

Abstract
The generation of platelets from megakaryocytes in the steady state is regulated by a variety of cytokines and transcription factors, including thrombopoietin (TPO), GATA-1, and NF-E2. Less is known about platelet production in the setting of stress thrombopoiesis, a pivotal event in the context of cytotoxic chemotherapy. Here we show in mice that the transcription factor Scl is critical for platelet production after chemotherapy and in thrombopoiesis induced by administration of TPO. Megakaryocytes from these mice showed appropriate increases in number and ploidy but failed to shed platelets. Ultrastructural examination of Scl-null megakaryocytes revealed a disorganized demarcation membrane and reduction in platelet granules. Quantitative real-time polymerase chain reaction showed that Scl-null platelets lacked NF-E2, and chromatin immunoprecipitation analysis demonstrated Scl binding to the NF-E2 promoter in the human megakaryoblastic-cell line Meg-01, along with its binding partners E47, Lmo2, and the cofactors Ldb1 and GATA-2. These findings suggest that Scl acts up-stream of NF-E2 expression to control megakaryocyte development and platelet release in settings of thrombopoietic stress.
Introduction
Megakaryocyte maturation and platelet production are complex processes that involve cellular mechanisms unique to the lineage, including polyploidization via nuclear replication in the absence of cellular cytokinesis, a process termed endomitosis. Platelet shedding from megakaryocytes occurs by extension of long pseudopods, termed proplatelets, from the megakaryocyte cytoplasm, and formation of anucleate platelets by budding at the ends of these structures.1,2 This process requires a reserve of cytoplasmic membrane, which exists in the cytoplasm of mature megakaryocytes within a structure termed the demarcation membrane system (DMS).3 There is also dynamic reorganization of the cytoskeleton, bringing about the assembly of the marginal band, a ring of microtubules primarily composed of β1-tubulin, a divergent β-tubulin isoform, which is restricted to the megakaryocyte lineage and is essential for platelet discoid shape.4-6
The primary cytokine regulator of megakaryocyte development is thrombopoietin, which binds to its cognate receptor c-Mpl to regulate megakaryocyte development. Thrombopoietin functions primarily at the progenitor level to promote megakaryocyte proliferation and development; however, it is not required for the terminal phase of platelet shedding.7
While the transcriptional control of platelet production is less well understood, a number of key regulators have been described including NF-E2, GATA-1, FOG-1, and Fli-1.8 The generation of mice lacking these genes has shed light on the transcriptional regulation of megakaryocyte development and platelet shedding. The most important identified regulator of platelet shedding is NF-E2, as mice lacking this factor display severe thrombocytopenia (< 5% the normal platelet count) despite increased numbers of megakaryocytes.9,10 NF-E2–null megakaryocytes display numerous morphologic abnormalities including an aberrant DMS, reduced cytoplasmic granules, and a complete absence of proplatelet formation.11 Several target genes of NF-E2 have been identified, including β-1 tubulin,5,6 caspase 12,12 thromboxane synthase,13 Rab27b,14 and 3β-hydroxysteroid dehydrogenase (3β-HSD), a mediator of autocrine biosynthesis of estradiol within megakaryocytes, which is absent in NF-E2–null megakaryocytes and sufficient to restore proplatelet formation when ectopically expressed in these cells.15
One potential regulator of NF-E2 is GATA-1. Mice lacking GATA-1 in the megakaryocyte compartment display marked thrombocytopenia (around 10% of normal platelet numbers) despite grossly increased numbers of immature megakaryocytes both in vitro and in vivo.16,17 Similar to NF-E2–null megakaryocytes, GATA-1–null megakaryocytes display abnormal ultrastructure including a reduction in platelet granules and disorganized DMS. Transcriptional profiling has shown a reduced expression of a number of genes, which also are decreased in NF-E2–null megakaryocytes, including β1-tubulin, 3β-HSD, and caspase-12.12,15,17,18 These findings may be explained by the findings that the expression of NF-E2 is reduced in GATA-1–null megakaryocytes,17,18 and that efficient transcriptional activity of the major NF-E2 promoter active in the erythroid/megakaryocytic lineages requires the presence of tandem GATA motifs, implying that GATA-1 may be a regulator of NF-E2 expression in megakaryocytes.19,20
Another potential regulator of platelet production is the basic helix-loop-helix (bHLH) transcription factor Scl (also known as Tal1), the expression of which is predominantly restricted to the megakaryocyte and erythroid lineages.21 Scl forms obligate heterodimers with ubiquitous E proteins (E12 and E47, products of the E2A gene, HEB and E2-2), which bind to E-box motifs (sequence CANNTG) and mediate either transcriptional activation or repression. In erythroid cells, these heterodimers are found in larger multimeric complexes containing at least the GATA-1, Lmo2, and Ldb1 proteins, which bind to bipartite E-box–GATA motifs.22 While several target genes of this complex have been described in the erythroid lineage, no megakaryocytic target genes have yet been identified.23
Mice lacking Scl die at embryonic day 9.5 due to a complete failure of hemopoietic specification24,25 and defective vasculogenesis.26 To overcome this and allow the analysis of the function of Scl in adult hemopoiesis, conditional Scl-null mice have been generated.27,28 Surprisingly, absence of Scl in adult hematopoiesis is not lethal, although there are demonstrable effects on hematopoietic stem cell function and erythropoiesis.29,30 Despite this, these mice survive long-term with persistent but stable anemia and thrombocytopenia. The aim of this study was to determine the mechanism of the thrombocytopenia in Scl-null mice. We show that Scl is a key regulator of platelet production, particularly during settings of hemopoietic stress. The mechanism for this appears to be via direct regulation of NF-E2, as this factor is reduced in Scl-null platelets, and its promoter is occupied by Scl in megakaryocytic cells.
Materials and methods
Mice
Mice with a loxP-targeted Scl allele (SclloxP),27 a lacZ reporter gene knocked into the Scl locus, generating a nonfunctional allele (herein termed Scl–),31 and Mx-Cre transgenic32 mice were described previously. Deletion of the SclloxP allele to generate the nonfunctional SclΔ allele was achieved by administration of polyinosinic-polycytidylic acid (PIPC) in adult mice expressing the Mx-Cre transgene as described.27 Mice were used for analysis more than 1 month after treatment, at which point the hematologic effects of PIPC treatment had resolved.
Hemopoietic analyses
For whole-blood counts, 250 μL blood was collected from the retro-orbital plexus into tubes containing potassium EDTA (ethylenediaminetetraacetic acid) (Sarstedt, Nümbrecht, Germany) and blood counts analyzed using an Avidia 120 automated hematologic analyzer (Bayer, Tarrytown, NY).
Stress thrombocytosis
To induce thrombocytosis, mice were injected with either 2 μg pegylated recombinant human thrombopoietin (PEG-rhTPO) in 200 μL phosphate-buffered saline (PBS) intraperitoneally, or 150 mg/kg 5-fluorouracil (5FU) (10 mg/mL in PBS) (Sigma, St Louis, MO) intraperitoneally. To serially analyze platelet counts, 1 μL whole blood harvested from the tail vein was diluted into 199 μL BSGC buffer (116 mM NaCl, 13.6 mM trisodium citrate, 8.6 mM Na2HPO4, 1.6 mM KH2PO4, 0.9 mM Na2 EDTA, 11.1 mM d-glucose, pH 6.8) containing 0.2 μL anti-CD41–fluorescein isothiocyanate (FITC) antibody (BD Pharmingen, San Diego, CA) and 100 000 allophycocyanin (APC) beads (Calibrite, BD Pharmingen), and the number of CD41+ platelets (relative to APC beads) was determined using a FACSCalibur flow cytometer (BD Pharmingen).
