

Abstract
The aberrant fusion protein NPM-ALK plays an important pathogenetic role in ALK+ anaplastic large-cell lymphoma (ALCL). We previously demonstrated that Jak3 potentiates the activity of NPM-ALK. Jak3 activation is restricted to interleukins that recruit the common γ chain (γc) receptor, including IL-9. NPM-ALK was previously shown to promote widespread lymphomas in IL-9 transgenic mice by unknown mechanisms. We hypothesized that IL-9 plays an important role in ALK+ ALCL via Jak3 activation. Our studies demonstrate the expression of IL-9Rα and IL-9 in 3 ALK+ ALCL-cell lines and 75% and 83% of primary tumors, respectively. IL-9 was detected in serum-free culture medium harvested from ALK+ ALCL-cell lines, supporting autocrine release of IL-9. Treatment of these cells with an anti–IL-9–neutralizing antibody decreased pJak3 and its kinase activity, along with pStat3 and ALK kinase activity. These effects were associated with decreased cell proliferation and colony formation in soft agar and cell-cycle arrest. Evidence suggests that cell-cycle arrest can be attributed to up-regulation of p21 and down-regulation of Pim-1. Our results illustrate that IL-9/Jak3 signaling plays a significant role in the pathogenesis of ALK+ ALCL and that it represents a potential therapeutic target for treating patients with ALK+ ALCL.
Introduction
Anaplastic lymphoma kinase-positive (ALK+) anaplastic large-cell lymphoma (ALCL) is defined by the World Health Organization (WHO) classification of hematologic malignancies as a subtype of T/null-cell non-Hodgkin lymphoma that is characterized by the consistent expression of CD30.1 In approximately 80% of ALK+ ALCL tumors, the aberrant expression of ALK occurs as a result of a t(2;5)(p23;q35) translocation, which leads to the fusion of the nucleophosmin (NPM) gene on 5q35 and ALK gene on 2p23.2,3 Characteristically, ALK+ ALCL occurs more frequently in children and young adults with an initial 5-year overall survival rate of approximately 70% following conventional cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP)–based therapy.4 Nonetheless, the prognosis for 30% to 40% of the patients is relatively poor.5-7 The oncogenic potential of NPM-ALK has been demonstrated by its transforming ability in vitro and by its ability to induce different types of malignant lymphomas in vivo.8-11 Previous studies showed that NPM-ALK mediates tyrosine phosphorylation and activation of various SH- or PTB-containing signaling molecules, such as GRB-2, PLC-γ, PI3K/Akt, IRS-1, Ras, SHC, FOXO, and Stats, that are directly involved in the regulation of cell survival and growth.12-18 However, the exact mechanisms by which NPM-ALK induces its oncogenic effects are not completely understood.
Janus kinase 3 (Jak3) is the final member identified of a family of protein tyrosine kinases that includes Jak1, Jak2, and tyrosine kinase 2 (Tyk2).19 Jaks reside in the cytoplasm; however, they can be recruited to certain cell-surface receptors on cytokine-induced receptor engagement. This process results in tyrosine phosphorylation and activation of Jaks (pJaks). Thereafter, pJaks phosphorylate receptor residues that act as docking sites for effector molecules including signal transducers and activators of transcription (Stats).20 pJaks subsequently tyrosine phosphorylate and activate Stats (pStats), which dissociate to the cytoplasm, dimerize, and translocate to the nucleus where they induce the transcription of a wide array of genes that can ultimately promote cell survival and proliferation. We and others have previously shown that Jak3 and ALK are physically associated in ALK+ ALCL cells and that selective pharmacologic inhibition of Jak3 reduces ALK tyrosine kinase activity and pStat3 levels in ALK+ ALCL cells.21,22
Jak3 activation is primarily restricted to interleukins (ILs) that possess the common γ chain (γc) in their respective receptors, namely IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. IL-9 is known to induce proliferation and antiapoptotic effects in T cells.23-25 Previous studies showed that enforced expression of NPM-ALK in IL-9 transgenic mice induces widespread lymphoma.26 The mechanism by which IL-9 promotes NPM-ALK activity is not known. In the present study, we hypothesized that IL-9 potentiates the oncogenic potential of NPM-ALK via Jak3. Herein we present evidence that IL-9Rα and IL-9 are frequently expressed in ALK+ ALCL-cell lines and primary tumors. Additional data demonstrate autocrine release of IL-9 by ALK+ ALCL-cell lines. More importantly, specific blockade of IL-9 using a neutralizing antibody resulted in decreased pJak3 and pStat3 levels as well as Jak3 and ALK tyrosine kinase activity. These effects resulted in decreased cell proliferation and colony formation in soft agar and cell-cycle arrest. Our findings implicate IL-9/Jak3 signaling as a potential therapeutic target for the treatment of patients with ALK+ ALCL.
Materials and methods
Cell lines, cell culture, and treatment with anti–human IL-9–neutralizing antibody
Three previously characterized ALK+ ALCL-cell lines, Karpas 299, SU-DHL-1, and SUP-M2, were used in the present study. In some of the experiments, the Hodgkin lymphoma–cell line L1236 and the colon carcinoma-cell line HT29 were used as positive and negative controls, respectively. Cells were cultured in RPMI 1640 (DMEM/F-12 for HT29 cells) medium (Life Technologies, Grand Island, NY) supplemented with 10% (15% for L1236 cells) heat-inactivated (56°C for 30 minutes) fetal bovine serum (FBS; Sigma, St Louis, MO), penicillin (10 000 U/mL; Sigma), streptomycin (10 mg/mL; Sigma), and L-glutamine (200 mM, 29.2 mg/mL; Life Technologies). Cell cultures were maintained at 37°C in 95% oxygen, 5% carbon dioxide, and 98% humidity. To detect autocrine release of IL-9, the cell-culture supernatants were concentrated for 1 hour at 4°C by using Centricon YM-10 centrifugal filter devices, which can elute protein complexes of 10 kDa or larger (Millipore, Billerica, MA), and then frozen at –80°C until used in Western blot studies for the detection of IL-9 as explained in the following experiments. For treatment of the cells as denoted in some of the experiments, cells were maintained in FBS-free RPMI for 12 hours before being incubated with goat anti–human IL-9–neutralizing antibody (catalog no. AB-209-NA; lot number DV064030; R&D Systems, Minneapolis, MN). Goat IgG (R&D Systems) was used as a negative control. To demonstrate effective binding of anti–IL-9–neutralizing antibody with IL-9, the cell-culture supernatants were concentrated twice by passing through Centricon YM-10 filters as previously described. The second concentrate was then frozen at –80°C until used in Western blot studies to demonstrate lack of IL-9.
[3H]-thymidine incorporation assay
To detect changes in cell proliferation, [3H]-thymidine incorporation was performed using standard techniques. Briefly, cell samples (20 × 103) in triplicate were suspended in serum-free medium for 12 hours and then treated with anti–IL-9–neutralizing antibody or control goat IgG for 6 hours (24 hours for L1236). To demonstrate the specificity of the antibody, recombinant human IL-9 (rhIL-9, 209-IL; R&D Systems) was incubated at 5 ng/mL with different concentrations of the antibody for 1 hour at 37°C in a 96-well plate. The mixture in a total of 100 μL, containing the antibody, rhIL-9, and Karpas 299 cells (1 × 105 cells/mL) was incubated at 37°C for 6 hours in a humidified CO2 incubator. Thereafter, [3H]-thymidine (0.2 μCi/mL [0.0074 MBq]; Sigma) was incubated with the cells for 4 hours. Incorporation of [3H]-thymidine was measured using a liquid scintillation counter (Packard Biosciences, Meriden, CT).
