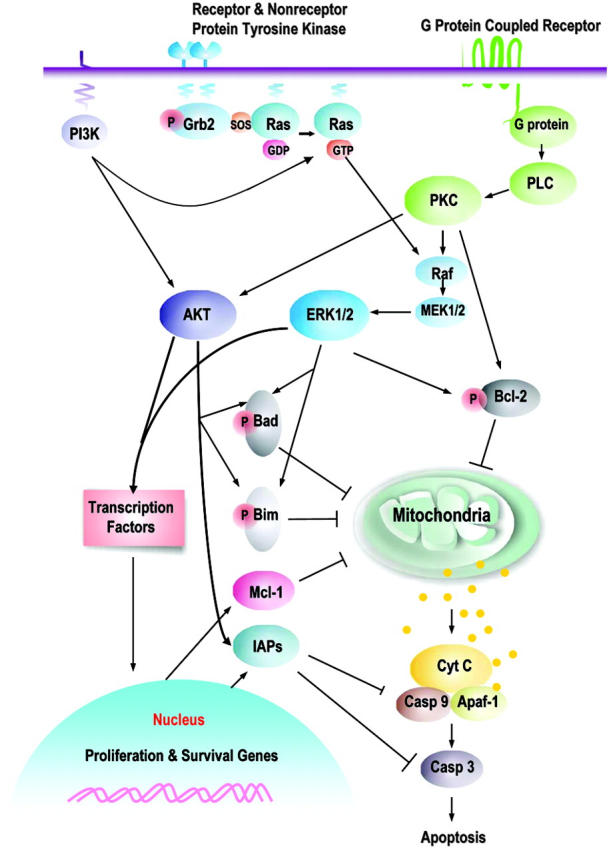
Prosurvival signaling pathways and their downstream targets. This is a schematic illustration of the principal pathways that are discussed in this paper. Growth factors induce, or membrane-associated molecules dimerize, spontaneously to activate mitogen-activated protein kinases (Raf/MEK/ERK) and PI3K/AKT signaling pathways. Transmembrane G protein–coupled receptors can activate PKC, which modulates the activity of Raf and MAPK pathway; both ERK1/2 and PKCα can serve as Bcl-2 kinases at serine 70. AKT and ERK induce phosphorylation of Bim (AKT, at serine 87 and ERK, at serines 55, 65, and 100), which attenuates the proapoptotic function of Bim, thereby promoting cell survival. Phosphorylation of Bad by MAPK (serine 112) or AKT (serine 136) inhibits apoptosis due to loss of the ability of Bad to heterodimerize with the survival proteins Bcl-2 and Bcl-XL. Hence, prosurvival phosphorylation of Bcl-2 family proteins modulates their antiapoptotic or proapoptotic activity at the mitochondrial membrane. This is complemented by AKT- or MAPK-driven gene transcription, which includes cyclins D and E (MAPK), cyclin-dependent kinase inhibitor p21, c-myc (AKT), which cause an increase in cell proliferation; and antiapoptotic proteins of the Bcl-2 (Mcl-1) and IAP (XIAP, survivin) families that regulate apoptosis at the level (Mcl-1) or downstream of mitochondria (IAPs). It is apparent that simultaneous activation of multiple signaling pathways might synergistically enhance prosurvival and proliferative potential of leukemic cells and the redundant downstream pathways negatively affect an ability of a particular signal transduction inhibitor to eliminate leukemia.
The functional effects of modulating the expression or activation of STP proteins have been evaluated in leukemia-cell lines and primary leukemia samples.
One of these pathways, the PI3K/AKT pathway, has been extensively studied in normal and malignant cells. The pathway is activated when threonine 308 is phosphorylated by PDK1 and serine 473 (thought to be more crucial for full activation) is phosphorylated by PDK2. Indeed, constitutive phosphorylation at serine 473 was found in 21 of 22 samples from patients with acute myelogenous leukemia (AML) with high blast counts and was an adverse prognostic factor for survival among 55 patients with AML.4 Phosphorylated AKT (pAKT), in turn, phosphorylates Bad, NF-κB, GSK/β-catenin, mTOR, caspases, and waf1, resulting in increased glucose flux and cell proliferation, and protection against apoptosis.1 In 2 studies, activated AKT was observed to promote leukemogenesis in non-transformed hematopoietic precursors.5,6 Conversely, in short-term cultures of cells from 10 cases of primary AML with high blast percentages, the blockade of AKT phosphorylation by LY294002 decreased cell survival and significantly reduced clonogenic cell growth by inducing apoptosis,7,8 whereas other AKT inhibitors have been shown to slow the progression of tumors.9
The RAS/Raf/MEK/ERK pathway is a key STP that has been demonstrated to result in increased cell proliferation, differentiation, and survival along with angiogenesis and metastasis.1,10 Activation of this pathway has been demonstrated in numerous human malignancies, including leukemia.1,10-12 We have demonstrated that conditional activation of this pathway in murine hematopoietic cells results in an AML-like phenotype.13 Mutations of upstream RAS and Raf are frequent, but mutations in MEK and ERK are uncommon, suggesting that activation of this pathway is triggered by changes elsewhere in the cell,1 possibly including changes in the level or function of ERK phosphatases including PAC112 and MKP-1.14 Conversely, the blockade of ERK activation in leukemia-cell lines was observed to result in cell death and, when combined with chemotherapy, to synergistically increase cell kill.15-18 In a retrospective study, we have demonstrated that the constitutive activation of ERK in primary AML samples is common and an adverse prognostic feature.19 Elegant studies using conditionally active kinase constructs indicate a requirement for the activation of the PI3K pathway during MEK-mediated transformation of certain hematopoietic cells20 and provide evidence that expression of activated Raf/MEK/ERK and PI3K/Akt pathways could synergize and result in the abrogation of cytokine dependence of hematopoietic cells.21
PKCα is a member of a family of cytoplasmic serine/threonine kinases with varying dependence on phospholipids and Ca2+ as cofactors and is widely expressed in normal tissue, including myeloid cells.22,23 PKCα must also be phosphorylated to be activated, and this is triggered by many growth factors, including vascular endothelial growth factor, platelet-derived growth factor, fibroblast growth factor, and epidermal growth factor, and by phorbol esters, phosphatidylserine, and diacylglycerol and has numerous interactions with the STPs, thereby regulating diverse cellular responses, including proliferation, differentiation, and apoptosis.22 It has been implicated in malignant transformation, and indeed, it is expressed at elevated levels in many human tumors.22,24 In fact, we have previously demonstrated that high levels of PKCα were an adverse prognostic factor for patients with AML.25 In B-cell chronic lymphocytic leukemia, activation of PKCα decreased chemosensitivity, and its inhibition induced apoptosis.26 Conversely, the inhibition of PKCα activity caused decreased phosphorylation and activation of ERK1/2, resulting in decreased cell survival and increased levels of apoptosis.27
