

Abstract
We have previously reported that an anti-human transferrin receptor IgG3-avidin fusion protein (anti-hTfR IgG3-Av) inhibits the proliferation of an erythroleukemia-cell line. We have now found that anti-hTfR IgG3-Av also inhibits the proliferation of additional human malignant B and plasma cells. Anti-hTfR IgG3-Av induces internalization and rapid degradation of the TfR. These events can be reproduced in cells treated with anti-hTfR IgG3 cross-linked with a secondary Ab, suggesting that they result from increased TfR cross-linking. Confocal microscopy of cells treated with anti-hTfR IgG3-Av shows that the TfR is directed to an intracellular compartment expressing the lysosomal marker LAMP-1. The degradation of TfR is partially blocked by cysteine protease inhibitors. Furthermore, cells treated with anti-hTfR IgG3-Av exhibit mitochondrial depolarization and activation of caspases 9, 8, and 3. The mitochondrial damage and cell death can be prevented by iron supplementation, but cannot be fully blocked by a pan-caspase inhibitor. These results suggest that anti-hTfR IgG3-Av induces lethal iron deprivation, but the resulting cell death does not solely depend on caspase activation. This report provides insights into the mechanism of cell death induced by anti-TfR Abs such as anti-hTfR IgG3-Av, a molecule that may be useful in the treatment of B-cell malignancies such as multiple myeloma.
Introduction
The primary function of transferrin (Tf) is to transport iron through the blood. After binding to the transferrin receptor (TfR) on the cell surface, Tf is internalized into an acidic compartment where the bound iron is released. The Tf-TfR complex then returns to the cell surface and the ligand dissociates from the receptor.1
Studies have shown that the TfR is expressed more abundantly in malignant tissues than their normal counterparts.2-7 This difference in expression level, in addition to its ability to internalize and its central roles in cell growth and division, makes the TfR an attractive target for cancer therapeutics. In fact, both anti-TfR antibodies (Abs) and Tf-toxin conjugates have shown efficacy against cancers in preclinical and clinical settings.8-12 We have previously demonstrated that anti-rat TfR IgG3-Av forms strong noncovalent interactions with different biotinylated molecules and delivers them into cancer cells through receptor-mediated endocytosis.13 This novel molecule can be used as a universal delivery system for a wide range of therapeutic agents without the need to make a different chemical conjugate or genetic fusion protein for every targeted therapeutic.
We also unexpectedly discovered that anti-rat TfR-IgG3-Av, but not an irrelevant IgG3-Av, inhibited the growth of a rat myeloma and a T-cell lymphoma-cell line. However, it did not inhibit the growth of either a carcinoma or a gliosarcoma-cell line.13 Treatment with anti-rat TfR-IgG3 containing the same variable regions did not inhibit growth. Furthermore, we found that anti-rat TfR-IgG3-Av exists as a noncovalent dimer with 4 antigen-binding sites, probably due to the interaction among the 4 avidins located on 2 separate fusion proteins, since avidin in solution forms a tetrameric structure.13 Thus, the inhibitory effect of the fusion protein may be due, at least in part, to its ability to cross-link cell-surface TfRs. Furthermore, we reported that a similar fusion protein specific for the human TfR (anti-hTfR IgG3-Av), but not a murine anti-TfR IgG1 (128.1) sharing the same variable regions, inhibited the growth of the erythroleukemia-cell line K562.13 However, the mechanism of growth inhibition by these 2 fusion proteins, as well as the therapeutic potential of anti-hTfR IgG3-Av, was not explored.
Now we report that anti-hTfR IgG3-Av inhibits the growth of malignant B- and plasma-cell lines and cells isolated from patients with multiple myeloma (MM), a malignancy that is generally regarded as incurable. Using 2 of the most sensitive cell lines, ARH-77 and IM-9, we show that anti-hTfR IgG3-Av induces rapid TfR degradation, iron deprivation, mitochondrial damage, and cell death. Among different cancers, hematopoietic tumors are particularly suitable for treatment using TfR-targeting therapeutics since they both express high levels of TfR14-17 and are known to be more sensitive to the inhibitory effect of anti-TfR Abs than other malignancies.18 Increased understanding of the mechanism of cell death induced by anti-hTfR IgG3-Av may make it possible to design improved therapeutics for the treatment of hematopoietic malignancies.
Materials and methods
Antibodies and antibody fusion proteins
Recombinant anti-hTfR IgG3, constructed by substituting the variable regions of anti-dansyl IgG319,20 with those of the murine IgG1 anti-human TfR monoclonal Ab (mAb) 128.1,21 was expressed in the murine myeloma-cell line NS0/1. Anti-hTfR IgG3-Av and anti-dansyl IgG3-Av have been previously described.13,22 Abs and Ab fusion proteins were purified and characterized as described previously.13 The murine anti-human IgG3 mAb HP6050 was a kind gift from Robert Hamilton (Johns Hopkins University, Baltimore, MD). Murine anti-human TfR mAb (H68.4) and goat anti-mouse IgG-horseradish peroxidase (HRP) conjugate were from Zymed (South San Francisco, CA). Rabbit polyclonal anti-human TfR1 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Murine anti-β-actin mAb was from Sigma (St Louis, MO); the biotinylated mouse anti-LAMP-1 mAb and goat anti-mouse IgG-FITC were from BD Pharmingen (Becton Dickinson, Franklin Lakes, NJ). The mouse IgG1/κ isotype control mAbs were from eBioscience (San Diego, CA).