Histology
Mouse sternal sections and spleens were fixed in 10% formaldehyde in PBS, sectioned, and stained with hematoxylin/eosin. Megakaryocyte counts per high power (400 ×) field of view (HPF) were calculated from at least 15 adjacent HPFs per mouse studied.
Platelet turnover studies
Measurement of platelet lifespan was performed according to Ault and Knowles.33 Briefly, mice were injected twice, 2 hours apart, with 150 μL of 4 mg/mL biotin-N-hydroxysuccinamide (a 40 mg/mL stock solution in DMSO [dimethyl sulphoxide] freshly diluted 1:10 in PBS). To measure the proportion of biotinylated platelets, 1 μL blood was stained with anti-CD41–FITC and streptavidin APC (BD Pharmingen) and analyzed by flow cytometry.
Measurement of megakaryocyte DNA content
Measurement of megakaryocyte DNA content was performed as described by Jackson et al.34 Briefly, bone marrow was flushed from femurs using CATCH buffer and stained with anti-CD41–FITC antibody (BD Pharmingen). Cells were resuspended in Hypotonic propidium iodide (PI) buffer (0.1% sodium citrate, 0.056% sodium chloride, 50 μg/mL DNase-free RNase A [Roche, Milan, Italy], 50 μg/mL propidium iodide) and analyzed by flow cytometry. Large CD41-positive cells were gated and analyzed for PI content.
Tail-bleeding studies
Tail-bleeding times were performed in anaesthetized mice (sodium pentobarbitone) as described previously.35 Briefly, a uniform incision 5-mm long and 1-mm deep was made using a template, 10 mm from the base of the tail. Bleeding was monitored every 30 seconds by blotting with filter paper, until cessation.
Platelet aggregation studies
Washed platelets (2.0 × 108/mL) prepared as previously described35 were stimulated with the indicated concentration of agonist (thrombin) in the presence of calcium (1 mM). All aggregations were initiated by stirring the suspensions at 950 rpm for 10 minutes at 37°C in a 4-channel automated platelet analyzer (Packs 4; Helena Laboratories, Beaumont, TX).
Electron microscopy
To analyze megakaryocytes using electron microscopy, mice were anesthetized before being perfused with 4 mL warmed 2% glutaraldehyde, 2.5% paraformaldehyde in 0.1 M sodium cacodylate buffer with 2 mM calcium chloride, PBS (pH 7.4), at a rate of 20.4 mL/h (Braun “perfusor compact S”) into the descending aorta. The vena cava was cut to release pressure. The femurs were excised and immersion-fixed for a further 4 hours before rinsing in PBS and decalcifying the bone in 10% EDTA (pH 7.4) for 7 days at 4°C. The bones were washed 3 times in PBS plus 5% sucrose for 15 minutes, then postfixed in 2% osmium tetroxide in PBS for 1 hour. Washing in distilled water preceded dehydration through a graded series of ethanol (50%, 70%, 90%, 95%, 2 × 100%, 90 minutes each) before infiltration and embedding in Spurrs resin as per standard electron microscopy (EM) protocol.
To analyze platelets, washed platelets isolated as described35 were fixed in 2% glutaraldehyde, 2.5% paraformaldehyde in PBS for 60 minutes at room temperature. Following 2 washes in 0.08 M PBS, 5% sucrose, platelets were postfixed in 2% osmium tetroxide in 0.1 M PBS for 60 minutes before processing into resin as described in the preceding paragraph. Dehydration steps were reduced to 15 minutes each.
Ultrathin sections were cut with a diamond knife (Diatome, Wien, Austria) using a Leica Ultracut S ultra-microtome (Leica Microsystems, Wetzlar, Germany) stained with both methanolic uranyl acetate and lead citrate before viewing in a transmission electron microscope, JEOL 1011 (JEOL, Tokyo, Japan) at 60 kV. Images were recorded with a MegaView III CCD cooled digital camera (Soft Imaging Systems, Münster, Germany) using analysis software (Olympus, Tokyo, Japan), and were processed with Adobe Photoshop CS2 (Adobe Systems, San Jose, CA). Platelet granularity was determined by visual examination of at least 100 platelets in each group.
RT-PCR
Platelets were purified as described35 from 2 mice in each group and used to generate total RNA using Trizol (Invitrogen, Carlsbad, CA). cDNA was generated using the Omniscript reverse transcriptase (Qiagen, Hilden, Germany) according to the manufacturer's recommendations. Real-time polymerase chain reaction (RT-PCR) was carried out in 20-μL reactions using SYBR green fluorescent DNA labeling (Molecular Probes, Eugene, OR) and the Rotorgene 2000 PCR machine (Corbett Research, Sydney, Australia). Relative quantitation was performed using the Standard Curve method, which was generated for each amplicon using 5-fold serial dilutions of normal mouse bone marrow cDNA. All values were normalized to those obtained via amplification with primers specific for 18s RNA. The primer sequences used were (forward and reverse, 5′-3′): NF-E2, GATGCCCCCGTGTCCTCCTCA and TCCTCCGCCTGCCAGCCTCCATAC; β-1 tubulin, GATATCGCTGCGCTGGTCGTC and ACGCAGCTCATTGTAGAAGGTGTGG; CoagX, Calp2 as in Muntean et al18; GATA-1, CTTACGGGGGAGCTGACTTTC and GATCTCGCGTGGCATTCCTC; Mpl, CACCTGGGAGAAATGTGAAGAGGA and ACCCGGTGTAGGTCTGGAAGCG; PF4, GACATGAGCGTCGCTGCGGTGTTT and CCATCGCTTTCTTCGGGACCA; β-actin, GATATCGCTGCGCTGGTCGTC and ACGCAGCTCATTGTAGAAGGTGTGG; and 18s, GTAACCCGTTGAACCCCATT and CCATCCAATCGGTAGTAGCG.
Chromatin immunoprecipitation
Chromatin immunoprecipitation assays were performed as described previously,36,37 using antibodies directed against Scl (3014, a kind gift of Dr C. Glenn Begley [Amgen, Thousand Oaks, CA]), Ldb1 (a kind gift of Dr Jane Visvader [Walter and Eliza Hall Institute, Melbourne, Australia]), anti-HA (12CA5, Roche), and Lmo2 (N-16), E47 (N-649), GATA-1 (N6), and GATA-2 (H-116 all from Santa Cruz Biotechnology, Santa Cruz, CA). Isolated DNA fragments were purified with a QIAquick spin kit (Qiagen), and 2 μL from a 40-μL DNA extraction was amplified quantitatively by PCR using the following primers: forward 5′-GTTAACCTATGGCCCAAATGACCCT-3′ and reverse 5′-AAGTTGTGGAAAGAGGCAAGCAGAC-3′. Where indicated, the immunoprecipitated chromatin was quantitated by real-time PCR as described in the preceding section.
Luciferase reporter assays
HEK-293T cells (2 × 105) were plated in triplicate in 24-well plates. The next day, combinations of 25 ng pEF-PGK-PURO-Scl (a gift of Dr Jane Visvader), MSCV-IRES-GFP (MIG)-HA-Scl or MIG-HA-SclΔb,38 25 ng pEF-BOS-GATA1(myc) (a gift of Dr Warren Alexander), 20 ng pEF-BOS-Ldb1, 20 ng pEF-BOS-Lmo2, and 5 ng pEF-BOS-E12 were transfected along with 130 ng of a pGL3 reporter plasmid containing the 232-bp fragment of the human NF-E2 promoter 1b (as in Figure 6B) and 13 ng of pRL-TK, using lipofectamine 2000 (Life Technologies, Bethesda, MD). Transfections were made up to a total of 750 ng using pEF-PGK-PURO. Forty-eight hours after transfection, cell extracts were prepared and luciferase activity measured using the Dual Luciferase Reporter Assay System (Promega, Madison, WI). Firefly luciferase activities were normalized to the corresponding levels of Renilla luciferase activity in each sample.