In general, the level of activation of each of these pathways has been ascertained in only small cohorts of patients, and these studies have not determined the activation state of the other STPs. The frequency of the concurrent activation of multiple STPs in AML and the prognostic relevance of STP activation individually or in combination in AML is unknown. If multiple pathways are frequently simultaneously activated in leukemia, then targeting a single upstream pathway activator may not decrease downstream activation because of cross-talk activation from other STPs. This would imply that therapeutic agents targeting single pathways that are being developed may be deemed clinically ineffective.1,10,22 To learn more about the simultaneous activation of STPs, we studied the level of expression and the activation status of 3 of these pathways by measuring PKCα, ERK2 and pERK2, and AKT and pAKT in a prospective fashion in samples from patients with primary leukemia. The prognostic impact of each protein was evaluated individually, and the combined prognostic impact of the activation of multiple pathways was also investigated. We observed that these pathways are activated in most cases of leukemia, that the simultaneous activation of multiple pathways is common, and that the prognosis worsens as more STPs are activated.
Patients, materials, and methods
Study group
Peripheral-blood or leukapheresis specimens or bone marrow specimens were collected prospectively from 188 patients with newly diagnosed AML evaluated at the University of Texas M. D. Anderson Cancer Center (MDACC) between September 1, 1999, and January 1, 2004. Samples were acquired during routine diagnostic assessments in accordance with the regulations and protocols sanctioned by the Investigational Review Board (IRB) of MDACC. Informed consent was obtained in accordance with the Declaration of Helsinki.
These patients were treated within a variety of IRB-approved protocols open at the MDACC during the collection period. The majority (81.5%) were treated with high dose ara-C (HDAC)–based regimens in combination with idarubicin (n = 89), daunorubicin (n = 20), fludarabine (n = 20), clofarabine (n = 10), cyclophosphamide and topotecan (n = 8), or other agents (n=5), and 2 patients were treated with a traditional idarubicin and standard-dose ara-C (3 + 7) regimen. All 3 patients with acute promyelocytic leukemia (APL) received regiments containing all-trans-retinoic acid (ATRA). Gemtuzumab ozogamicin was used alone or in combination in 8.5% and other investigational agents (deoxyazacytidine [DAC], suberoylanilide hydroxamic acid [SAHA], tipifarnib [Zarnestra]) were used in 6.3%, with all of these patients being older than 68 years. Stem-cell transplantation (SCT) was performed in 32 cases (allogeneic in 22, matched unrelated donor in 10), including 6 primary refractory, 5 first remission, and 21 after first relapse or in second remission.
Sample processing
Samples were placed on ice immediately after collection and were processed fresh within 2 hours of collection to generate a leukemia-cell– enriched fraction by isolating the mononuclear-cell fraction by Ficoll-Hypaque separation (Mediatech, Herndon, VA) followed by the depletion of CD3+/CD19+ B and T cells by magnetic antibody-conjugated sorting (Miltenyi Biotec, Auburn, CA), as previously described.28 The cells were used fresh to make whole-cell lysates for Western blotting.
Western immunoblotting analyses
Western immunoblotting analyses were performed using material from 4.5 × 105 cells and the Bio-Rad criterion system (Bio-Rad, Hercules, CA), with bone marrow and peripheral blood samples loaded on the same blot with control cell lines (K562 and Jurkat cells) and molecular-weight markers as previously described.28 Assays for AKT and pAKT were added after analysis of several apoptosis and STP proteins had already been performed and insufficient material remained to measure these proteins in 42 cases.
Triplicate membranes were created for each sample. The first blot was probed with anti-pERK2 monoclonal antibody (at a 1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) and anti-BCL2 (at a 1:2000 dilution; DAKO, Carpinteria, CA) and then stripped and reprobed for total AKT (at a 1:1000 dilution; Cell Signaling Technologies, Beverly, MA). The second blot was initially probed for pAKT (at a 1:1000 dilution; Cell Signaling Technologies) and then stripped and reprobed for total ERK2 using a monoclonal antibody (at a 1:1000 dilution; Santa Cruz Biotechnology). The third blot was probed for PKCα (at a 1:500 dilution; Santa Cruz Biotechnology) and Bax (at a 1:1000 dilution; Cell Signaling Technologies) and then stripped and reprobed for actin (at a 1:2000 dilution; Sigma-Aldrich, St Louis, MO). Because antibodies that specifically recognize only pPKCα were not available when this study was initiated, we studied total PKCα instead.
Following incubation with the primary antibody, the membranes were washed, conjugated to a secondary antibody, washed, and imaged using chemiluminescence as described.28 Results were normalized against the expression of the same protein in a standardized protein preparation from an AML-cell line (K562 or Jurkat cells) and against the level of actin expression in the patient's sample.