Colony formation in soft agar
Soft agar (0.54% wt/vol) was prepared by autoclaving Bacto agar (Difco, Detroit, MI) in distilled water prior to use. Cells harvested in serum-fee medium were treated for 6 hours with anti–IL-9–neutralizing antibody (80 μg/mL) or IgG, then resuspended in cooled 0.33% agar in RPMI medium supplemented with 10% FBS at a density of 200 cells/35-mm plate, and seeded onto solidified 0.54% agar-containing culture medium; plates were kept at 4°C for 30 minutes and then 1 mL RPMI medium supplemented with 10% FBS was added to each well. Plates were cultured for 2 weeks as described. The colonies were stained with 0.5% crystal violet, counted, and photographed with FluorChem 8800 Imaging System (Alpha Innotech, San Leandro, CA). Triplicate samples were used in the experiment.
Cell-cycle analysis
For flow cytometric analysis of the cell cycle, cells (10 × 105) were fixed in 70% ice-cold ethanol and stored overnight at –20°C. Cells were then washed twice in PBS and incubated for 5 minutes with 100 U/mL ribonuclease A containing 0.1% Triton X in PBS. The cells were stained with propidium iodide (PI; Sigma) at a final concentration of 50 μg/mL for 20 minutes and analyzed using a flow cytometer after suspension in 7-AAD (Becton Dickinson, San Jose, CA).
Tyrosine kinase activity assay
Tyrosine kinase activity was measured using standard techniques and the Universal Tyrosine Kinase Assay Kit (Takara Bio, Otsu, Japan). Briefly, cells treated with anti–IL-9–neutralizing antibody or IgG were lysed in a buffer containing protease and phosphatase inhibitors. To limit nonspecific immunoglobulin binding, 20 μL/reaction of protein A-agarose (Santa Cruz Biotechnology, Santa Cruz, CA) was added to the cell lysates and then removed. Precleared cell lysates were then incubated with 10 μg anti-Jak3 (Santa Cruz Biotechnology) or anti-ALK antibody (DakoCytomation, Carpinteria, CA) overnight at 4°C. To capture the immune complex, 20 μL/reaction of protein A agarose was added to the lysates at room temperature for 20 minutes. The lysates were briefly centrifuged at 500g and the precipitate resuspended in 50 μL kinase-reacting buffer. Kinase reactions were initiated with 10 μL of 40 μM ATP-2Na in immobilized wells, incubated for 30 minutes at 37°C, and blocked by blocking solution according to manufacturer's recommendations. After the blocking solution was discarded, 50 μL horseradish peroxidase-conjugated antiphosphotyrosine (PY20) antibody (Takara Bio) was added to each well for 30 minutes and developed by addition of 100 μL horseradish peroxidase and substrate solution (TMBZ) for 15 minutes at 37°C. The reaction was stopped with 1 N H2SO4 and absorbance measured at 450 nm in a microplate reader (MRX II; Dynex, Frankfurt, Germany). The activity of Jak3 or ALK was normalized by an internal tyrosine kinase control.
Immunoprecipitation and Western blotting
Briefly, cells were lysed in lysis buffer and centrifuged at 14 000g for 10 minutes at 4°C. The supernatant was collected and 50 to 80 μg protein was electrophoresed on a 6% to 12% SDS polyacrylamide gel or immunoprecipitated with a specific antibody. Cell lysates were incubated with anti-γc antibody (BAF-284; R&D Systems) overnight at 4°C. Agarose beads conjugated with protein A/C were then added and incubated for 2 hours at 4°C. Immunocomplexes were harvested by centrifugation at 500g, washed 3 times with cold PBS and once with lysis buffer, and then subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Western blotting was then performed using primary antibodies, including Jak3 and pJak3, diluted 1:1000 in blocking buffer. Specific antibodies were purchased for other Western blot studies. Excluding IL-9 (R&D Systems) and β-actin (A5316; Sigma), other antibodies including Jak3 (sc-1080), pJak3 (sc-16567-R), Stat3 (sc-482), pStat3 (sc-8059), p21 (sc-6246), p27 (sc-1641), cyclin D3 (sc-6283), and Pim-1 (sc-13513) were from Santa Cruz Biotechnology. All blots were developed with a horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) and the enhanced chemiluminescence detection kit (Amersham Life Sciences, Arlington Heights, IL).
RT-PCR
Total cellular RNA was extracted from the ALK+ ALCL, L1236, and HT29-cell lines using RNeasy Mini Kit (Qiagen, Valencia, CA). Reverse transcription (RT) was performed by using 2 μg total RNA in a first-strand cDNA synthesis reaction with superscript reverse transcriptase as recommended by the manufacturer (Invitrogen Life Technologies, Carlsbad, CA). Polymerase chain reaction (PCR) was performed by adding 1 μL RT product into 50 μL total volume reaction containing 1 × buffer, 200 μMof each dNTPs, 20 pM of each oligonucleotide primer, and 0.2 U AmpliTaq polymerase. Oligonucleotides specific for IL-9Rα sequences were used in the PCR. Sequences of primer pairs were as follows: IL-9Rα, 5′-ACCTGCCTCACCAACAACATTCTCA-3′ and 5′-TCAAATCCAACAAAGTCTGGCTTA-3′27; IL-9, 5′-GGGATCCTGGACATCAACTTC-3′ and 5′-GAAGCATGGTCTGGTGCAGTT-3′28; β-actin, 5′-GCTCCTCCTGAGCGCAAGT-3′ and 5′-TCGTCATACTCCTGCTTGCTGAT-3′.28 For DNA amplification, cDNA was denatured at 94°C for 1 minute, subjected to primer annealing at 60°C (IL-9 and IL-9Rα) or 54°C (β-actin) for 1 minute, and then subjected to DNA extension at 72°C for 1 minute for 35 cycles in a thermal cycler (PTC100; MJ Research, Watertown, MA). Amplified products were analyzed by DNA gel electrophoresis in 2% agarose and visualized by FluorChem 8800 Imaging System (Alpha Innotech).
Immunofluorescence staining and confocal microscopy
To visualize the expression of IL-9Rα and IL-9 in the ALK+ ALCL-cell lines, cytospin slides were prepared and fixed with 4% paraformaldehyde at 4°C. The slides then were incubated with 10 μg/mL anti–IL-9Rα (1:100, sc-1030, Santa Cruz Biotechnology), anti-IL-9 (1:100; R&D Systems), or isotype-matching control in a dilution buffer (DakoCytomation) overnight at 4°C and then washed twice with PBS. Thereafter, the slides were incubated with Cy3-conjugated donkey anti–rabbit IgG antibody (1:50; Jackson ImmunoResearch Laboratories) for 2 hours at room temperature. The slides were extensively washed with PBS and counterstained with 4′,6-diamino-2-phenylindole for 2 minutes. Fluorescence signals were detected at 460 (DAPI), 490 (FITC; IL-9Rα), and 595 nm (Texas red; IL-9), by using a 60 ×/1.40 NA oil-immersion objective and confocal microscopy (Zeiss LSM 510; Carl Zeiss MicroImaging, Thornwood, NY). Images were captured and analyzed by Adobe Photoshop software (Version 7.0; Adobe, San Jose, CA).