Figure 6.

Scl regulates NF-E2 expression in platelets. (A) Quantitative PCR on platelet cDNA. Platelets were purified from mice of the indicated genotypes and used to generate cDNA, which was amplified by real-time PCR using primers specific for the indicated genes. (B) Partial sequence of the NF-E2 internal 1b promoter. Nucleotides 420-652 of the human promoter/exon 1b region are shown. E boxes present in the region are shown in bold. Tandem GATA motifs, which have been shown to be important in regulation, are underlined. (C) Chromatin immunoprecipitation using Meg-01 cells. Cell extracts were prepared from the human megakaryoblastic-cell line Meg-01 and either 10% input (input), or immunoprecipitates using the indicated antibodies, were used to PCR amplify the region of the NF-E2 promoter/exon 1b sequence shown in panel C. “None” indicates no antibody; control Ig, normal rabbit serum. (D) Chromatin immunoprecipitation using Meg-01 cells. As in panel C, using an anti–GATA-2 antibody as indicated. (E) Chromatin immunoprecipitation using K562 cells. Cell extracts were prepared from the human erythroleukemic-cell line K562 and subjected to chromatin immunoprecipitation as in panel C. (F) Transactivation of the NF-E2 promoter. A luciferase reporter construct containing the 232-bp NF-E2 promoter region as in panel B was transfected into 293T cells along with expression plasmids for the indicated genes. The activities shown are normalized to a cotransfected Renilla luciferase expression plasmid. Data are mean + SD of triplicate determinations.
Retrovirus production and bone marrow reconstitution
Retroviral supernatants were prepared using transient transfection of HEK-293T cells with MSCV-IRES-GFP (MIG) retroviral plasmids containing hemagglutinin epitope-tagged (HA-)Scl, HA-SclΔb,38 and the pGag-Pol and pCAG-Eco (a kind gift of Dr Derek Persons [St Jude Children's Research Hospital, Memphis, TN]) packaging plasmids. K562 cells were infected by coculture with retroviral supernatants and sorted for GFP expression using a FACS-Vantage cell sorter (Becton Dickinson, San Jose, CA).
For bone marrow reconstitutions SclloxP/– mice were treated with 150 mg/kg 5FU, and 4 days later bone marrow was harvested and infected with 3 rounds of MIG-Δb viral supernatant, 12 hours apart, using Retronectin (Takara Bio, Shiga, Japan). Cells (106) were used to reconstitute lethally irradiated (2 × 550 rads, 4 hours apart) BL/6 mice, mice that were treated with PI-PC (4 weeks after reconstitution) and PEG-rhTPO (8 weeks after reconstitution) as described in “Mice” and “Stress thrombocytosis.”
Results
Defective platelet production in Scl–/Δ mice
We have previously generated mice in which a loxP-flanked allele of Scl (SclloxP) can be conditionally deleted through the use of the Mx-Cre transgene. In this transgenic line, Cre recombinase is induced throughout the hemopoietic system by release of interferon-α/β (IFN-α/β) in response to injection of the double-stranded RNA analog PI-PC, resulting in more than 90% deletion of the floxed Scl allele in the bone marrow.27 The resultant deleted Scl allele (herein termed SclΔ) is paired with either a wild-type allele (termed Scl+) or a nonfunctional allele into which the bacterial beta-galactosidase gene has been inserted (termed Scl–), resulting in adult mice that are either heterozygous (Scl+/Δ) or null (Scl–/Δ) for Scl function in hemopoiesis. Previous expression analyses confirmed no detectable Scl mRNA expression in the bone marrow of Scl–/Δ mice.27
Scl–/Δ mice have approximately 50% normal peripheral-blood platelet levels (Figure 1A). To examine the cause of thrombocytopenia in these mice, platelet survival was measured using in vivo biotinylation. This showed that the half-life of Scl–/Δ platelets was normal (57 hours), while the production of new, nonbiotinylated platelets was significantly impaired (Figure 1B). The impaired platelet production was not explained by reduced megakaryocyte numbers, as megakaryocyte numbers in the marrow of Scl–/Δ mice were normal (Figure 1C). In addition, megakaryocyte endomitosis as measured by DNA content (ploidy) was not impaired. Indeed, the ploidy of megakaryocytes from Scl–/Δ mice was significantly increased, with the modal ploidy elevated to 32N compared to the normal value of 16N (Figure 1D). Hence, the defective platelet production in Scl–/Δ mice was not due to defects in megakaryocyte number or polyploidization and was most likely due to a defect in platelet release by mature megakaryocytes.
Figure 1.
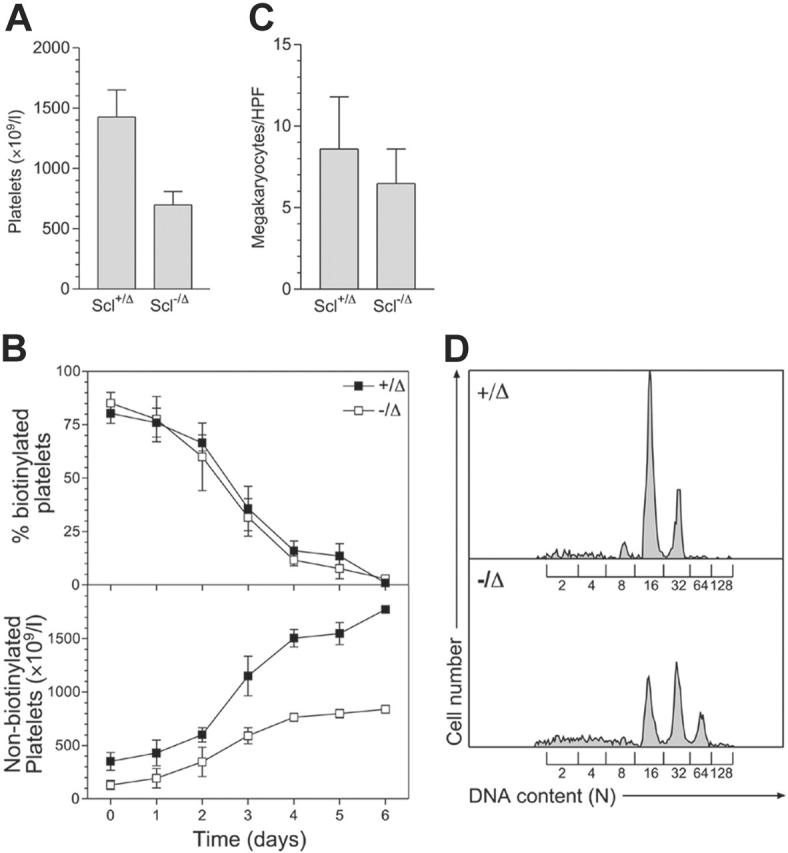
Scl–/Δ mice show defective platelet production and megakaryocyte hyperploidy. (A) Thrombocytopenia seen in Scl–/Δ mice. Mice of the indicated Scl genotypes were bled more than 1 month following PI-PC treatment, and platelet counts were determined using a blood analyzer. Results are the mean + standard deviation of 5 mice in each group. (B) Measurement of platelet lifespan. Mice were treated with N-hydroxy-succinomidyl biotin on day 0 and the percentage of biotinylated platelets determined daily using flow cytometry (top panel). Relating this value to the determined platelet count yielded the number of nonbiotinylated platelets (bottom panel). Data are mean ± standard deviation of quadruplicate determinations. (C) Megakaryocyte frequency in the bone marrow of Scl+/Δ and Scl–/Δ mice. Sternal bone marrow sections were prepared and analyzed using light microscopy. The number of megakaryocytes per 400 × microscopic HPF is indicated for each genotype. Results are the mean + standard deviation of 15 adjacent fields analyzed for 4 separate mice. (D) Hyperploidy of Scl–/Δ megakaryocytes. The DNA content of bone marrow megakaryocytes was determined using propidium iodide staining and flow cytometry. The level of staining corresponding to the numbers of haploid genomes (N) contained in each cell is indicated. Data are representative of 3 separate mice of each genotype analyzed.