Statistical analysis
Pair-wise associations between patients' covariates were assessed by performing 2-sample t tests for continuous variables (age) and by performing Fisher exact test29 and its generalizations30 for categorical variables (sex, cytogenetic characteristics, performance status [PS], antecedent hematologic disorder [AHD], complete remission [CR], and relapse). Pearson correlation was used to assess correlation between protein expression. If pathway cross-activations were occurring, then the frequency of cases with multiple pathway activation would be expected to be higher than anticipated based on the frequency of individual pathway activation (Freqhigh-PKCα*Freqhigh-ERK2&pERK2*Freqhigh-pAKT). To assess this, patients were grouped according to whether they had no, 1, 2, or 3 pathways activated and the χ2 test was used for comparing the frequency of events. Unadjusted survival and remission duration analyses were performed using Kaplan-Meier plots.31 Unadjusted comparisons of survival between patient subgroups were made using the log-rank test.32 The Cox proportional hazards model33 and its generalizations34 using stepwise logistical regression were used to assess the ability of the treatment indicators, including PKCα, ERK2 and pERK2, AKT and pAKT, and other patient characteristics, to independently predict survival. Variables were selected for inclusion in the model if they had previously been recognized as important prognostic factors35-37 and there was evidence of their association with survival in this population. The final model was obtained by performing backward selection with a P cutoff of .05. All computations were carried out using Statistica version 6 software (Stat Soft, Tulsa, OK).
Results
Peripheral-blood or leukophoresis specimens (n = 118) and bone marrow specimens (n = 108) were collected from patients with newly diagnosed AML during the study period. Simultaneous blood and marrow specimens were available from 37 of these patients. Previous analysis of this same sample set had shown that the mean expression levels and the range of expression and of these same proteins, determined in leukemia-cell–enriched fractions, were statistically identical regardless of whether the source is blood or marrow.28 Consequently, the data for marrow and blood samples were pooled for this analysis, with the blood sample used whenever both blood and marrow samples were available (the total blast yield from blood samples generally exceeds that from marrow samples, so the use of blood samples permits analysis of more proteins from the same sample). Demographic and clinical details for the 188 patients are shown in Table 1. The prognostic impact of each protein was evaluated individually and in combination reflecting the activation of multiple pathways.
Table 1.
Demographic and clinical outcome data for 188 patients
| Demographic variable | Value |
|---|---|
| Age, y | |
| Average | 57 ± 16.56 |
| Median | 60 |
| Minimum | 18 |
| Maximum | 86 |
| FAB | |
| MO | 7% |
| M1 | 14% |
| M2 | 29% |
| M3 | 2% |
| M4 | 27% |
| M5 | 11% |
| M6 | 2% |
| M7 | 2% |
| Unknown | 6% |
| Cytogenetics | |
| Favorable | 11% |
| Intermediate | 48% |
| Unfavorable | 41% |
| AHD more than 2 mo | 29% |
| Etiology | |
| De novo | 68% |
| MDS evolution | 12% |
| TX-related | 20% |
| Zubrod Ps = 3 or 4 | 7% |
| Response | |
| CR | 54% |
| Failure | 12% |
| Resistant | 34% |
| Relapse | 57% |
| Median CR duration, wk | 47 |
| Median survival, wk | 42 |
| Alive | 32% |
| Med follow-up, wk | 123 |
Men accounted for 50.5% of the patients.
Expressions of ERK and pERK2 are adverse prognostic factors in AML
Higher expression of total ERK2 did not significantly affect the CR (53.6% versus 56.3%) or relapse rates (58.2% versus 52.8%) in patients with AML, but it was associated with a trend toward shorter CR durations (41.7 versus 74.1 weeks), which resulted in an inferior overall median survival duration (35.2 versus 67.7 weeks; P ≤ .02; for additional details, please refer to Tables S1 and S2, available on the Blood website by clicking on the Supplemental Tables link at the top of the online article). In a prior retrospective study, we observed that the activation of ERK2, as shown by the presence of pERK2, was absent in 25% of AML patient samples, weak in 37.5%, and strong in 37.5%. Patients showing a high expression had a poor prognosis. In this prospective study, we observed nearly identical patterns of expression, with pERK2 undetectable in 27% of the patient samples, weakly expressed in 39%, and strongly expressed in 33%. Strong expression of pERK2 was associated with trends toward lower CR rates (48% versus 58%) and shorter overall median survival times (31.4 versus 46.6 weeks; P = .11). The adverse effect of high levels of ERK was more prominent in patients lacking high pERK expression (Table 2; Figure 2). Because both high ERK2 and pERK2 levels were adverse prognostic factors, generating a ratio of pERK2 to ERK2 obscured their prognostic relevance. In contrast, creating 4 groups on the basis of whether both were low in comparison with groups in which one or both were high revealed that survival was best in patients with low levels of both and similar in patients with high levels of one or both (72, 39, and 30 weeks for those with no, one, or both high [ERK2 and pERK2], respectively; P = .012).
Table 2.