Cell lines and primary cells
MM.1S, S6B45, OCI-My5, U266, 8226/S, and 8226/DOX40 (doxorubicin-resistant variant of 8226/S) are human MM-cell lines. ARH-77 and IM-9 are Epstein-Barr virus (EBV)-transformed lymphoblastoid-cell lines established from cells isolated from IgG plasma-cell leukemia and MM patients, respectively. When injected into SCID mice, ARH-77 behaves like human MM, with mice developing hypercalcemia, lytic bone lesions, and hind limb paralysis.23,24 U266, 8226, ARH-77, and IM-9 cell lines were purchased from ATCC (Rockville, MD). The MM.1S, S6B45, and OCI-My5 cell lines were kindly provided by Drs Kenneth Anderson and Darminder Chauhan (Harvard University), and the 8226/DOX40-cell line was provided by Alan Lichtenstein (UCLA). The human erythroleukemia-cell line K562 was kindly provided by J. Larrick (Palo Alto Institute of Molecular Medicine, Mountain View, CA). Cells were cultured at 37°C, 5% CO2 in RPMI 1640 medium (GIBCO BRL, Grand Island, NY), with 5% fetal bovine serum (FBS; HyClone, Logan, UT) in all the experiments unless stated otherwise. Serum complement was inactivated by incubation at 56°C for 30 minutes.
Bone marrow aspirates from a patient with MM and another with plasma-cell leukemia (PCL), a more aggressive variant of MM,25 were obtained with informed consent following the standards of the institutional review board of UCLA. Mononuclear cells were separated by Ficoll-Paque Plus density gradient centrifugation (Amersham Pharmacia Biosciences, Uppsala, Sweden). CD138+ cells were separated from bone marrow mononuclear cells by positive selection using EasySep Human CD138+ Selection magnetic nanoparticles following the manufacturer's instructions (StemCell Technologies, Vancouver, BC, Canada).
Proliferation assays using cell lines
Cells (5000/well of a 96-well plate) were treated with the indicated reagents for 72 hours at 37°C. The cells were then cultured with 4 μCi (0.148 MBq)/mL [3H]-thymidine (ICN Biomedicals, Irvine, CA) for an additional 24 hours. Cells were harvested and radioactivity was counted as previously described.13 In the assay with 9 different cell lines, cells were cultured in Dulbecco modified Eagle medium (GIBCO BRL) supplemented with 2.5% FBS. Similar results were obtained using cells cultured in RPMI medium with 5% FBS. In the cross-linking assay, anti-hTfR IgG3 was mixed with a 5-fold excess of anti-hIgG3 mAb for 2 hours at 4°C before being added to the cells. In the metal supplement assay, cells were incubated with or without ferric ammonium citrate or zinc sulfate (Sigma) during the entire study.
Proliferation assays using primary cells
IM-9, U266, or primary cells (20 000/well of a 96-well plate) were cultured in Iscoves modified Dulbecco medium (GIBCO BRL) supplemented with nonessential amino acids (Invitrogen, Carlsbad, CA) and 10% FBS and were treated with buffer or 100 nM anti-hTfR IgG3-Av for 48 hours at 37°C. The cells were then pulsed with 4 μCi (0.148 MBq)/mL [3H]-thymidine for an additional 48 hours. Cells were harvested and radioactivity was counted as previously described.13
Detection of cell-surface TfR
Cells (5 × 105) treated with buffer, Ab, or Ab fusion protein were harvested, washed, and incubated with 5 μg Tf-FITC (Molecular Probes, Eugene, OR) in medium containing 1% BSA for 1 hour on ice. Cells were then washed and analyzed with a BD-LSR Analytic Flow Cytometer (Becton Dickinson).
Immunoblotting
Cells (5 × 105) were incubated with the indicated reagents, washed, and resuspended in lysis buffer (0.125% NP-40, 0.875% Brij 97, 10 mM Tris, 150 mM NaCl, 2 mM EDTA, 1 mM PMSF, 2.5 μM aprotinin and leupeptin, pH 7.5). Lysate (20 μg) was reduced and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred onto PVDF membranes (Millipore, Bedford, MA) using a GeneMate semidry blotter (ISC BioExpress, Kaysville, UT). The membranes were blocked with 5% milk and incubated overnight with anti-human TfR (H68.4) in 5% milk, 0.2% Tween 20 in PBS, followed by washing and incubation with anti-mouse IgG-HRP. The membranes were then incubated with SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL) before being exposed to film.