Defective response to thrombopoietin and 5FU in Scl–/Δ mice
The role of Scl in platelet production was further analyzed in the setting of pharmacologic doses of TPO. This cytokine acts on megakaryocyte progenitors in the bone marrow to increase their number and maturation, subsequently leading to thrombocytosis in the blood.39 Mice were administered TPO for 5 consecutive days and platelet counts measured during the subsequent 2 weeks. Strikingly, platelet levels in Scl–/Δ mice did not rise at any point during or following TPO administration (Figure 2A). Thus, Scl was required for thrombocytosis in response to TPO.
Figure 2.
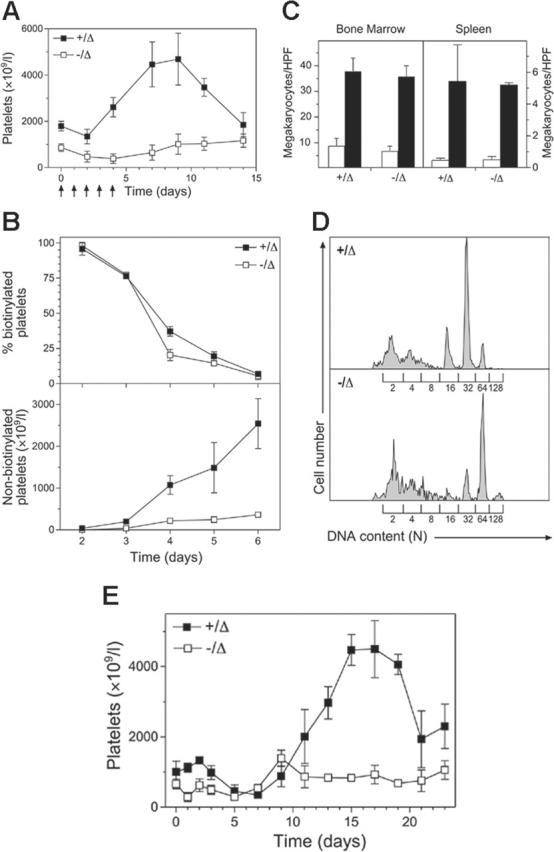
Scl–/Δ mice show abnormal stress thrombopoiesis. (A) Response of Scl–/Δ mice to thrombopoietin. Mice of the indicated Scl genotypes were injected intraperitoneally with 2 μg pegylated recombinant human thrombopoietin (PEG-rhTPO) daily for 5 days as indicated (arrows), and platelet counts were determined periodically. Results are the mean ± standard deviation of 4 mice analyzed. (B) Determination of platelet turnover during TPO administration. Mice were administered 2 μg/mL PEG-rhTPO on days 0 to 5 and N-hydroxy-succinomidyl biotin on day 2 and the percentage of biotinylated platelets determined daily using flow cytometry (top panel). Relating this value to the determined platelet count yielded the number of nonbiotinylated platelets (bottom panel). Data are mean ± standard deviation of 3 mice analyzed. (C) Normal expansion of Scl–/Δ megakaryocytes in response to thrombopoietin. Mice of the indicated Scl genotypes were either untreated (□) or treated with 2 μg PEG-rhTPO for 5 days, then analyzed on day 8 (▪). Sternal bone marrow and spleen sections were stained with hematoxylin and eosin and analyzed using light microscopy. The number of megakaryocytes per 400 × microscopic HPF is indicated for each genotype. Results are the mean + standard deviation of 15 adjacent fields analyzed for 4 mice. (D) Increased ploidization of megakaryocytes in response to TPO. Mice of the indicated Scl genotypes were treated with 2 μg/mL PEG-rhTPO for 5 days and the DNA content of megakaryocytes determined using propidium iodide staining and flow cytometry. The level of staining corresponding to the numbers of haploid genomes (N) contained in each cell is indicated. Data are representative of 3 separate mice of each genotype analyzed. (E) Response of Scl–/Δ mice to 5FU. Mice of the indicated Scl genotypes were injected intraperitoneally with 150 mg/kg 5FU, and platelet counts were determined periodically. Results are the mean ± standard deviation of 3 mice.
To confirm that the abnormal response to TPO was due to defective platelet production rather than increased destruction, platelet turnover was studied in Scl–/Δ mice in the setting of TPO treatment. Mice were treated with TPO for 5 days, as previously, with injection of NHS-biotin on the third day of TPO administration. Platelet number and biotinylation were subsequently determined daily. While loss of circulating, labeled platelets in TPO-treated Scl+/Δ and Scl–/Δ mice displayed similar kinetics, the production of nascent, nonbiotinylated platelets in TPO-treated Scl–/Δ mice was markedly reduced compared to that observed in the Scl+/Δ animals (Figure 2B), confirming that the absence of thrombocytosis in response to TPO was due to a defect in platelet production.
Platelet production in response to TPO is preceded by an increase in the number of megakaryocytes in the bone marrow and spleen.39 To determine whether this occurred in Scl–/Δ mice, bone marrow and splenic megakaryocytes were enumerated histologically. Surprisingly, megakaryocyte numbers increased appropriately in the absence of Scl, indicating that Scl was not required for megakaryocyte proliferation in response to TPO (Figure 2C). We found that spleen megakaryocytes in long-term deleted Scl mice were normal, in contrast to the low levels seen previously in short-term deleted mice.27
In addition to proliferation, TPO administration also promotes increased ploidization of megakaryocytes via endomitosis. To assess the role of Scl in TPO-induced megakaryocyte endomitosis, mice were administered TPO for 4 consecutive days and megakaryocyte ploidy assessed on day 5. The ploidy of megakaryocytes in Scl–/Δ mice increased to a similar extent to controls (modal ploidy shifting from 32N to 64N compared with 16N to 32N in controls) (Figure 2D). Hence, the inability of Scl–/Δ mice to mount a thrombocytotic response TPO was not explained by defects in TPO-induced proliferation or endomitosis of megakaryocytes.
To examine the specificity of the abnormal TPO response, platelet recovery in Scl–/Δ mice was examined following treatment with the chemotherapeutic agent 5FU. This drug ablates mature megakaryocytes and cycling progenitors, leading to thrombocytopenia followed by a “rebound” thrombocytosis, which occurs later than that induced by TPO, implying that it is derived from immature, noncycling progenitors such as hemopoietic stem cells.40 Following 5FU treatment, platelet levels in control Scl+/Δ mice reduced to 35% of starting levels over 7 days, which was followed by a progressive rebound thrombocytosis (Figure 2E). In Scl–/Δ animals a markedly different response was seen. While platelet levels fell appropriately in response to 5FU and recovered by day 9, the typical rebound thrombocytosis was never observed in Scl–/Δ mice in 3 independent experiments. Other responses to 5FU such as leukocyte recovery were normal, indicating that this aberrant response was specific to the platelet lineage (data not shown).