Patients, tissue microarray, and immunohistochemical staining
Archival tissue samples from 12 lymph node biopsies from patients with ALK+ ALCL were collected prior to therapeutic interventions following approval by the ethics research committee at Cross Cancer Institute (Edmonton, AB, Canada). All patients provided informed consent in accordance with the Declaration of Helsinki. The diagnosis of these cases was based on the criteria established by the WHO.1 Detailed clinical and follow-up data were available for all patients. The tumor samples were fixed in formalin, routinely processed, and embedded in paraffin. Representative cores were selected from each of the paraffin blocks and included in a tissue microarray. Cores from a reactive lymph node were also included in the tissue microarray as internal controls. The tissue microarray was constructed using a manual tissue arrayer (Beecher Instruments, Sun Prairie, WI). Immunohistochemical staining was performed on sections from the tissue microarray as well as on formalin-fixed and paraffin-embedded sections from cell blocks prepared from the ALK+ ALCL, L1236, and HT29-cell lines. Tissue sections were first deparaffinized in xylene and rehydrated using a graded series of ethanol. A 3-step streptavidin-biotin-horseradish peroxidase method was used after heat-induced epitope retrieval. Briefly, endogenous peroxidase activity was blocked for 30 minutes in 3% hydrogen peroxide and, subsequently, the slides were incubated with protein blocking solution (DakoCytomation) for 15 minutes. Thereafter, the slides were incubated overnight with the primary antibodies diluted in 0.1% bovine serum albumin, 50 mM Tris-HCl buffer, pH 7.6. The dilutions of the antibodies used in the study were 1:100 for IL-9 (R&D Systems) and 1:250 for IL-9Rα (Santa Cruz Biotechnology). Detection of the immunoreaction was achieved using the LSAB+ kit (DakoCytomation), which contains the secondary biotinylated antibody (incubation time, 20 minutes) and the streptavidin/horseradish peroxidase complex (incubation time, 20 minutes). 3,3′-Diaminobenzidine/H2O2 (DakoCytomation) was used as the chromogen and hematoxylin (DakoCytomation) as the counterstain. Application of mouse IgG1 antibody (DakoCytomation) was used as a negative control to exclude unspecific cross-reactions of the primary antibodies in all experiments. The photomicrographs were obtained using a Nikon Microphot FXA microscope (Nikon Instruments, Melville, NY) and an Olympus DP70 camera (Olympus America, Melville, NY).
Statistical analysis
Statistical analysis was performed using t test for paired data and Statview software (Abacus Concepts, Berkeley, CA).
Results
Previous studies showed that enforced expression of NPM-ALK in IL-9 transgenic mice induces widespread malignant lymphomas.26 To determine whether a correlation exists within ALK+ ALCL-cell lines and patients' tumors, we initiated the following studies to probe the role of IL-9 in this disease.
Expression of IL-9Rα, IL-9, and γc in ALK+ ALCL
RT-PCR studies demonstrated the presence of IL-9Rα mRNA in 3 ALK+ ALCL-cell lines including Karpas 299, SU-DHL-1, and SUP-M2 as well as in the positive control Hodgkin lymphoma cells L1236. The colon carcinoma cells HT29 were negative for IL-9Rα mRNA (Figure 1A). We further confirmed the expression of IL-9Rα protein in the ALK+ ALCL by using standard immunohistochemical staining. L1236 and HT29 cells were used as positive and negative controls, respectively (Figure 1B). Additionally, immunofluorescence staining validated this study (Figure 1C). As shown in the lower panel, all 3 cell lines stained positively for IL-9Rα, as compared to isotype control shown in the upper panel (Figure 1C). Because IL-9Rα requires γc for Jak3-mediated signaling, we observed via immunoprecipitation and Western blotting that γc is present and physically associated with pJak3 in Karpas 299 cells (Figure 1D). Similar results were obtained for Jak3 (data not shown).
Figure 1.

ALK+ ALCL-cell lines express IL-9Rα. (A) RT-PCR studies show the presence of IL-9Rα mRNA in all ALK+ ALCL cells. The Hodgkin lymphoma-cell line L1236 was used as positive control and the colon carcinoma HT29-cell line as negative control. β-Actin shows equal loading. (B) Immunohistochemical staining of paraffin-embedded tissue sections from cell blocks confirmed the expression of IL-9Rα in all the ALK+ ALCL-cell lines. Similar to RT-PCR studies, L1236 and HT29 cells were used as positive and negative controls, respectively (original magnification × 200). (C) Confocal microscopy after immunofluorescence staining of cytospin slides with antibodies directed against IL-9Rα (bottom panel) or control IgG (top panel) demonstrates the expression of IL-9Rα in the ALK+ ALCL-cell lines, Karpas 299, SU-DHL-1, and SUP-M2 (original magnification × 600). (D) Immunoprecipitation and Western blotting show the physical association between γc and pJak3 in Karpas 299 cells. As shown in the right panel, immunoprecipitation was first performed on the cell lysate using anti-γc antibody followed by Western blotting using anti-pJak3 antibody. The left panel demonstrates a simultaneous control study where the lysate was analyzed only by Western blotting using an anti-pJak3 antibody. pJak3 bands are present at the expected molecular weight of 118 kDa in the 2 panels. Similar findings were noted when anti-Jak3 antibody was used instead of anti-pJak3 antibody (data not shown).
Because ALK+ ALCL-cell lines expressed IL-9Rα and γc, we next sought to determine whether these cells express IL-9. This cytokine was most likely derived from the cells because RT-PCR analysis showed that IL-9 mRNA was present in all ALK+ ALCL-cell lines as compared with positive and negative control cell lines L1236 and HT29, respectively (Figure 2A). The same cells were stained with an antibody to IL-9 using immunohistochemical staining techniques. Similar to RT-PCR studies, L1236 and HT29 cells were used as positive and negative controls, respectively (Figure 2B). We confirmed these findings by using immunofluorescence staining. Indeed all of the ALK+ ALCL cells demonstrated the expression of IL-9, as compared with cells stained with isotype control (Figure 2C lower and upper panels, respectively).
Figure 2.

ALK+ ALCL-cell lines express IL-9. (A) RT-PCR studies demonstrate the presence of IL-9 mRNA in ALK+ ALCL cells. L1236 and HT29 cells were used as positive and negative controls, respectively. β-Actin demonstrates equal loading. (B) Immunohistochemical staining of the ALK+ ALCL-cell lines confirmed the expression of IL-9. L1236 and HT29 were used as positive and negative controls, respectively (original magnification × 200). (C) Confocal microscopy and immunofluorescence staining using specific antibody (bottom panel) or IgG (top panel) show the expression of IL-9 in ALK+ ALCL-cell lines (original magnification × 600).
We also used standard immunohistochemical staining techniques to evaluate the expression of IL-9Rα and IL-9 in primary tumors collected from 12 patients with ALK+ ALCL and included in a tissue microarray. The 5 female and 7 male patients ranged in age from 6 to 74 years (median, 26.5 years). IL-9Rα and IL-9 were expressed in the neoplastic cells of 75% (9 of 12) and 83% (10 of 12) of the primary tumors, respectively. Figure 3 shows the pattern of expression of IL-9Rα (A) and IL-9 (B) in a reactive lymph node. Figure 3 also illustrates 2 examples of the ALK+ ALCL tumors that expressed IL-9Rα (C,E) and IL-9 (D,F).