Clinical outcome data for the 3 STP components individually or in combination
|
PKCα
|
ERK2 & pERK2
|
pAKT
|
PKCa-ERK2&pERK2-pAKT
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low | High | P | Low-low | Low-high | High-low | High-high | P | Low | High | P | None | 1 high | 2 + high | 3 + high | P | |
| No. of patients | 89 | 99 | — | 47 | 15 | 76 | 50 | — | 78 | 70 | 18 | 44 | 39 | 45 | — | |
| Response | .14 | .69 | — | .55 | ||||||||||||
| CR | 60% | 49% | 60% | 53% | 57% | 48% | 60% | 50% | .21 | 56% | 59% | 64% | 49% | |||
| Fail | 8% | 15% | 6% | 13% | 16% | 12% | 8% | 16% | 0% | 11% | 10% | 16% | ||||
| Resistant | 31% | 35% | 34% | 33% | 28% | 40% | 32% | 34% | 44% | 30% | 26% | 36% | ||||
| Relapse | 51% | 63% | .21 | 57% | 38% | 60% | 54% | .68 | 51% | 54% | .77 | 40% | 46% | 52% | 64% | .55 |
| Median CR duration, wk | 50 | 45 | .1 | 63.4 | 107.7 | 37.8 | 57.1 | .3 | 72.57 | 40.3 | .13 | 76.4 | 71.57 | 53.3 | 25.7 | .125 |
| Median survival, wk | 59 | 34 | .02 | 72 | 39 | 38.5 | 30.93 | .05 | 61.21 | 29.85 | .028 | 78.57 | 57.86 | 42.3 | 23.4 | .012 |
| Alive | 38% | 27% | — | 47% | 27% | 28% | 28% | — | 42% | 27% | — | 61% | 39% | 33% | 24% | — |
| Median follow-up, wk | 112 | 134.5 | — | 153 | 135 | 88 | 121 | — | 135.6 | 104 | — | 152 | 106 | 104 | 104 | — |
— indicates not applicable.
Figure 2.

The effect of ERK2 and pERK2 expression on survival. Patients were divided into 2 groups on the basis of ERK2 expression, lower one third versus higher two thirds, on the basis of retrospective study data. Likewise, patients were divided into 2 groups, lower two thirds versus higher one third, on the basis of the level of pERK2 expression and retrospective study data. These were combined to form 4 groups for ERK2 and pERK2 (low/low, low/high, high/low, and high/high), and the Kaplan-Meier survival curves are shown here. The P value shown is for the plot of all 4 curves. P = .002 for the comparison of those with low/low ERK2 and pERK2 with those with one or both high.
Expression of high levels of PKCα is an adverse prognostic factor in AML
In our prior retrospective study, we observed a trend toward an inferior prognosis in patients with high levels of total PKCα.25 Our observations in the present study were very similar, in that the CR rate was lower in patients whose AML cells expressed high levels of PKCα (49% versus 60%; P = .14), the relapse rates tended to be higher (63% versus 51%; P = .21), and the CR duration was somewhat shorter (45 versus 50 weeks; P = .1), which together resulted in a lower overall median survival duration (34 versus 60 weeks; P = .02) as shown in Table 2 and Figure 3.
Figure 3.

The effect of PKCα expression on survival. Patients were divided into 2 groups, low and high, according to whether they were above or below the median PKCα expression observed in all 226 samples in 188 patients. Patients with a PKCα greater than the median level (P = .02) or in the highest third (P = .016) or the highest one sixth (P < .001) of this group had an inferior survival duration compared with those with levels of PKCα below the median, suggesting a progressively adverse prognostic effect of increasingly higher levels of PKCα.
Total AKT levels are not prognostic in AML
The total AKT levels did not prove to be a prognostic factor for remission, CR duration, or survival (P = .78).
High level of pAKT is an adverse prognostic factor in AML
pAKT was not detected in 9% of the patient samples and was weakly, moderately, and strongly expressed in 27%, 38%, and 32% of the samples, respectively. When patients were dichotomized into 2 groups according to the median level of expression, patients with low levels of pAKT had somewhat better CR rates (60% versus 50%; P = .21), longer median CR durations (71 versus 32 weeks; P = .13), and statistically significant longer median survival times (59 versus 30 weeks; P = .02) compared with those with high levels of pAKT (Table 2; Figure 4).
Figure 4.

The effect of pAKT expression on survival. Patients were divided into 2 groups, low and high, according to whether they were above or below the median pAKT expression observed in all 185 samples in 148 patients. Patients with high pAKT expression, greater than the median, had a survival duration inferior to that in patients with low pAKT expression, less than or equal to the median (P = .02).
Multiple pathway activation is more frequent than expected
If pathway cross-activation is occurring, then the frequency of cases with multiple pathway activation would be anticipated to be higher than expected based on the frequency of individual pathway activation (Freqhigh-PKCα*Freqhigh-ERK2&pERK2*Freqhigh-pAKT). The expected and observed frequencies for each possible combination and of having 0, 1, 2, or 3 pathways activated is shown in Table 3. In this data set, the observed-expected ratio for having all 3 pathways activated was 1.67 and for having no pathways activated was 1.9 (χ2 = 23.3, P < .001), strongly suggesting that cross-activation was occurring. Of note, no significant correlation was found between the level of expression of any of these proteins when compared individually (Table 4).
Table 3.
Observed and expected frequencies for the 8 possible combinations of low and high pAKT, ERK2&pERK2, and PKCα
|
PKCα low; 0.47
|
PKCα high; 0.53
|
|||||||
|---|---|---|---|---|---|---|---|---|
|
ERK2&pERK2 low; 0.26
|
ERK2&pERK2 high; 0.74
|
ERK2&pERK2 low 0.26
|
ERK2&pERK2 high; 0.74
|
|||||
| Expected | Observed | Expected | Observed | Expected | Observed | Expected | Observed | |
| pAKT low; 0.53 | 9.5 | 18 | 26.9 | 32 | 10.5 | 7 | 30 | 21 |
| pAKT high; 0.47 | 8.5 | 5 | 24.1 | 15 | 9.5 | 3 | 26.9 | 45 |
The population frequency is shown after the semicolon following each criteria. The expected or observed numbers of patients are shown in each cell. n = 146.
Table 4.
Correlation coefficients (R2) between the proteins measured
| ERK | PKCα | pERK | AKT | |
|---|---|---|---|---|
| pAKT | 0.07 | 0.12 | 0.08 | 0.11 |
| AKT | 0.11 | 0.11 | 0.0001 | — |
| pERK | 0.03 | 0.013 | — | — |
| PKCα | 0.37 | — | — | — |
— indicates not applicable.