Ultrastructural abnormalities of Scl–/Δ megakaryocytes
To further elucidate the mechanism of the defective platelet production in Scl–/Δ mice, bone marrow megakaryocytes were analyzed by transmission electron microscopy. In order to observe megakaryocytes at a point in which the consequences of loss of Scl are most apparent, mice were pretreated with TPO for 4 days and analyzed on day 5. At this point, the DMS of Scl–/Δ megakaryocytes was highly disorganized and was accompanied by increased rough endoplasmic reticulum and an almost complete absence of both primary and dense platelet granules in the cytoplasm (compare Figure 3A,C with Figure 3B,D). These structural defects also were found in untreated Scl–/Δ megakaryocytes but to a lesser extent (data not shown). In contrast, treatment with TPO did not affect the ultrastructure of control Scl+/Δ megakaryocytes (data not shown). Scl–/Δ megakaryocytes also displayed prominent emperipolesis (the presence of other cells within the megakaryocytes), with 18% of megakaryocytes studied containing between 1 and 3 cells in the cytoplasm compared with none in control megakaryocytes (n = 39 and 29, respectively) (Figure 3E). Together, these results suggest that the platelet production defect of Scl–/Δ mice is due at least in part to defects in cytoplasmic maturation of megakaryocytes, including a failure to properly specify platelet demarcation membranes prior to platelet shedding.
Figure 3.

Ultrastructural defects of Scl–/Δ megakaryocytes. Transmission electron microscopic (TEM) images of representative megakaryocytes present in the bone marrow of Scl+/Δ (A) and Scl–/Δ (B) mice treated for 4 days with 2 μg PEG-rhTPO and analyzed on day 5. Panels C and D show magnified regions of panels A and B, respectively, as indicated by the boxed regions. Note the presence of alpha granules (g) in the cytoplasm of the Scl+/Δ megakaryocyte in panel C and depleted granules (g) in the cytoplasm of the Scl–/Δ megakaryocyte in panel D. (E) Example of a megakaryocyte from a TPO-treated Scl–/Δ mouse showing emperipolesis (3 cells contained within the cytoplasm). Nuclei (n) and demarcation membrane systems (dms) are indicated. Scale bars represent 10 μM (A-B, E) and 2 μM (C-D).
Ultrastructural abnormalities of Scl–/Δ platelets
In addition to thrombocytopenia, the platelets in Scl–/Δ mice were larger and more heterogeneous in size (mean platelet volume, 6.7 ± 1.8 versus 4.9 ± 0.2 in controls; platelet distribution width, 55.8 ± 16.6 versus 40.5 ± 15.6 in controls). Upon examination using transmission electron microscopy, the most notable abnormality of Scl–/Δ platelets was a loss of elongated forms, which are evidence of the normal discoid shape, indicating that the platelets were spherical, a finding that was confirmed using differential interference contrast microscopy (Figure 4B,D). Other abnormalities included decreased numbers of both primary granules (an average of 1.9 per platelet versus 3.1 in controls) and dense granules (0.45 per platelet versus 0.71 in controls) and a corresponding increase in “depleted” granules (granules with little or no content, 1.7 per platelet compared with 1.0 in controls), defects that were mildly exacerbated following treatment of the mice with TPO (data not shown). There also was increased vacuolation of the cytoplasm (Figure 4B). Hence, in addition to the platelet production defect present in Scl–/Δ mice, platelets produced by these mice were structurally abnormal.
Figure 4.

Ultrastructural defects of platelets from Scl–/Δ mice. TEM images of platelets from Scl+/Δ (A) and Scl–/Δ (B) mice. Primary (g) and dense (d) granules are indicated in panel A. Depleted granules (g) and dilated canalicular membrane system (c) are indicated in panel B. Scale bars represent 5 μM. Differential interference contrast (DIC) images of platelets from a Scl+/Δ (C) and Scl–/Δ (D) mice. Arrows in panel C show elongated forms indicative of the normal discoid shape (d), which are absent in panel D, indicating that Scl–/Δ platelets are round. Images are at 100 × magnification.
Platelets from Scl–/Δ mice are functional
To test the hemostatic function of platelets in the absence of Scl, tail-bleed assays were performed. There were slightly longer bleed times in Scl–/Δ mice, which may be due in part to the lower platelet count and hematocrit in these animals (Figure 5A). To further examine platelet function in the absence of Scl, in vitro platelet aggregation assays were performed. Consistent with the spherical nature of Scl–/Δ platelets, thrombin-induced platelet shape change (measured by an initial decrease in light transmission following addition of agonist) was markedly reduced (Figure 5B). However, Scl–/Δ platelets were capable of a normal aggregation in response to thrombin, adenosine diphosphate, and collagen (Figure 5B and data not shown). These results indicate that Scl–/Δ platelets were functional despite numerous structural abnormalities.
Figure 5.

Platelets derived from Scl–/Δ mice are functional. (A) Tail skin bleed time analysis of mice of the indicated Scl genotypes. Horizontal bars denote the mean. (B) Washed platelets derived from mice of the indicated Scl genotypes were stimulated with 0.5 U/mL thrombin (arrow) and aggregation monitored as described under “Materials and methods.” Scale bars show the magnitude of change in light transmission due to platelet shape change. This trace is taken from 1 experiment representative of 3 independent experiments.
Defective gene expression in Scl–/Δ platelets
To examine the molecular basis for the defective platelet production in Scl–/Δ mice, the expression of several genes implicated in platelet shedding was studied. Large numbers of Scl–/Δ megakaryocytes for expression analysis could not be obtained because these cells do not grow or survive in vitro,27 despite the normal in vivo response to TPO shown here. Therefore, purified platelets were used as a source of RNA for real-time PCR analysis. A small residual expression of Scl (≤ 10%) was found in some platelet RNA preparations from Scl–/Δ mice; however, its presence did not appear to affect any of the phenotypes detailed in this study (Figure 6A). The NF-E2 transcription factor is critical for platelet shedding.9 RT-PCR analysis revealed 8-fold reduced expression of NF-E2 in Scl–/Δ platelets (Figure 6A). Furthermore, β-1 tubulin, a proposed target gene of NF-E2, was reduced 10-fold in platelets derived from Scl–/Δ mice (Figure 6A). Of the other genes studied, coagulation factor X, calpain 2, and platelet factor 4 were only marginally reduced in Scl–/Δ platelets. Interestingly, the expression of GATA-1 was reduced 2-fold, which is consistent with the binding of Scl to a tandem E-box GATA motif upstream of this gene.41 Expression of the TPO receptor, Mpl, was normal, indicating that abnormal platelet production in response to TPO was not due to reduced receptor expression.
Scl binds the NF-E2 promoter in megakaryocytic cells
NF-E2 has been proposed to be regulated by GATA-1 binding to tandem GATA motifs located in the proximal 1b promoter located in the first intron of the gene.19,20 Interestingly, an Scl DNA binding motif (E-box) is located 10 base pairs from a GATA site within this motif (Figure 6B). To determine whether Scl can bind to the NF-E2 promoter, chromatin immunoprecipitation (ChIP) analysis was performed using antibodies directed against Scl, GATA-1, and other factors identified in a multimeric complex in erythroid cells.22 Scl bound the NF-E2 promoter in the human megakaryoblastic-cell line Meg-01 (Figure 6C). In addition, Lmo2, E47, and Ldb1 also were bound, suggesting the presence of a transcriptional complex. However, an anti–GATA-1 antibody was unable to immunoprecipitate the NF-E2 promoter in Meg-01 cells, which may be due to the fact that this cell line is developmentally immature and expresses low levels of GATA-1.42 However, we found that Meg-01 cells express high levels of GATA-2 protein by Western blotting and that GATA-2 protein bound to the NF-E2 promoter in these cells in the ChIP assay (data not shown, Figure 6D).
To confirm that GATA-1 can bind the NF-E2 promoter, an identical ChIP assay was performed using the human erythroleukemic-cell line K562, which has both erythroid and megakaryocytic potential, as these cells expresses higher levels of GATA-1. In these cells, GATA-1 was found to bind the NF-E2 promoter along with Scl, Lmo2, Ldb1, and E47 (Figure 6E). Thus, GATA-1 and Scl both bind to the NF-E2 promoter in K562 cells. However, in immature megakaryocytic cells GATA-2 binds the promoter and not GATA-1.