Figure 3.
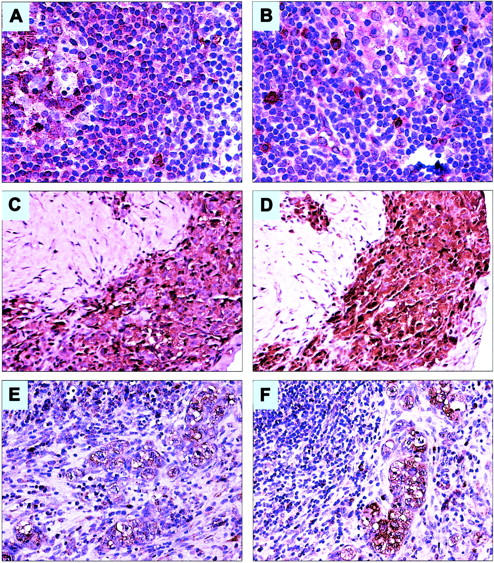
Expression of IL-9Rα and IL-9 in a reactive lymph node and in primary ALK+ ALCL tumors from patients. (A) IL-9Rα is strongly expressed in a significant number of cells within the germinal center (left side) and the mantle zone of a reactive lymph node. The frequency and intensity of expression of IL-9Rα are relatively diminished in the marginal zone and interfollicular areas (original magnification × 200). (B) IL-9 is strongly expressed in scattered small lymphoid cells and mast cells in a reactive lymph node. Most of these cells are localized in the interfollicular areas as well as around and within the lymph node sinuses (original magnification × 200). (C-D) An example of a lymph node showing sclerotic tissue with dense infiltration by ALK+ ALCL cells that are strongly positive for IL-9Rα (C) and IL-9 (D; original magnification × 200). (E-F) Another example of a lymph node involved by ALK+ ALCL demonstrating large neoplastic cells that are positive for IL-9Rα (E) and IL-9 (F). The large neoplastic cells are confined to the lymph node sinuses, a characteristic morphologic feature of ALK+ ALCL (original magnification × 200). In all of the tissue samples, the staining for both IL-9Rα and IL-9 was membranous and cytoplasmic, whereas the nuclei were negative for the 2 proteins.
Taken together, these data demonstrate that ALK+ ALCL cells express IL-9 and its receptor suggesting a signaling cascade that may promote tumor progression in an autocrine fashion. To further validate that IL-9 is produced and secreted by ALK+ ALCL cells, we performed the following set of experiments.
Autocrine release of IL-9 by ALK+ ALCL cells
IL-9 was detected in cell lysates from the 3 ALK+ ALCL-cell lines (Figure 4). Importantly, high levels of IL-9 also were readily detectable in the supernatants of FBS-free tissue culture medium from these cells (Figure 4). Similar studies showed no evidence of IL-9 in the cell lysate or supernatant from the negative control cells HT29 (Figure 4). As shown in Figure 4, treating the ALK+ ALCL cells with 80 μg/mL anti–IL-9–neutralizing antibody completely depleted IL-9 from the cell-culture supernatant after protein concentration and filtration.
Figure 4.

Autocrine release of IL-9 by ALK+ ALCL cells. Western blot studies show the presence of IL-9 protein in cell lysates from Karpas 299, SU-DHL-1, and SUP-M2 cells. Anti–IL-9–neutralizing antibody had no significant effect on IL-9 levels in the cell lysates. β-Actin shows equal loading of the proteins. Also, these studies show the presence of IL-9 in the culture medium from the same cells after being maintained in FBS-free medium for 12 hours. The anti–IL-9–neutralizing antibody (80 μg/mL) effectively bound to IL-9, as demonstrated by the lack of IL-9 in the culture medium after protein complexes were eluted. The negative control cells HT29 demonstrate lack of IL-9 in the cell lysate and the cell-culture medium. The experiment was repeated 2 times with consistent results.
We previously showed that inhibition of Jak3 decreases pStat3 levels and ALK kinase activity in ALK+ ALCL cells.21 Because IL-9 signaling induces tyrosine phosphorylation and activation of Jak3, we sought to study the effects of blockade of IL-9 on Jak3, Stat3, and ALK in these cells via the following studies.
Specific blockade of IL-9 decreases pJak3 and its tyrosine kinase activity, along with pStat3 and ALK tyrosine kinase activity in ALK+ ALCL cells
Using Western blotting, we measured protein levels of pJak3 and pStat3 after treatment of the ALK+ ALCL cells with anti–IL-9–neutralizing antibody. Indeed, specific blockade of IL-9 decreased pJak3 and pStat3 levels, without notable changes in the total levels of Jak3 or Stat3 (Figure 5A). To confirm the specificity of this antibody, we used HT29 cells that were previously shown to express Jak3, Stat3, and their phosphorylated forms.29 Significant changes in these proteins were not noted after treating HT29 cells with the anti–IL-9–neutralizing antibody using similar protocols (Figure 5B).
Figure 5.

Effects of specific blockade of IL-9 on Jak3, Stat3, and ALK. (A) Western blot studies show that increasing concentrations of anti–IL-9–neutralizing antibody (80 and 160 μg/mL) decreases pJak3 in the 3 ALK+ ALCL-cell lines. The decrease in pStat3 levels in SU-DHL-1 and SUP-M2 cells was more pronounced at a concentration of 160 μg/mL. Despite the slight increase in pStat3 levels in Karpas 299 cells at a concentration of 80 μg/mL, it decreased to the baseline level at 160 μg/mL. Total levels of Jak3 and Stat3 were not affected. β-Actin confirmed equal loading of the proteins. The experiment was performed 3 times with consistent results. (B) Western blot studies did not demonstrate similar changes in pJak3, Jak3, pStat3, and Stat3 levels after treating the negative control cells HT29 with similar concentrations of the anti–IL-9–neutralizing antibody. β-Actin confirmed equal loading of the proteins. (C) After treating the ALK+ ALCL-cell lines with anti–IL-9–neutralizing antibody (160 μg/mL), tyrosine kinase activity of Jak3 and ALK were measured. Normalized data reveal reduction to 60% or less of the control levels of the kinase activity of the 2 enzymes. Control cells were treated with IgG. Shown are averaged data of 2 consistent experiments.
Because specific blockade of IL-9 signaling decreased tyrosine phosphorylated levels of Jak3 and Stat3, we next investigated whether this treatment inhibited the tyrosine kinase activity of Jak3 and ALK. Presumably, IL-9 engages its receptor resulting in generation of catalytically primed Jak3. To monitor their catalytic activity, lysates from ALK+ ALCL-cell lines were assayed for tyrosine kinase activity following pretreatment with the anti–IL-9–neutralizing antibody. Interestingly, specific blockade of IL-9 reduced both Jak3 and ALK tyrosine kinase activity to approximately 60% or less of the baseline levels (Figure 5C).
As mentioned, Jak3, ALK, and Stat3 promote cell growth and survival. To determine the effect of specific blockade of IL-9 on cell survival of ALK+ ALCL, we performed the following studies.