FLT-3 abnormalities are not associated with increased STP activation
Because these STPs are downstream of FLT-3, the presence of a FLT-3 internal tandem duplication (ITD) or mutation might correlate with STP activation status. The presence of the FLT3-ITD or FLT3-D835 mutation (routinely assessed at diagnosis) was found in 27 and 7 of 101 cases, respectively, but did not correlate with the level of ERK, pERK, PKCα, or pAKT; however, the level of total AKT tended to be higher among patients with either mutation than in those lacking both mutations (P = .09). Similarly, the distribution of patients with none, 1, 2, or 3 STPs activated did not differ based on FLT-3 status (χ2 = .43; P = .93).
Prognosis worsens with increasing activation of multiple signalling pathways
When patients were grouped according to whether they had no, 1, 2, or 3 pathways activated, prognosis proved to be worse for those patients who had multiple pathways activated (Figure 5), with a median survival of 24, 42, and 58 weeks in those with 3, 2, and 1 pathway activated, respectively; the median was not reached in those who had no pathway activated (59% alive beyond 78 weeks; P = .01).
Figure 5.

Simultaneous activation of multiple STPs confers a poor prognosis. Patients were divided into 3 groups (all low, 1 or 2 high, or all 3 pathways highly activated) according to the activation of PKCα, ERK2 and pERK2, and pAKT and on the basis of the dichotomizations discussed in Figures 2, 3, 4. Patients were statistically more likely to have pan-activation or no activation than would be expected from the individual frequencies of low/high PKCα, ERK2 and pERK2, or pAKT. Survival was worse for those with 1 or 2 highly activated pathways (P = .02) and those with all 3 highly activated pathways (P < .001) than in those with all 3 pathways showing low levels of activation.
Univariate and multivariate analyses, with survival as the end point, were conducted using traditional prognostic factors and terms for low or high PKCα, ERK2 and pERK2, pAKT, and the combination variable PKCα-ERK2&pERK2-pAKT. As expected, in the univariate analysis (Table 5), the following prognostic variables were significant: age, cytogenetic characteristics, PS, AHD, lactate dehydrogenase, albumin, blood urea nitrogen, creatinine, β2-microglobulin, international normalized ratio (INR), SCT as well as PKCα, ERK, pERK, pAKT, and the combination variable. A list of nonsignificant variables is included in the text accompanying Table 5. Stepwise logistical regression analysis was performed with the add-back of all variables once a final model was fitted. The final model, excluding the STP variables, included age, PS, and favorable and unfavorable cytogenetic characteristics. When the terms for high PKCα (P = .03), ERK2&pERK2 (P = .04), pAKT (P = .05), or PKCα-ERK2p%ERk2-pAKT (P = .02) were individually added to this, they remained significant independent predictors of outcome. No other variables proved to be significant predictors of outcome when added back to the original model or to the models with the STP variables.
Table 5.
Univariate and multivariate analysis for survival
|
Multivariate analysis P
|
|||||||
|---|---|---|---|---|---|---|---|
| Parameter | Univariate P* | Base model | Nonsignificant† | ERK&pERK | PKCα | pAKT | Combination |
| Age | ≤.001 | <.001 | — | .001 | <.001 | <.001 | <.001 |
| Performance status | <.001 | <.001 | — | <.001 | <.001 | .002 | <.001 |
| Favorable cytogenetics | ≤.001 | .025 | — | .029 | .024 | .026 | .03 |
| Unfavorable cytogenetics | ≤.001 | <.001 | — | <.001 | <.001 | <.001 | <.001 |
| ERK2, low/high | .017 | — | .15 | — | — | — | — |
| ERK2 & pERK2, 2 groups | .01 | — | — | .04 | — | — | — |
| PKCα, low/high | .009 | — | — | — | .03 | — | — |
| pAKT, low/high | .006 | — | — | — | — | .05 | — |
| PKCα-ERK2&pERK2-pAKT, 4 groups | .002 | — | — | — | — | — | .013 |
| AHD equal to 2 or higher | .019 | — | .24 | — | — | — | — |
| %C-Kit (CD117) + by flow | .025 | — | .61 | — | — | — | — |
| LDH | .005 | — | .25 | — | — | — | — |
| Albumin | .007 | — | .22 | — | — | — | — |
| BUN | .001 | — | .97 | — | — | — | — |
| CR | .002 | — | .22 | — | — | — | — |
| β2-microglobulin | <.001 | — | .07 | — | — | — | — |
| INR | .05 | — | .18 | — | — | — | — |
| SCT‡ | <.001 | — | .18 | — | — | — | — |
LDH indicates lactate dehydrogenase; BUN, blood urea nitrogen.
Variables that were analyzed but found to be nonsignificant at the P = .05 level in univariate analysis included chemotherapy treatment protocol, continuous or categorized white blood cell count, hemoglobin, platelet count, percentage of blood blasts, percentage of marrow blasts, percentage of CD34+ cells by flow cytometry, serum bilirubin, fibrinogen, fibrin split products, D-dimers, calcium, prothrombin time, activated partial thromboplastin time, sample source (blood versus marrow, P = .47), and diagnosis date, or sample date, the latter two of which were included as controls.
Variables listed in this column were shown to be significant by univariate analysis but were not significant when added back one at a time into the final model consisting of these variables: age, performance status, favorable cytogenetics, and unfavorable cytogenetics. The P shown is for the addition of that parameter alone to the aforementioned four parameters.
Stem-cell transplantation is not significant if the data set is limited to a population similar to those who underwent transplantation, that is, those younger than 67 years of age and surviving longer than 20 weeks. Since SCT is not a variable available for consideration at diagnosis, it was not included in the final model using variables available at the time of diagnosis. The multivariate results do not change significantly if SCT is included, with the combination terms having similar statistical significance, but the term for age dropping out of the final models.