To examine the consequences of Scl binding to the NF-E2 promoter, the proximal 1b promoter region was cloned into a luciferase reporter construct and used in transient transfection experiments in HEK-293T cells along with expression vectors for the various transcription factors found associated with this promoter. We found that while Scl and its partner proteins E12, Lmo2, and Ldb1 had no effect on transcription driven by the NF-E2 promoter, these factors were able to augment transcriptional activation of the promoter by GATA-1 (Figure 6F). This effect required Scl; however, a DNA binding mutant of Scl (Δb) could substitute for Scl, indicating that direct DNA binding by Scl was not required (Figure 6F).
The DNA binding region of Scl is not required for NF-E2 promoter binding or platelet production
As the ability of Scl to promote transcriptional activation of the NF-E2 promoter did not require its DNA binding activity, we tested whether this region was required to bind the NF-E2 promoter region in K562 cells. After expression of hemagglutinin epitope (HA-)–tagged Scl and Δb in these cells by retroviral vectors, the cells were subjected to ChIP assays using an anti-HA antibody. We found that both Scl and Δb were associated with the NF-E2 promoter at levels comparable with those obtained using and anti-E47 antibody (Figure 7A). Therefore, the DNA binding activity of Scl was not required to bind to the NF-E2 promoter.
Figure 7.

Binding of the NF-E2 promoter and TPO-induced platelet production by a DNA binding mutant of Scl (Δb). (A) NF-E2 promoter binding by HA-Δb. K562 cells expressing MIG-HA-Scl and MIG-HA-Δb were subjected to chromatin immunoprecipitation using antibodies against the HA epitope tag and E47. The left panel shows gel electrophoresis of the NF-E2 promoter PCR products as in Figure 6, while the right panel shows quantitation of the immunoprecipitated chromatin using real-time PCR. (B) Platelet production by an MIG-Δb reconstituted mouse in response to TPO. The left panel shows histograms depicting the percentage of GFP-expressing platelets in MIG-Δb reconstituted mice before (top) and after (bottom) treatment with 2 μg PEG-rhTPO for 5 days and analysis on day 8. The right panel shows the absolute numbers of GFP-negative (GFPneg) and GFP-positive (GFPpos) platelets in the same mouse, taking into account the platelet counts. Data are representative of 2 MIG-Δb reconstituted mice analyzed.
To test whether the DNA-binding mutant of Scl is able to perform its functions in platelet production, mice were reconstituted with Scl-targeted (Scl–/loxP) bone marrow infected with a green fluorescent protein (GFP)–encoding retroviral vector expressing the Δb mutant. Following reconstitution, recipient mice were treated with PIPC to remove Scl from the transplanted bone marrow (resulting in an Scl–/Δ genotype). After subsequent treatment with TPO, a 3-fold expansion of GFP+ platelets was seen, indicating that retrovirally encoded Δb could compensate for the loss of Scl in TPO-induced platelet production (Figure 7B). Therefore, the DNA binding activity of Scl was not required for platelet production in response to TPO.
Discussion
The transcriptional regulation of megakaryocyte development, including platelet shedding, remains poorly understood. The most critical identified regulator is NF-E2, which is absolutely required during the terminal phase of platelet production.9 Here we have used a conditionally deleted allele of Scl to demonstrate that this transcription factor is essential for platelet production from mature megakaryocytes, particularly in the setting of platelet recovery following the administration of the chemotherapeutic agent 5FU or in response to pharmacologic doses of TPO. One role of Scl in platelet production appears to be regulation of NF-E2, as expression of NF-E2 was reduced 8-fold in the platelets of Scl–/Δ mice, Scl bound to the major NF-E2 promoter in megakaryocytic cells, and Scl could augment transcriptional activation of the NF-E2 promoter.
Defective platelet production in Scl–/Δ mice appears to be due to a late defect in megakaryocyte maturation, as megakaryocyte numbers were normal and ploidy increased, but these showed a disordered demarcation membrane system and, following TPO administration, reduced platelet granules in the cytoplasm. These defects are similar to those seen in mice lacking NF-E2.9 NF-E2–null megakaryocytes are unable to form proplatelets,11 and we propose that this is also the mechanism for the reduced platelet production of Scl–/Δ mice, although we are unable to test this in vitro using the proplatelet assay due to a separate survival defect of Scl–/Δ megakaryocytes in vitro.27 In contrast, we have shown here that Scl was not required for megakaryocyte proliferation in vivo, either in the resting state or in response to TPO. While megakaryocyte numbers often are increased in settings of thrombocytopenia, this was not seen in Scl–/Δ mice. This may be explained by the increased platelet volume in these mice (35%), which produces a relatively normal platelet mass, which is a proposed negative feedback regulator of TPO signaling via ligand binding to the Mpl receptor on platelets.43
A surprising finding was the inability of Scl–/Δ mice to develop thrombocytosis in response to TPO or 5FU, despite the relatively mild thrombocytopenia seen in the resting state. There are at least 2 possible explanations for this defect. First, Scl may be a critical downstream mediator of the TPO signaling pathway. Scl is a nuclear target of the erythropoietin signaling pathway through activation of a mitogen-activated protein kinase.44 TPO shares a similar signal transduction pathway with erythropoietin, suggesting that Scl also might be phosphorylated by TPO signaling.43 However, as we show here that Scl is not required for TPO-induced megakaryocyte proliferation and ploidization (Figure 2), the major functions of TPO in megakaryopoiesis, it is unlikely that Scl is an important target of TPO signaling. Second, Scl function may be absolutely required for stress thrombopoiesis, whereas it is partially redundant in the resting state or its loss is able to be compensated for. Consequently, platelet production in Scl–/Δ mice may be at maximal capacity in the resting state, such that a stress response is not possible. Indeed, megakaryopoiesis in Scl–/Δ mice displayed several features of stress, including increased ploidy and large platelets. It may be that the reduced NF-E2 levels seen in Scl–/Δ mice are sufficient to produce the moderate platelet levels seen in the resting state, but insufficient to induce stress thrombocytosis.
A key implication of this work is that NF-E2 is a direct target of Scl. This assertion is supported by 3 observations: that NF-E2 expression was markedly reduced in Scl–/Δ platelets, that Scl occupied the NF-E2 promoter in a megakaryocytic-cell line, and that Scl can augment transcriptional activation of the NF-E2 promoter. To date no Scl target genes have been described in the megakaryocyte lineage. Consistent with NF-E2 being a target of Scl, the phenotype of Scl–/Δ mice bears many similarities to NF-E2–null mice. In both cases, megakaryocytes expand and develop in response to TPO, in the absence of platelet production.9 Moreover, the ultrastructural morphology of NF-E2–null megakaryocytes is similar to that of TPO-treated Scl megakaryocytes, in that the cells are developmentally mature but show a loss of platelet granules and a disordered demarcation membrane system. These findings imply that a critical function of Scl in the megakaryocyte lineage is the regulation of NF-E2. While NF-E2 also is expressed in the erythroid lineage, its loss has relatively minor consequences in this lineage.30,45 So too does loss of Scl, with the erythroid stress responses of Scl–/Δ mice being normal.30 Given that NF-E2 is a target of Scl and that NF-E2 knockout mice lack platelets almost entirely,9 it is surprising that the thrombocytopenia seen in Scl–/Δ mice in the resting state was not more severe. The residual platelet formation in these mice is likely to be due to the low levels of NF-E2 still present in Scl–/Δ platelets (13%, Figure 6). This residual expression of NF-E2 is unlikely to be due to residual Scl expression, as similar platelet counts were found in mice in which no residual Scl expression could be detected (data not shown). Alternatively, regulation of NF-E2 by other transcriptional mechanisms, or compensation for the loss of Scl by other bHLH transcription factors, may account for the residual NF-E2 expression.