Specific blockade of IL-9 decreases ALK+ ALCL-cell proliferation and colony formation in soft agar due to cell-cycle arrest
The ALK+ ALCL cells were treated with the anti–IL-9–neutralizing antibody. Thereafter, cell proliferation was measured by [3H]-thymidine incorporation (Figure 6A). Anti–IL-9–neutralizing antibody at a concentration of 80 μg/mL decreased cell proliferation to approximately 40% of the baseline levels (P < .001). A similar effect on cell proliferation was not detected when the negative control cells HT29 were used in similar experiments (Figure 6A). In addition, preincubation of anti–IL-9–neutralizing antibody with rhIL-9 also abolished the decrease in proliferation of Karpas 299 cells (Figure 6A). The anti–IL-9–neutralizing antibody induced a gradual concentration-dependent decrease in the proliferation of the Hodgkin lymphoma cells L1236 to 69% of its baseline levels at a concentration of 80 μg/mL (P < .01; Figure 6A). Of note is that the decrease in L1236 cell proliferation was relatively less pronounced, albeit statistically significant from the baseline level, than the one observed in ALK+ ALCL cells. Most likely, this difference can be explained by the release of relatively higher levels of IL-9 by L1236 cells compared with ALK+ ALCL cells, as illustrated in the RT-PCR studies (Figure 2A).
Figure 6.

Specific blockade of IL-9 decreases ALK+ ALCL-cell proliferation and colony formation potential. (A) anti–IL-9–neutralizing antibody induces concentration-dependent decrease in ALK+ ALCL-cell proliferation as revealed by the [3H]-thymidine incorporation assay. The decrease in cell proliferation reached approximately 40% of the baseline level at a concentration of 80 μg/mL (P < .001). Similar effects were not detected when the anti–IL-9–neutralizing antibody was preincubated with rhIL-9 before treating Karpas 299 cells, and cell proliferation remained stable with a very slight decrease to 90% of its baseline level at 80 μg/mL concentration of the antibody. Similarly, changes in cell proliferation were not seen in the negative control cells HT29 after treatment with anti–IL-9–neutralizing antibody. When the Hodgkin lymphoma cells L1236 were used as a positive control, a significant and gradual decrease in cell proliferation was observed, which became 69% of the baseline level at a concentration of 80 μg/mL (P < .01). The results are shown as means ± SE of at least 3 consistent experiments. *Statistically significant compared with baseline cells (0) treated with IgG. (B) anti–IL-9–neutralizing antibody (80 μg/mL) induces marked decrease in colony formation of Karpas 299 cells in soft agar. The top panel shows the means ± SD of 3 consistent experiments. Compared to a control level of 60 ± 8 colonies/plate in control cells treated with IgG, cells treated with the anti–IL-9–neutralizing antibody developed only 24 ± 6 colonies/plate. The bottom panel shows examples of the cultured plates. The plate on the right side contains Karpas 299 cells treated with anti–IL-9–neutralizing antibody before being cultured for 2 weeks. The plate on the left side contains control cells treated with IgG under the same experimental conditions.
To monitor the effect of blockade of IL-9 on cell growth in soft agar, Karpas 299 cells were treated as above with anti–IL-9–neutralizing antibody (80 μg/mL) and colonies were observed. The anti–IL-9–neutralizing antibody limited colony formation of ALK+ ALCL cells to only 40% of the control levels (Figure 6B).
To explore possible explanations of the decrease in cell proliferation and growth after specific blockade of IL-9 in ALK+ ALCL, we studied changes in the cell cycle using flow cytometric analysis after staining with PI and 7-ADD. Anti–IL-9–neutralizing antibody induced G1 cell-cycle arrest as demonstrated by a significant decrease in ALK+ ALCL cells in the S phase (Figure 7A). Anti–IL-9–neutralizing antibody-induced cell-cycle arrest could be explained by a significant increase in p21 and decrease in Pim-1 kinase levels (Figure 7B). There were no significant changes in p27 or cyclin D3 levels (Figure 7B).
Figure 7.

Specific blockade of IL-9 induces G1 cell-cycle arrest associated with increased p21 and decreased Pim-1 kinase levels in ALK+ ALCL cells. (A) Analysis of the cell cycle using flow cytometry and PI/7-ADD staining. The right panel shows histograms of the ALK+ ALCL cells treated with 80 μg/mL anti–IL-9–neutralizing antibody compared with control cells treated with equivalent concentrations of IgG and shown in the left panel. The anti–IL-9–neutralizing antibody induces G1 cell-cycle arrest as demonstrated by the marked decrease of cells in the S phase. The number of the cells in the S phase decreased to 59%, 40%, and 36% of their corresponding baseline levels in Karpas 299, SU-DHL-1, and SUP-M2 cells, respectively. The experiment was repeated twice with consistent findings. (B) Western blot studies showing concentration-dependent increase in p21 levels after treating the ALK+ ALCL cells with 80 and 160 μg/mL anti–IL-9–neutralizing antibody. There was a simultaneous concentration-dependent decrease in Pim-1 levels in Karpas 299 and SUP-M2 cells. Whereas Pim-1 level in SU-DHL-1 cells slightly increased at a concentration of 80 μg/mL anti–IL-9–neutralizing antibody, it decreased to the baseline level at a concentration of 160 μg/mL. Significant changes were not detected in p27 and cyclin D3. β-Actin confirmed equal loading of the proteins. The results represent 1 of 2 consistent experiments.
Discussion
ALK+ ALCL is a unique type of non-Hodgkin lymphoma characterized by several chromosomal aberrations of which t(2;5)(p23;q35) is the most common. This translocation leads to the aberrant expression of the fusion protein NPM-ALK.2,3 It is believed that NPM-ALK plays a major role in the pathogenesis of ALCL. Previous in vitro studies showed that NPM-ALK possesses significant transformation potential.8,9 In addition, enforced expression of NPM-ALK gene induces malignant lymphoma in mice.10,11,30-32 Notably, a significant number of the NPM-ALK–induced tumors in the mice models, including tumors driven by T-cell–specific CD2 or CD4 promoters,11,32 demonstrated B-cell immunoblastic/plasmablastic morphologic and immunophenotypic features, were CD30–, and were confined to the mediastinum. Considering that the vast majority of ALK+ ALCL tumors in humans demonstrate T/null-cell immunophenotype, express CD30, and present as a widespread systemic disease, the findings from the animal models suggest that NPM-ALK is not the only factor that drives the biologic sequences that determine the characteristic features of ALK+ ALCL in humans. To further support this concept, previous studies demonstrated the presence of NPM-ALK in nonneoplastic cells.33-35 These observations implicate that additional events and signaling pathways are most likely required to drive the oncogenic events that lead to the characteristic histopathologic, immunophenotypic, and clinical features of ALK+ ALCL in humans.
The biologic processes that lead to lymphomagenesis are complex but likely differ among the cell lineages and different lymphoma histotypes. Numerous cytokines are thought to induce or support lymphomagenesis. IL-9 is a multifunctional cytokine secreted by TH2 lymphocytes.36 Despite the lack of significant effects on freshly isolated T cells,24,37 IL-9 induces significant proliferative effects on stimulated T cells.23,24 Nonetheless, Renauld et al showed that only 7% of IL-9 transgenic mice develop T-cell thymic lymphomas.38 Importantly, a subcarcinogenic dose of N-methyl-N-nitrosourea induced thymic lymphomas in all of the treated IL-9 transgenic mice.38 These findings demonstrate that dysregulated IL-9 requires additional factors for the induction and progression of malignant lymphomas. Recently, NPM-ALK was shown to induce massive and widespread lymphomas in IL-9 transgenic mice.26 Of these tumors, 46% demonstrated T-cell immunophenotype.