Discussion
We conclude from our data that activation of the STP components PKCα, pERK2, and pAKT is very frequent in AML and that each has an adverse prognostic impact that is significant and independent of the traditional prognostic factors of age, PS, and cytogenetics. The statistically significant difference in survival arises from the combined effect of nonsignificant trends for lower remission rates, shorter remission durations, and higher relapse rates observed in patients with STP activation. The higher-than-expected frequency of the concurrent activation of either all pathways or none of the pathways demonstrates that there is cross-activation of the pathways when STPs are activated. The importance of this is suggested by the more adverse prognosis in patients whose AML has all 3 pathways activated. Because activation of individual STP has been reported in numerous types of malignancies, it is highly likely that these observations may generalize to other types of cancer beyond AML.
Our study has several advantages. First, the large number of patients analyzed in this prospective study (146 for AKT) significantly exceeds the 55 patients analyzed by Min et al,4 and the 188 cases for which PKCα and ERK2 were analyzed in this study complements our similarly sized retrospective studies. In addition, to our knowledge, this is the first time that multiple STP activation has been shown to confer a more adverse prognosis than the isolated activation of a single STP.
The question immediately arises regarding the nature of the observed activation of multiple STPs. Analysis of the spectrum of mutations identified in human leukemias has indicated that activating kinase mutations that confer a proliferative or survival advantage to hematopoietic progenitors collectively account for as many as 50% of cases of AML, but only very rarely is more than one such mutation present in the same patient.38 These epidemiologic data suggest that simultaneous activation of multiple STPs is triggered by other mechanisms. Possible scenarios include mutations in phosphatases, which commonly control activation of several signaling pathways, aberrant expression or mutations in recently discovered microRNA (miRs), such as let-739, and activation of STPs in response to microenvironmental stimuli. The first 2 possibilities are currently under active investigation, but recent evidence by our group (Tabe, manuscript submitted) and others,40,41 indeed demonstrates robust activation of multiple signaling pathways on the contact of leukemic with bone marrow stromal cells. This exemplifies the apparent advantages of proteomic approaches based on the expression patterns of phosphorylated substrates, which might reveal different phosphoprotein patterns in response to the tumor's microenvironment despite identical genetic profiles of the tumor cells themselves.
From a therapeutic perspective, the finding that activation of each of these pathways is both common and affects prognosis makes them highly attractive targets for emerging therapies directed at preventing or reversing STP activation. The clinical relevance is that the effects of agents that target upstream activators of these STPs are likely to be blunted by the downstream cross-activation of other members of that same pathway by activated signaling proteins in different signaling pathways. Thus, combinations of agents targeting separate STPs are likely to be required for these novel agents to be efficacious.
Indeed, evidence from experiments with cell lines has demonstrated that combined blockade is often necessary for synergistic or effective cell killing to occur. The prototypes of the combined blockade of several pathways include inhibition of the parallel signaling pathways (such as PI3K/AKT and RAS/Raf/MEK/ERK42,43), intrapathway inhibition of the upstream and downstream kinases (combined use of the RAS and MEK inhibitors44), and simultaneous targeting of signal transduction and apoptosis pathways (MAPK and Bcl-216,45). We consider the last strategy most promising, based on the extensive evidence of intimate relationships between STP and apoptosis pathways, the obvious examples being redundant prosurvival phosphorylation of Bcl-2 family proteins (Bcl-2 itself, BAD, or BIM)46-49 or transcriptional regulation of apoptosis regulators (such as IAPs, Bcl-2, Mcl-1).41,50,51 Hence, an attractive approach to rational combinations is to select agents targeting different pathways that impinge on the regulation of key targets critical for the survival of leukemic cells. Clearly, high-throughput proteomic approaches that allow simultaneous analysis of high numbers of regulators of both signal transduction and apoptosis signaling will assist in characterization of signaling networks and identification of such critical prosurvival targets, guiding in constructing mechanism-based combinations of signal transduction-related therapies.52
A potential limitation of our study was that we looked at PKCα levels instead of pPKCα levels because pPKCα antibodies were not available when the study started. However, using a pPKCα antibody with high specificity (Upstate Biologicals, Charlottesville, VA) that recently became available, we performed reversephase protein array (RPPA) experiments and probed 90 of the specimens from patients in the present study for pPKCα.53 This showed a very high correlation between pPKCα levels and total PKCα levels, and confirmed the prognostic impact of pPKCα levels in support of the data presented here. Likewise, correlation with other proteins with prognostic impact in AML, such as NPM, CEBP-α, and BAALC, along with other STP proteins, such as JNK and p38, is now being evaluated in RPPA experiments to determine if they also have prognostic or additive value when considered concurrently with STPs.
The prognostic impact of multiple STP activation was predominantly observed in patients with intermediate or unfavorable cytogenetic characteristics, because only 3 of 20 patients with favorable cytogenetic findings in this cohort have died. Of note, 2 of these 188 patients had the DEK-CAN fusion protein, t(6:9), and both showed low levels of all 3 pathways, suggesting that STP activation does not explain the profound resistance to therapy of patients with this translocation. Similarly increased STP activation was not associated with the FLT3-ITD or FLT3-D835 mutation, perhaps reflecting the effects of cross-activation by other activated STPs.
In summary, our results suggest that leukemia treatment targeting multiple signal transduction pathways may be more efficacious than therapy aimed at inhibiting a single pathway. Because agents are traditionally evaluated individually during drug development, the important implication of our findings is that there is a high likelihood that new STP-blocking agents will be discarded as ineffective on the basis of the results from studies evaluating their use either alone or in combination with conventional chemotherapy (eg, FLT-3 inhibitors CEP701, PKC412, or MEK inhibitor PD184352) due to cross-activation. Thus, if the full potential of STP blockade is to be realized in AML and potentially in other malignancies, this may require the unprecedented cooperation of different pharmaceutical firms and academic investigators holding rights to agents active in different pathways.
Supplementary Material
Prepublished online as Blood First Edition Paper, June 8, 2006; DOI 10.1182/blood-2006-02-003475.