NF-E2 is thought to be regulated by GATA-1, primarily through binding to a GATA-1 repeat sequence in the proximal intronic promoter 1b element of NF-E2 and, consequently, it has been inferred that these factors act in a linear fashion to regulate megakaryocyte development.8,19,20 While this previously has been studied using electrophoretic mobility shift assays, our finding that an anti–GATA-1 antibody could immunoprecipitate this element in K562 cells provides further evidence that GATA-1 binds this element in vivo. Scl also bound the NF-E2 promoter in erythroid and megakaryocytic cells, implying that Scl and GATA-1 cooperate in the regulation of NF-E2 through the formation of a pentameric complex with Lmo2, Ldb1, and E47, all of which were found bound to this element. Such complexes have been described previously in erythroid cells, and it is notable that the spacing between an E box and the second (inverted) GATA motif in this element is 10 base pairs, within the 9 to 11 base-pair range required for this complex to function in in vitro binding assays.22 Interestingly, Scl, along with E47, Lmo2, and Ldb1, was found to occupy the promoter 1b element in the immature megakaryoblastic-cell line Meg-01 in the absence of GATA-1. This is consistent with the recent finding that the major Scl-containing complex present in a megakaryocytic-cell line contains E47, Lmo2, and Ldb1, but not GATA-1.46 However, in Meg-01 cells GATA-2 was found bound to the promoter. The up-regulation of GATA-1 later in megakaryocyte development coincides with an up-regulation of NF-E2 expression, implying that expression of GATA-1 may be required to potentiate transcriptional activation by the NF-E2 promoter, as has been shown in in vitro studies.22,47 This suggests a situation in which GATA-2 is replaced later in development by GATA-1, as has been shown in erythroid development.48
Interestingly, a DNA binding mutant of Scl could compensate for the major functions of Scl described in this study, namely, binding and transactivation of the NF-E2 promoter, and platelet production in response to TPO. Moreover, the E-box sites present in the NF-E2 promoter are not consensus sites for Scl binding in conjunction with GATA factors (CAGGTG).49 These findings indicate that Scl may be recruited to the NF-E2 promoter by the binding of GATA factors to the tandem GATA motif present in this region.
Among the defined target genes of GATA-1 and NF-E2 is β-1 tubulin, a platelet-specific isoform of tubulin that is essential for normal platelet production and function. Loss of this factor leads to defective proplatelet formation, thrombocytopenia, and platelet spherocytosis.5,6,50 Scl–/Δ platelets showed a severe reduction in β-1 tubulin expression, providing a molecular explanation for their spherocytosis, and resultant lack of shape change in response to thrombin (Figures 4, 5). Of the other GATA-1 target genes tested, coagulation factor X and calpain 2 were only mildly affected, and Mpl and platelet factor 4 expression were not affected. This implies that Scl is not required for expression of all GATA-1 target genes in megakaryocyte development. This assertion is supported by the numerous differences in the megakaryocyte development of Scl–/Δ and GATA-1–deficient mice. Namely, GATA-1–deficient megakaryocytes are hyperproliferative in vitro and in vivo and are blocked in maturation, and GATA-1–deficient mice show a thrombocytopenia that is more severe than that observed in Scl–/Δ mice.16
Our findings indicate that Scl is critical for platelet production in response to thrombopoietin or 5FU treatment. Future studies will aim to determine whether NF-E2 is the key target gene of Scl required for these processes. These studies will aid our understanding of platelet shedding and may lead to the development of new strategies to augment platelet production in human platelet disorders or following myeloablative therapy.
Acknowledgments
We gratefully acknowledge the assistance of Sumitha Vasudevan for genotyping of mice, Lan Ta for animal husbandry, Jason Corbin for blood analysis, and Dr Sharelle Sturgeon for tail-bleed analyses.
Prepublished online as Blood First Edition Paper, June 8, 2006; DOI 10.1182/blood-2006-02-002188.
S.M.J. and S.P.J. are Principal Research Fellows of the Australian National Health and Medical Research Council (NHMRC). S.M.S. is a Monash University Logan Research Fellow. This work was supported by PO1 HL53749-03 from the National Institutes of Health, Project Grants 282400 (D.J.C.) and 237011 (S.P.J.) from the NHMRC, and G03M1131 (S.M.S.) from the National Heart Foundation (NHF) of Australia.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Choi ES, Nichol JL, Hokom MM, Hornkohl AC, Hunt P. Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood. 1995;85: 402-413. [PubMed] [Google Scholar]
- 2.Italiano JE Jr, Lecine P, Shivdasani RA, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. 1999;147: 1299-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radley JM, Haller CJ. The demarcation membrane system of the megakaryocyte: a misnomer? Blood. 1982;60: 213-219. [PubMed] [Google Scholar]
- 4.Wang D, Villasante A, Lewis SA, Cowan NJ. The mammalian beta-tubulin repertoire: hematopoietic expression of a novel, heterologous beta-tubulin isotype. J Cell Biol. 1986;103: 1903-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lecine P, Italiano JE Jr, Kim SW, Villeval JL, Shivdasani RA. Hematopoietic-specific beta 1 tubulin participates in a pathway of platelet biogenesis dependent on the transcription factor NF-E2. Blood. 2000;96: 1366-1373. [PubMed] [Google Scholar]
- 6.Schwer HD, Lecine P, Tiwari S, Italiano JE Jr, Hartwig JH, Shivdasani RA. A lineage-restricted and divergent beta-tubulin isoform is essential for the biogenesis, structure and function of blood platelets. Curr Biol. 2001;11: 579-586. [DOI] [PubMed] [Google Scholar]
- 7.Ishida Y, Ito T, Kuriya S. Effects of c-mpl ligand on cytoplasmic maturation of murine megakaryocytes and on platelet production. J Histochem Cytochem. 1998;46: 49-57. [DOI] [PubMed] [Google Scholar]
- 8.Shivdasani RA. Molecular and transcriptional regulation of megakaryocyte differentiation. Stem Cells. 2001;19: 397-407. [DOI] [PubMed] [Google Scholar]
- 9.Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, et al. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell. 1995;81: 695-704. [DOI] [PubMed] [Google Scholar]
- 10.Levin J, Peng JP, Baker GR, et al. Pathophysiology of thrombocytopenia and anemia in mice lacking transcription factor NF-E2. Blood. 1999;94: 3037-3047. [PubMed] [Google Scholar]
- 11.Lecine P, Villeval JL, Vyas P, Swencki B, Xu Y, Shivdasani RA. Mice lacking transcription factor NF-E2 provide in vivo validation of the proplatelet model of thrombocytopoiesis and show a platelet production defect that is intrinsic to megakaryocytes. Blood. 1998;92: 1608-1616. [PubMed] [Google Scholar]
- 12.Kerrigan SW, Gaur M, Murphy RP, Shattil SJ, Leavitt AD. Caspase-12: a developmental link between G-protein–coupled receptors and integrin alphaIIbbeta3 activation. Blood. 2004;104: 1327-1334. [DOI] [PubMed] [Google Scholar]
- 13.Deveaux S, Cohen-Kaminsky S, Shivdasani RA, et al. p45 NF-E2 regulates expression of thromboxane synthase in megakaryocytes. EMBO J. 1997;16: 5654-5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiwari S, Italiano JE Jr, Barral DC, Mules EH, et al. A role for Rab27b in NF-E2–dependent pathways of platelet formation. Blood. 2003;102: 3970-3979. [DOI] [PubMed] [Google Scholar]