Jak3 is a protein tyrosine kinase whose activation/phosphorylation is limited to a small number of interleukins that recruit the IL-2 common γ chain (γc) in their receptors, including IL-9.19,39-41 Several studies have demonstrated the important role that Jak3 plays in T-cell development, homeostasis, and survival.42-46 We and others have previously shown that Jak3 is physically associated with ALK in ALK+ ALCL-cell lines.21,22 In addition, we demonstrated that selective inhibitors of Jak3 induce apoptotic-cell death and cell-cycle arrest and decrease pStat3 levels and ALK tyrosine kinase activity in these cells.21 Furthermore, we found that Jak3 is constitutively activated and significantly associated with ALK expression in human primary ALCL tumors.47 In the present study, using immunoprecipitation, we further demonstrated that Jak3 is physically associated with γc in Karpas 299 cells. These findings suggest that Jak3 signaling is an important oncogenic pathway in ALK+ ALCL.
The aim of the present study was to test the hypothesis that IL-9 plays a significant role in the pathogenesis of ALK+ ALCL through a Jak3-dependent pathway. Using immunofluorescence and immunohistochemical staining and RT-PCR, we showed that IL-9Rα and IL-9 are expressed in 3 ALK+ ALCL-cell lines: Karpas 299, SU-DHL-1, and SUP-M2. In addition, IL-9Rα and IL-9 were expressed in a majority representing 75% and 83% of the ALK+ ALCL tumors that we studied, respectively. Importantly, our results demonstrated the presence of IL-9 in supernatants of ALK+ ALCL cells harvested in serum-free medium. Blockade of this pathway inhibited oncogenic growth of these cells. These findings suggest that the ALK+ ALCL cells release and respond to IL-9 in an autocrine fashion.
To our knowledge, these findings are the first to demonstrate constitutive expression of IL-9Rα and IL-9 and document autocrine release of IL-9 by ALK+ ALCL cells. Merz et al have previously studied 6 cases of large-cell lymphoma with anaplastic morphology.48 These cases included malignant lymphomas of T-, B-, and null-cell immunophenotypes, of which 2 cases with T-cell immunophenotype showed the presence of IL-9 mRNA. However, the significance of ALK expression was not yet established. Therefore, these 2 cases may have represented peripheral T-cell lymphoma or ALK– ALCL, 2 types of non-Hodgkin lymphoma with known biologic and clinical heterogeneity. In contrast to our findings, Gaiser et al found that IL-9 mRNA is down-regulated in ALK+ ALCL-cell lines obtained from cDNA expression profiling.49 It is of note that gene expression of the ALK+ ALCL-cell lines was compared to the gene expression of activated T cells, which are expected to produce a significant amount of IL-9. Furthermore, no confirmation to elucidate the expression of IL-9 or IL-9Rα in ALK+ ALCL-cell lines was used in their study.49
To examine the contribution of IL-9 to the pathogenesis of ALK+ ALCL cells, we used a commercially available IL-9–neutralizing antibody, which readily down-regulated pJak3 levels that corresponded to a marked decrease in Jak3 tyrosine kinase activity. Concomitantly, there was as a decrease in pStat3 levels. Total protein levels of Jak3 and Stat3 were not affected. Treating ALK+ ALCL cells with IL-9–neutralizing antibody also decreased ALK tyrosine kinase activity. These results are in agreement with our previous findings21 and provide further evidence supporting the concept that Jak3 plays an important role in the pathogenesis of ALK+ ALCL via interaction with ALK and Stat3, and further support the crosstalk between the 2 enzymes, Jak3 and ALK. Functionally, anti–IL-9–neutralizing antibody significantly abrogated the growth potential of ALK+ ALCL cells as demonstrated by a marked decrease in [3H]-thymidine incorporation and colony formation in soft agar. Anti–IL-9–neutralizing antibody also caused cell-cycle arrest in ALK+ ALCL cells. Previous studies showed that p21 plays a significant role in cell-cycle regulation in ALK+ ALCL via a CD30-dependent pathway.50 Indeed, we observed a concentration-dependent increase in p21 after treatment with anti–IL-9–neutralizing antibody. Pim-1 is a serine/threonine kinase involved in several important biologic functions including mitosis and cell-cycle progression.51-54 Previous studies showed that Pim-1 possesses a significant oncogenic potential in malignant neoplasms, including malignant lymphomas.55-58 A recent study demonstrated that Pim-1 kinase phosphorylates p21 and leads to its sequestration in the cytoplasm.59 The expression of Pim-1 appears to be regulated, at least in part, via IL-9/Jak/Stat signaling pathway.60,61 Our results demonstrated that anti–IL-9–neutralizing antibody reduces the expression of Pim-1 in ALK+ ALCL cells. This is the first report to demonstrate the expression of Pim-1 in ALK+ ALCL cells. These results also suggest that Pim-1 is a downstream target of the IL-9/Jak3/Stat3 signaling pathway in ALK+ ALCL and that it may have a role in the pathogenesis of this type of malignant lymphoma. We have previously demonstrated that the occurrence of cell-cycle arrest in ALK+ ALCL due to inhibition of Jak3 or other signaling pathways was associated with significant alterations in cyclin D3 and p27, 2 downstream targets of Jak/Stat signaling pathway.21,62 Therefore, we also sought to explore possible changes in these 2 proteins. The lack of significant changes in cyclin D3 and p27 levels after treatment with anti–IL-9–neutralizing antibody could be due to the effects of other signaling pathways known to affect the expression of these cell-cycle regulators, such as PI3K/Akt, FOXO, FAK, and PKC.18,62,63 Another possible explanation is that the experimental conditions in the present study were not sufficient to induce notable changes in cyclin D3 and p27 levels.
In conclusion, the present study provides evidence that the IL-9/Jak3 signaling plays a major role in the pathogenesis of ALK+ ALCL via activation of ALK and Stat3. These findings have important clinical implications because they identify the IL-9/Jak3 signaling pathway as a potential therapeutic target to treat this type of malignant lymphoma. Considering the effects of IL-9/Jak3 on T-cell development and T-cell lymphomagenesis, our results suggest that, in addition to ALK, the constitutive activation of IL-9/Jak3 signaling may represent a secondary biologic event that leads to the development of the distinct features that characterize ALK+ ALCL.
Prepublished online as Blood First Edition Paper, June 8, 2006; DOI 10.1182/blood-2006-04-020305.
H.M.A. is supported by a K08CA114395 grant from the National Cancer Institute, the Physician Scientist Program Award at M. D. Anderson Cancer Center, and a Career Development Award from the National Institutes of Health (NIH) Leukemia Specialized Program of Research Excellence (SPORE) grant to M. D. Anderson Cancer Center. R.A.K. is supported by grants AI053566 and SG12RR008124 from NIH.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Delsol G, Ralfkiaer E, Stein H, Jaffe ES. Anaplastic large cell lymphoma. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, eds. Pathology and Genetics of Tumors of Haematopoietic and Lymphoid Tissues: World Health Organization Classification of Tumours. Lyon, France: IARC Press; 2001: 230-235.