Supported by grant 6089 from the Leukemia Society of America and grant PO1 CA-55164 from the National Institutes of Health (S.M.K.). The online version of this article contains a data supplement.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Steelman LS, Pohnert SC, Shelton JG et al. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18: 189-218. [DOI] [PubMed] [Google Scholar]
- 2.Reuter CW, Morgan MA, Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood. 2000;96: 1655-1669. [PubMed] [Google Scholar]
- 3.Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1: 417-420. [DOI] [PubMed] [Google Scholar]
- 4.Min YH, Cheong JW, Kim JY, et al. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res. 2004;64: 5225-5231. [DOI] [PubMed] [Google Scholar]
- 5.Elstrom RL, Bauer DE, Buzzai M, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64: 3892-3899. [DOI] [PubMed] [Google Scholar]
- 6.Karnauskas R, Niu Q, Talapatra S, et al. Bcl-x(L) and Akt cooperate to promote leukemogenesis in vivo. Oncogene. 2003;22: 688-698. [DOI] [PubMed] [Google Scholar]
- 7.Zhao S, Konopleva M, Cabreira-Hansen M, et al. Inhibition of phosphatidylinositol 3-kinase dephosphorylates BAD and promotes apoptosis in myeloid leukemias. Leukemia. 2004;18: 267-275. [DOI] [PubMed] [Google Scholar]
- 8.Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102: 972-980. [DOI] [PubMed] [Google Scholar]
- 9.Luo Y, Shoemaker AR, Liu X, et al. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol Cancer Ther. 2005;4: 977-986. [DOI] [PubMed] [Google Scholar]
- 10.Sebolt-Leopold JS. MEK inhibitors: a therapeutic approach to targeting the Ras-MAP kinase pathway in tumors. Curr Pharm Des. 2004;10: 1907-1914. [DOI] [PubMed] [Google Scholar]
- 11.Towatari M, lida H, Tanimoto M, et al. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia. 1997;11: 479-484. [DOI] [PubMed] [Google Scholar]
- 12.Kim S-C, Hahn J-S, Min Y-H,et al. Constitutive activation of extracellular signal-regulated kinase in human acute leukemias: combined role of activation of MEK, hyperexpression of extracellular signal-regulated kinase, and downregulation of a phosphatase, PAC1. Blood. 1999;93: 3893-3899. [PubMed] [Google Scholar]
- 13.Konopleva M, Shi Y, Steelman LS,. et al. Development of a conditional in vivo model to evaluate the efficacy of small molecule inhibitors for the treatment of Raf-transformed hematopoietic cells. Cancer Res. 2005;65: 9962-9969. [DOI] [PubMed] [Google Scholar]
- 14.Loda M, Capodieci P, Mishra R, et al. Expression of mitogen-activated protein kinase phosphatase-1 in the early phases of human epithelial carcinogenesis. Am J Pathol. 1996;149: 1553-1564. [PMC free article] [PubMed] [Google Scholar]
- 15.Milella M, Kornblau SM, Estrov Z, et al. Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J Clin Invest. 2001;108: 851-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milella M, Estrov Z, Kornblau SM, et al. Synergistic induction of apoptosis by simultaneous disruption of the Bcl-2 and MEK/MAPK pathways in acute myelogenous leukemia. Blood. 2002;99: 3461-3464. [DOI] [PubMed] [Google Scholar]
- 17.Morgan MA, Dolp O, Reuter CW. Cell-cycle-dependent activation of mitogen-activated protein kinase kinase (MEK-1/2) in myeloid leukemia cell lines and induction of growth inhibition and apoptosis by inhibitors of RAS signaling. Blood. 2001;97: 1823-1834. [DOI] [PubMed] [Google Scholar]
- 18.Ricciardi M, McQueen T, Chism D, et al. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia. 2005;19: 1543-1549. [DOI] [PubMed] [Google Scholar]
- 19.Milella M, Kornblau SM, Andreeff M. The mitogen-activated protein kinase signaling module as a therapeutic target in hematologic malignancies. Rev Clin Exp Hematol. 2003;7: 160-190. [PubMed] [Google Scholar]
- 20.Blalock WL, Navolanic PM, Steelman LS, et al. Requirement for the PI3K/Akt pathway in MEK1-mediated growth and prevention of apoptosis: identification of an Achilles heel in leukemia. Leukemia. 2003;17: 1058-1067. [DOI] [PubMed] [Google Scholar]
- 21.Shelton JG, Steelman LS, Lee JT, et al. Effects of the RAF/MEK/ERK and PI3K/AKT signal transduction pathways on the abrogation of cytokine-dependence and prevention of apoptosis in hematopoietic cells. Oncogene. 2003;22: 2478-2492. [DOI] [PubMed] [Google Scholar]
- 22.Hanauske AR, Sundell K, Lahn M. The role of protein kinase C-alpha (PKC-alpha) in cancer and its modulation by the novel PKC-alpha-specific inhibitor aprinocarsen. Curr Pharm Des. 2004;10: 1923-1936. [DOI] [PubMed] [Google Scholar]
- 23.Devalia V, Thomas NS, Roberts PJ, Jones HM, Linch DC. Down-regulation of human protein kinase C alpha is associated with terminal neutrophil differentiation. Blood. 1992;80: 68-76. [PubMed] [Google Scholar]
- 24.Gavrielides MV, Frijhoff AF, Conti CJ, Kazanietz MG. Protein kinase C and prostate carcinogenesis: targeting the cell cycle and apoptotic mechanisms. Curr Drug Targets. 2004;5: 431-443. [DOI] [PubMed] [Google Scholar]
- 25.Kornblau SM, Vu HT, Ruvolo P, et al. BAX and PKCalpha modulate the prognostic impact of BCL2 expression in acute myelogenous leukemia. Clin Cancer Res. 2000;6: 1401-1409. [PubMed] [Google Scholar]
- 26.Barragan M, Campas C, Bellosillo B, Gil J. Protein kinases in the regulation of apoptosis in B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2003;44: 1865-1870. [DOI] [PubMed] [Google Scholar]
- 27.Leirdal M, Sioud M. Protein kinase Calpha isoform regulates the activation of the MAP kinase ERK1/2 in human glioma cells: involvement in cell survival and gene expression. Mol Cell Biol Res Commun. 2000;4: 106-110. [DOI] [PubMed] [Google Scholar]
- 28.Kornblau SM, Womble M, Cade JS, Lemker EM, Qiu YH. Comparative analysis of the effects of sample source and test methodology on the assessment of protein expression in acute myelogenous leukemia. Leukemia. 2005;19: 1550-1557. [DOI] [PubMed] [Google Scholar]
- 29.Fisher RA. The condition under which χ2 measures the discrepancy between observation and hypothesis. J Royal Stat Soc. 1924;87: 442-450. [Google Scholar]
- 30.Mehta CR. The exact analysis of contingency tables in medical research. Stat Methods Med Res. 1994;3: 135-156. [DOI] [PubMed] [Google Scholar]
- 31.Kaplan EL, Meier P. Nonparametric estimation from incomplete observation. J Am Stat Assn. 1958;53: 457-481. [Google Scholar]
- 32.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. J Royal Stat Soc. 1966;50: 63-170. [PubMed] [Google Scholar]
- 33.Cox DR. Regression models and life tables. J Royal Stat Soc. 1972;34: 187-220. [Google Scholar]
- 34.Fleming TR, Harrington DP. Counting Processes and Survival Analysis. New York, NY: Wiley; 1991.