- 15.Nagata Y, Yoshikawa J, Hashimoto A, Yamamoto M, Payne AH, Todokoro K. Proplatelet formation of megakaryocytes is triggered by autocrine-synthesized estradiol. Genes Dev. 2003;17: 2864-2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 1997;16: 3965-3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vyas P, Ault K, Jackson CW, Orkin SH, Shivdasani RA. Consequences of GATA-1 deficiency in megakaryocytes and platelets. Blood. 1999;93: 2867-2875. [PubMed] [Google Scholar]
- 18.Muntean AG, Crispino JD. Differential requirements for the activation domain and FOG-interaction surface of GATA-1 in megakaryocyte gene expression and development. Blood. 2005;106: 1223-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moroni E, Mastrangelo T, Razzini R, et al. Regulation of mouse p45 NF-E2 transcription by an erythroid-specific GATA-dependent intronic alternative promoter. J Biol Chem. 2000;275: 10567-10576. [DOI] [PubMed] [Google Scholar]
- 20.Toki T, Arai K, Terui K, et al. Functional characterization of the two alternative promoters of human p45 NF-E2 gene. Exp Hematol. 2000;28: 1113-1119. [DOI] [PubMed] [Google Scholar]
- 21.Begley CG, Green AR. The SCL gene: from case report to critical hematopoietic regulator. Blood. 1999;93: 2760-2770. [PubMed] [Google Scholar]
- 22.Wadman IA, Osada H, Grutz GG, et al. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 1997;16: 3145-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lecuyer E, Hoang T. SCL: from the origin of hematopoiesis to stem cells and leukemia. Exp Hematol. 2004;32: 11-24. [DOI] [PubMed] [Google Scholar]
- 24.Robb L, Lyons I, Li R, et al. Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Proc Natl Acad Sci U S A. 1995;92: 7075-7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature. 1995;373: 432-434. [DOI] [PubMed] [Google Scholar]
- 26.Visvader JE, Fujiwara Y, Orkin SH. Unsuspected role for the T-cell leukemia protein SCL/tal-1 in vascular development. Genes Dev. 1998;12: 473-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall MA, Curtis DJ, Metcalf D, et al. The critical regulator of embryonic hematopoiesis, SCL, is vital in the adult for megakaryopoiesis, erythropoiesis, and lineage choice in CFU-S12. Proc Natl Acad Sci U S A. 2003;100: 992-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikkola HK, Klintman J, Yang H, et al. Haematopoietic stem cells retain long-term repopulating activity and multipotency in the absence of stem-cell leukaemia SCL/tal-1 gene. Nature. 2003;421: 547-551. [DOI] [PubMed] [Google Scholar]
- 29.Curtis DJ, Hall MA, Van Stekelenburg LJ, Robb L, Jane SM, Begley CG. SCL is required for normal function of short-term repopulating hematopoietic stem cells. Blood. 2004;103: 3342-3348. [DOI] [PubMed] [Google Scholar]
- 30.Hall MA, Slater NJ, Begley CG, et al. Functional but abnormal adult erythropoiesis in the absence of the stem cell leukemia gene. Mol Cell Biol. 2005;25: 6355-6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elefanty AG, Begley CG, Metcalf D, Barnett L, Kontgen F, Robb L. Characterization of hematopoietic progenitor cells that express the transcription factor SCL, using a lacZ “knock-in” strategy. Proc Natl Acad Sci U S A. 1998;95: 11897-11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269: 1427-1429. [DOI] [PubMed] [Google Scholar]
- 33.Ault KA, Knowles C. In vivo biotinylation demonstrates that reticulated platelets are the youngest platelets in circulation. Exp Hematol. 1995;23: 996-1001. [PubMed] [Google Scholar]
- 34.Jackson CW, Brown LK, Somerville BC, Lyles SA, Look AT. Two-color flow cytometric measurement of DNA distributions of rat megakaryocytes in unfixed, unfractionated marrow cell suspensions. Blood. 1984;63: 768-778. [PubMed] [Google Scholar]
- 35.Jackson SP, Schoenwaelder SM, Goncalves I, et al. PI 3-kinase p110beta: a new target for anti-thrombotic therapy. Nat Med. 2005;11: 507-514. [DOI] [PubMed] [Google Scholar]
- 36.Johnson KD, Christensen HM, Zhao B, Bresnick EH. Distinct mechanisms control RNA polymerase II recruitment to a tissue-specific locus control region and a downstream promoter. Mol Cell. 2001;8: 465-471. [DOI] [PubMed] [Google Scholar]
- 37.Johnson KD, Grass JA, Boyer ME, Kiekhaefer CM, Blobel GA, Weiss MJ, Bresnick EH. Cooperative activities of hematopoietic regulators recruit RNA polymerase II to a tissue-specific chromatin domain. Proc Natl Acad Sci U S A. 2002;99: 11760-11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Porcher C, Liao EC, Fujiwara Y, Zon LI, Orkin SH. Specification of hematopoietic and vascular development by the bHLH transcription factor SCL without direct DNA binding. Development. 1999;126: 4603-4615. [DOI] [PubMed] [Google Scholar]
- 39.Arnold JT, Daw NC, Stenberg PE, Jayawardene D, Srivastava DK, Jackson CW. A single injection of pegylated murine megakaryocyte growth and development factor (MGDF) into mice is sufficient to produce a profound stimulation of megakaryocyte frequency, size, and ploidization. Blood. 1997;89: 823-833. [PubMed] [Google Scholar]
- 40.Radley JM, Hodgson GS, Levin J. Platelet production after administration of antiplatelet serum and 5-fluorouracil. Blood. 1980;55: 164-166. [PubMed] [Google Scholar]
- 41.Vyas P, McDevitt MA, Cantor AB, Katz SG, Fujiwara Y, Orkin SH. Different sequence requirements for expression in erythroid and megakaryocytic cells within a regulatory element upstream of the GATA-1 gene. Development. 1999;126: 2799-2811. [DOI] [PubMed] [Google Scholar]
- 42.Jackers P, Szalai G, Moussa O, Watson DK. Etsdependent regulation of target gene expression during megakaryopoiesis. J Biol Chem. 2004;279: 52183-52190. [DOI] [PubMed] [Google Scholar]
- 43.Kaushansky K, Drachman JG. The molecular and cellular biology of thrombopoietin: the primary regulator of platelet production. Oncogene. 2002;21: 3359-3367. [DOI] [PubMed] [Google Scholar]
- 44.Tang T, Prasad KS, Koury MJ, Brandt SJ. Mitogen-activated protein kinase mediates erythropoietin-induced phosphorylation of the TAL1/SCL transcription factor in murine proerythroblasts. Biochem J. 1999;343: 615-620. [PMC free article] [PubMed] [Google Scholar]
- 45.Shivdasani RA, Orkin SH. Erythropoiesis and globin gene expression in mice lacking the transcription factor NF-E2. Proc Natl Acad Sci U S A. 1995;92: 8690-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuh AH, Tipping AJ, Clark AJ, et al. ETO-2 associates with SCL in erythroid cells and megakaryocytes and provides repressor functions in erythropoiesis. Mol Cell Biol. 2005;25: 10235-10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Italiano JE Jr, Shivdasani RA. Megakaryocytes and beyond: the birth of platelets. J Thromb Haemost. 2003;1: 1174-1182. [DOI] [PubMed] [Google Scholar]
- 48.Anguita E, Hughes J, Heyworth C, Blobel GA, Wood WG, Higgs DR. Globin gene activation during haemopoiesis is driven by protein complexes nucleated by GATA-1 and GATA-2. EMBO J. 2004;23: 2841-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wadman I, Li J, Bash RO, et al. Specific in vivo association between the bHLH and LIM proteins implicated in human T cell leukemia. EMBO J. 1994;13: 4831-4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Italiano JE Jr, Bergmeier W, Tiwari S, et al. Mechanisms and implications of platelet discoid shape. Blood. 2003;101: 4789-4796. [DOI] [PubMed] [Google Scholar]