- 2.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263: 1281-1284. [DOI] [PubMed] [Google Scholar]
- 3.Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene. 1994;9: 1567-1574. [PubMed] [Google Scholar]
- 4.Falini B, Pileri S, Zinzani PL, et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood. 1999;93: 2697-2706. [PubMed] [Google Scholar]
- 5.Brugieres L, Deley MC, Pacquement H, et al. CD30+ anaplastic large-cell lymphoma in children: analysis of 82 patients enrolled in two consecutive studies of the French Society of Pediatric Oncology. Blood. 1998;92: 3591-3598. [PubMed] [Google Scholar]
- 6.Brugieres L, Quartier P, Le Deley MC, et al. Relapses of childhood large-cell lymphoma: treatment results in a series of 41 children—a report from the French Society of Pediatric Oncology. Ann Oncol. 2000;11: 53-58. [DOI] [PubMed] [Google Scholar]
- 7.Pellatt J, Sweetenham J, Pickering RM, Brown L, Wilkins B. A single-center study of treatment outcomes and survival in 120 patients with peripheral T-cell non-Hodgkin's lymphoma. Ann Hematol. 2002;81: 267-272. [DOI] [PubMed] [Google Scholar]
- 8.Fujimoto J, Shiota M, Iwahara T, et al. Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc Natl Acad Sci U S A. 1996;93: 4181-4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wellmann A, Doseeva V, Butcher W, et al. The activated anaplastic lymphoma kinase increases cellular proliferation and oncogene up-regulation in rat 1a fibroblasts. FASEB J. 1997;11: 965-972. [DOI] [PubMed] [Google Scholar]
- 10.Kuefer MU, Look AT, Pulford K, et al. Retrovirus-mediated transfer of NPM-ALK causes lymphoid malignancy in mice. Blood. 1997;90: 2901-2910. [PubMed] [Google Scholar]
- 11.Chiarle R, Gong JZ, Guasparri I, et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphoma and plasma cell tumors. Blood. 2003;101: 1919-1927. [DOI] [PubMed] [Google Scholar]
- 12.Bai RY, Dieter P, Peschel C, Morris SW, Duyster J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-γ to mediate its mitogenicity. Mol Cell Biol. 1998;18: 6951-6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J. Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood. 2000;96: 4319-4327. [PubMed] [Google Scholar]
- 14.Nieborowska-Skorska M, Slupianek A, Xue L, et al. Role of signal transducer and activator of transcription 5 in nucleophosmin/anaplastic lymphoma kinase-mediated malignant transformation of lymphoid cells. Cancer Res. 2001;61: 6517-6523. [PubMed] [Google Scholar]
- 15.Simonitsch I, Polgar D, Hajek M, et al. The cytoplasmic truncated receptor tyrosine kinase homodimer immortalizes and cooperates with ras in cellular transformation. FASEB J. 2001;15: 1416-1418. [DOI] [PubMed] [Google Scholar]
- 16.Zamo A, Chiarle R, Piva R, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21: 1038-1047. [DOI] [PubMed] [Google Scholar]
- 17.Cussac D, Greenland C, Roche S, et al. Nucleophosmin-anaplastic lymphoma kinase of anaplastic large-cell lymphoma recruits, activates, and uses pp60c-src to meditate its mitogenicity. Blood. 2004;103: 1464-1471. [DOI] [PubMed] [Google Scholar]
- 18.Gu TL, Tothova Z, Scheijen B, Griffin JD, Gilliland DG, Sternberg DW. NPM-ALK fusion kinase of anaplastic large-cell lymphoma regulates survival and proliferative signaling through modulation of FOXO3a. Blood. 2004;103: 4622-4629. [DOI] [PubMed] [Google Scholar]
- 19.Kawamura M, McVicar DW, Johnston JA, et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc Natl Acad Sci U S A. 1994;91: 6374-6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujitani Y, Hibi M, Fukada T, et al. An alternative pathway for STAT activation that is mediated by direct interaction between JAK and STAT. Oncogene. 1997;14: 751-761. [DOI] [PubMed] [Google Scholar]
- 21.Amin HM, Medeiros LJ, Ma Y, et al. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene. 2003;22: 5399-5407. [DOI] [PubMed] [Google Scholar]
- 22.Crockett DK, Lin Z, Elenitoba-Johnson KS, Lim MS. Identification of NPM-ALK interacting proteins by tandem mass spectrometry. Oncogene. 2004;23: 2617-2629. [DOI] [PubMed] [Google Scholar]
- 23.Uyttenhove C, Druez C, Renauld J-C, Herin M, Hoel H, van Snick J. Autonomous growth and tumorigenicity induced by P40/interleukin 9 cDNA transfection of a mouse P40-dependent T cell line. J Exp Med. 1991;173: 519-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houssiau FA, Renauld J-C, Stevens M, et al. Human T cell lines and clones respond to IL-9. J Immunol. 1993;150: 2634-2640. [PubMed] [Google Scholar]
- 25.Renauld J-C, Vink A, Louahed J, van Snick J. Interleukin-9 is a major anti-apoptotic factor for thymic lymphomas. Blood. 1995;85: 1300-1305. [PubMed] [Google Scholar]
- 26.Lange K, Uckert W, Blankenstein T, et al. Overexpression of NPM-ALK induces different types of malignant lymphoma in IL-9 transgenic mice. Oncogene. 2003;22: 517-527. [DOI] [PubMed] [Google Scholar]
- 27.De Smedt M, Verhasselt B, Kerre T, et al. Signals from the IL-9 receptor are critical for the early stages of human intrathymic T cell development. J Immunol. 2000;164: 1761-1767. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Aozasa K, Oshimi K, Takada K. Epstein-Barr virus (EBV)-encoded RNA promotes growth of EBV-infected T cells through interleukin-9 induction. Cancer Res. 2004;64: 5332-5337. [DOI] [PubMed] [Google Scholar]
- 29.Lin Q, Lai R, Chirieac LR, et al. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am J Pathol. 2005;167: 969-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miething C, Grundler R, Fend F, et al. The oncogenic fusion protein nucleophosmin-anaplastic lymphoma kinase (NPM-ALK) induces two distinct malignant phenotypes in a murine retroviral transplantation model. Oncogene. 2003;22: 4642-4647. [DOI] [PubMed] [Google Scholar]
- 31.Turner SD, Tooze R, Maclennan K, Alexander DR. Vav-promoter regulated oncogenic fusion protein NPM-ALK in transgenic mice causes B-cell lymphomas with hyperactive Jun kinase. Oncogene. 2003;22: 7750-7761. [DOI] [PubMed] [Google Scholar]
- 32.Turner SD, Alexander DR. What have we learned from mouse models of NPM-ALK-induced lymphomagenesis? Leukemia. 2005;19: 1128-1134. [DOI] [PubMed] [Google Scholar]
- 33.Pulford K, Lamant L, Morris SW, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood. 1997;89: 1394-1404. [PubMed] [Google Scholar]