- 35.Schiffer CA, Lee E, Tomiyasu T, Wiernik P, Testa J. Prognostic impact of cytogenetic abnormalities in patients with de novo acute nonlymphocytic leukemia. Blood. 1989;73: 263-270. [PubMed] [Google Scholar]
- 36.Smith TL, McCredie K. Prediction of survival during induction therapy in patients with newly diagnosed acute myeloblastic leukemia. Leukemia. 1989;3: 257-263. [PubMed] [Google Scholar]
- 37.Keating MJ, Cork A, Broach Y, et al. Toward a clinically relevant cytogenetic classification of AML. Leuk Res. 1987;11: 119-133. [DOI] [PubMed] [Google Scholar]
- 38.Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106: 1154-1163. [DOI] [PubMed] [Google Scholar]
- 39.Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120: 635-647. [DOI] [PubMed] [Google Scholar]
- 40.Bertrand FE, Spengemen JD, Shelton JG, McCubrey JA. Inhibition of PI3K, mTOR and MEK signaling pathways promotes rapid apoptosis in B-lineage ALL in the presence of stromal cell support. Leukemia. 2005;19: 98-102. [DOI] [PubMed] [Google Scholar]
- 41.Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9: 1158-1165. [DOI] [PubMed] [Google Scholar]
- 42.Yu C, Dai Y, Dent P, Grant S. Coadministration of UCN-01 with MEK1/2 inhibitors potently induces apoptosis in BCR/ABL+ leukemia cells sensitive and resistant to ST1571. Cancer Biol Ther. 2002;1: 674-682. [DOI] [PubMed] [Google Scholar]
- 43.Yang X, Galinsky I, DeAngelo DJ, Sternberg D, Stone RM. Apoptosis suppression by FLT3 activated via an internal tandem duplication mutation (ITD) requires both the MEK/ERK and PI3K/AKT signaling pathways [suppl 1]. Blood. 2003;100: 549a. [Google Scholar]
- 44.Morgan MA, Dolp O, Reuter CW. Cell-cycle-dependent activation of mitogen-activated protein kinase kinase (MEK-1/2) in myeloid leukemia cell lines and induction of growth inhibition and apoptosis by inhibitors of RAS signaling. Blood. 2001;97: 1823-1834. [DOI] [PubMed] [Google Scholar]
- 45.Benimetskaya L, Miller P, Benimetsky S, et al. Inhibition of potentially anti-apoptotic proteins by antisense protein kinase C-alpha (Isis 3521) and antisense bcl-2 (G3139) phosphorothioate oligodeoxynucleotides: relationship to the decreased viability of T24 bladder and PC3 prostate cancer cells. Mol Pharmacol. 2001;60: 1296-1307. [DOI] [PubMed] [Google Scholar]
- 46.Ito T, Deng X, Carr B, Strauss E. Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem. 1997;272: 11671-11673. [DOI] [PubMed] [Google Scholar]
- 47.Scheid MP, Schubert KM, Duronio V. Regulation of Bad phosphorylation and association with Bcl-XL by the MAPK/Erk kinase. J Biol Chem. 1999;274: 31108-31113. [DOI] [PubMed] [Google Scholar]
- 48.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278: 687-689. [DOI] [PubMed] [Google Scholar]
- 49.Dijkers PF, Birkenkamp KU, Lam EW, et al. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol. 2002;156: 531-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carter BZ, Milella M, Altieri DC, Andreeff M. Cytokine-regulated expression of survivin in myeloid leukemia. Blood. 2001;97: 2784-2790. [DOI] [PubMed] [Google Scholar]
- 51.Wang JM, Chao JR, Chen W, et al. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999;19: 6195-6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dancey J, Sausville EA. Issues and progress with protein kinase inhibitors for cancer treatment. Nat Rev Drug Discov. 2003;2: 296-313. [DOI] [PubMed] [Google Scholar]
- 53.Tibes R, Qiu YH, Coombes K, et al. Classification of acute myelogenous leukemia (AML) based on patterns of signal transduction pathway (STP) and apoptosis regulating protein activation determined by reverse phase proteins arrays (RPPA) [abstract]. Blood. 2005;106: 145a. Abstract 484. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.