- 34.Trumper L, Pfreundschuh M, Bonin FV, Daus H. Detection of the t(2;5)-associated NPM/ALK fusion cDNA in peripheral blood cells of healthy individuals. Br J Haematol. 1998;103: 1138-1144. [DOI] [PubMed] [Google Scholar]
- 35.Maes B, Vanhentenrijk BV, Wlodarska I, et al. The NPM-ALK and the ATIC-ALK fusion genes can be detected in non-neoplastic cells. Am J Pathol. 2001;158: 2185-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knoops L, Renauld J-C. IL-9 and its receptor: from signal transduction to tumorigenesis. Growth Factors. 2004;22: 207-215. [DOI] [PubMed] [Google Scholar]
- 37.Schmitt E, van Brandwijk R, van Snick J, Siebold B, Rude E. TCGF III/P40 is produced by naïve murine CD4+ T cells but is not a general T cell growth factor. Eur J Immunol. 1989;19: 2167-2170. [DOI] [PubMed] [Google Scholar]
- 38.Renauld J-C, van der Lugt N, Vink A, et al. Thymic lymphomas in interleukin 9 transgenic mice. Oncogene. 1994;9: 1327-1332. [PubMed] [Google Scholar]
- 39.Johnston JA, Kawamura M, Kirken RA, et al. Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2. Nature. 1994;370: 151-153. [DOI] [PubMed] [Google Scholar]
- 40.Witthuhn BA, Silvennoinen O, Miura O, et al. Involvement of the Jak-3 Janus kinase in signaling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature. 1994;370: 153-157. [DOI] [PubMed] [Google Scholar]
- 41.Yin T, Yang L, Yang YC. Tyrosine phosphorylation and activation of JAK family tyrosine kinases by interleukin-9 in MO7E cells. Blood. 1995;85: 3101-3106. [PubMed] [Google Scholar]
- 42.Thomis DC, Berg LJ. Peripheral expression of Jak3 is required to maintain T lymphocyte function. J Exp Med. 1997;185: 197-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomis DC, Lee W, Berg LJ. T cells from Jak3-deficient mice have intact TCR signaling, but increased apoptosis. J Immunol. 1997;159: 4708-4719. [PubMed] [Google Scholar]
- 44.Sohn SJ, Forbush KA, Nguyen N, et al. Requirement for Jak3 in mature T cells: its role in regulation of T cell homeostasis. J Immunol. 1998;160: 2130-2138. [PubMed] [Google Scholar]
- 45.Eynon EE, Livak F, Kuida K, Schatz DG, Flavell RA. Distinct effects of Jak3 signaling on αβ and γδ thymocyte development. J Immunol. 1999;162: 1448-1459. [PubMed] [Google Scholar]
- 46.Wen R, Wang D, McKay C, et al. Jak3 selectively regulates Bax and Bcl-2 expression to promote T-cell development. Mol Cell Biol. 2001;21: 678-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lai R, Rassidakis GZ, Lin Q, Atwell C, Medeiros LJ, Amin HM. Jak3 activation is significantly associated with ALK expression in anaplastic large cell lymphoma. Hum Pathol. 2005;36: 939-944. [DOI] [PubMed] [Google Scholar]
- 48.Merz H, Houssiau FA, Orscheschek K, et al. Interleukin-9 expression in human malignant lymphomas: unique association with Hodgkin's disease and large cell anaplastic lymphoma. Blood. 1991;78: 1311-1317. [PubMed] [Google Scholar]
- 49.Gaiser T, Thorns C, Merz H, Noack F, Feller AC, Lange K. Gene profiling in anaplastic large-cell lymphoma-derived cell lines with cDNA expression arrays. J Hematother Stem Cell Res. 2002;11: 423-428. [DOI] [PubMed] [Google Scholar]
- 50.Hubinger G, Muller E, Scheffrahn I, et al. CD30-mediated cell cycle arrest associated with induced expression of p21CIP1/WAF1 in the anaplastic large cell lymphoma cell line Karpas 299. Oncogene. 2001;20: 590-598. [DOI] [PubMed] [Google Scholar]
- 51.Mochizuki T, Kitanaka C, Noguchi K, Muramatsu T, Asai A, Kuchino Y. Physical and functional interactions between Pim-1 kinase and Cdc25A phosphatase: implications for the Pim-1-mediated activation of the c-Myc signaling pathway. J Biol Chem. 1999;274: 18659-18666. [DOI] [PubMed] [Google Scholar]
- 52.Bhattacharya N, Wang Z, Davitt C, McKenzie IFC, Xing P-X, Magnuson NS. Pim-1 associates with protein complexes necessary for mitosis. Chromosoma. 2002;111: 80-95. [DOI] [PubMed] [Google Scholar]
- 53.Roh M, Gary B, Song C, et al. Overexpression of the oncogenic kinase Pim-1 leads to genomic instability. Cancer Res. 2003;63: 8079-8084. [PubMed] [Google Scholar]
- 54.Bachmann M, Hennemann H, Xing PX, Hoffmann I, Moroy T. The oncogenic serine/threonine kinase Pim-1 phosphorylates and inhibits the activity of Cdc25C-assoicated kinase 1 (C-TAK1): a novel role for Pim-1 at the G2/M cell cycle checkpoint. J Biol Chem. 2004;279: 48319-48328. [DOI] [PubMed] [Google Scholar]
- 55.Breuer M, Slebos R, Verbeek S, van Lohuizen M, Wientjens E, Berns A. Very high frequency of lymphoma induction by a chemical carcinogen in pim-1 transgenic mice. Nature. 1998;340: 61-63. [DOI] [PubMed] [Google Scholar]
- 56.Yoshida S, Kaneita Y, Aoki Y, Seto M, Mori S, Moriyama M. Identification of heterologous translocation partner genes fused to the BCL6 gene in diffuse large B-cell lymphomas: 5′-RACE and LA-PCR analyses of biopsy samples. Oncogene. 1999;18: 7994-7999. [DOI] [PubMed] [Google Scholar]
- 57.Nieborowska-Skorska M, Hoser G, Kossev P, Wasik MA, Skorski T. Complementary functions of the antiapoptotic protein A1 and serine/threonine kinase pim-1 in the BCR/ABL-mediated leukemogenesis. Blood. 2002;99: 4531-4539. [DOI] [PubMed] [Google Scholar]
- 58.Chen WW, Chan DC, Donald C, Lilly MB, Kraft AS. Pim family kinases enhance tumor growth of prostate cancer cells. Mol Cancer Res. 2005;3: 443-451. [DOI] [PubMed] [Google Scholar]
- 59.Wang Z, Bhattacharya N, Mixter PF, Wei W, Sedivy J, Magnuson NS. Phosphorylation of the cell cycle inhibitor p21Cip1/WAF1 by Pim-1 kinase. Biochim Biophys Acta. 2002;1593: 45-55. [DOI] [PubMed] [Google Scholar]
- 60.Demoulin JB, van Roost E, Stevens M, Groner B, Renauld JC. Distinct roles for STAT1, STAT3, and STAT5 in differentiation gene induction and apoptosis inhibition by interleukin-9. J Biol Chem. 1999;274: 25855-25861. [DOI] [PubMed] [Google Scholar]
- 61.Demoulin JB, Uyttenhove C, Lejeune D, Mui A, Groner B, Renauld JC. STAT5 activation is required for interleukin-9-dependent growth and transformation of lymphoid cells. Cancer Res. 2000;60: 3971-3977. [PubMed] [Google Scholar]
- 62.Rassidakis GZ, Feretzaki M, Atwell C, et al. Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood. 2005;105: 827-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamamoto D, Sonoda Y, Hasegawa M, Funakoshi-Tago M, Aizu-Yokota E, Kasahara T. FAK overexpression upregulates cyclin D3 and enhances cell proliferation via the PKC and PI3-kinase-Akt pathways. Cell Signal. 2003;15: 575-583. [DOI] [PubMed] [Google Scholar]