Radiolabeling and immunoprecipitation of TfR fragments
Cells (2 × 106) labeled with [35S]-methionine for 18 hours were washed and cultured with anti-hTfR IgG3 or anti-hTfR IgG3-Av for an additional 6 hours. The culture supernatants were then immunoprecipitated using anti-human TfR polyclonal Ab and protein A-conjugated Sepharose beads (Sigma). Samples were reduced and resolved by SDS-PAGE. The gel was dried and exposed to film.
Apoptosis and mitochondrial membrane potential assays
Cells (75 000) were treated with the indicated reagents, washed, and stained with propidium iodide (PI) and annexin V Alexa Fluor 350 or 488 conjugates following procedures suggested by the manufacturer of the Vybrant Apoptosis kit (Molecular Probes). Up to 10 000 events were recorded for each flow cytometry measurement. To measure mitochondrial depolarization, 40 nM DiOC6(3) (Molecular Probes) was added to the culture for 30 minutes. Samples were analyzed by flow cytometry. DFO and Z-VAD-FMK were purchased from Calbiochem (La Jolla, CA).
Caspase activity assay
Cells (25 000 per well of a 96-well plate) were treated with buffer or anti-hTfR IgG3-Av, and caspase activity was measured at 12, 24, 36, 44, 48, and 52 hours using fluorogenic substrates specific for caspases 9 (Ac-LEHD-AMC), 8 (Ac-IETD-AMC), and 3 (Ac-DMQD-AMC) (Alexis Biochemicals, San Diego, CA). Ac-DMQD-AMC, instead of Ac-DEVD-AMC, was used because it has been shown to be more specific for caspase 3.26,27 At each time point, reaction buffer (1.5% NP-40, 0.3% CHAPS, 30% sucrose, 30 mM MgCl2, 150 mM KCl, 450 mM NaCl, 150 mM HEPES, 1.2 mM EDTA, 30 mM DTT, 3 mM PMSF, pH 7.4) containing 150 μM fluorogenic substrate was added to each well and incubated for 4 hours at 37°C. Fluorescence was measured by a Synergy HT Microplate Reader (Bio-Tek Instruments, Winooski, VT) at 360/460 nm. Substrate incubation for 4 hours yields greater differences between the control and experimental sample values when compared with other incubation times.
Confocal microscopy
Cells (2 × 106) treated with anti-hTfR IgG3-Av were fixed with 4% paraformaldehyde and permeabilized with 0.2% Tween 20 in PBS. After blocking for 2 hours on ice in confocal buffer (2% calf serum, 0.1% sodium azide in PBS) containing 10% human serum (Omega Scientific, Tarzana, CA), cells were incubated on ice for 2 hours with 1 μg anti-TfR (H68.4) or isotype control mAb, and for 1 hour with anti-mouse IgG-FITC (1:100). Cells were then fixed with 4% paraformaldehyde and incubated on ice for 2 hours with 0.1 μg biotinylated anti-LAMP-1 or isotype control, and for 1 hour with streptavidin-Alexa Fluor 568 (1:500; Molecular Probes). Cells were washed twice with confocal buffer after each incubation. Stained cells were resuspended in Prolong solution (Molecular Probes) and mounted onto slides. Images were taken using an MRC1024ES confocal system (Bio-Rad Laboratories, Hercules, CA) equipped with a Nikon E800 microscope (Nikon, Melville, NY), a Nikon 63×/1.4 numeric aperture oil objective lens, and LaserSharp 2000 acquisition software version 6 (Bio-Rad). Colocalization studies were performed using ImageJ software version 1.32 (National Institutes of Health, Bethesda, MD). The mean fluorescence intensity (MFI) of the red and green channels in each image field was determined and a threshold for positive staining of MFI + 2 SD set. A pixel was considered colocalized when the overlapping area of the 2 channels was at least 50% of the area of the pixel. The percent colocalization for each field was the number of colocalized pixels, divided by the total number of positive pixels. The significance of values compared to 0 minutes was determined using the Student t test.
Results
Anti-hTfR IgG3-Av inhibits the growth of a panel of malignant B- and plasma-cell lines, and primary cells from patients
We previously reported that anti-hTfR IgG3-Av is an antiproliferative/proapoptotic drug that strongly inhibits the human erythroleukemia-cell line K562.13 To explore the potential of anti-hTfR IgG3-Av as a therapeutic for plasma-cell malignancies, we measured the proliferation of a panel of human malignant B- and plasma-cell lines incubated with the fusion protein. K562 cells were included in the assay for comparison. Anti-hTfR IgG3-Av inhibits the growth of all the cell lines tested, but to different extents (Figure 1A). Anti-hTfR IgG3-Av also significantly inhibits the in vitro growth of primary cells isolated from 2 patients, one diagnosed with multiple myeloma and one with plasma-cell leukemia (Figure 1B). Further studies with a larger sample size are necessary to confirm the spectrum of sensitivities to treatment with anti-hTfR IgG3-Av in primary cells. The 2 most sensitive cell lines, ARH-77 and IM-9, were used in subsequent experiments to determine the mechanism of growth inhibition by anti-hTfR IgG3-Av.
Figure 1.
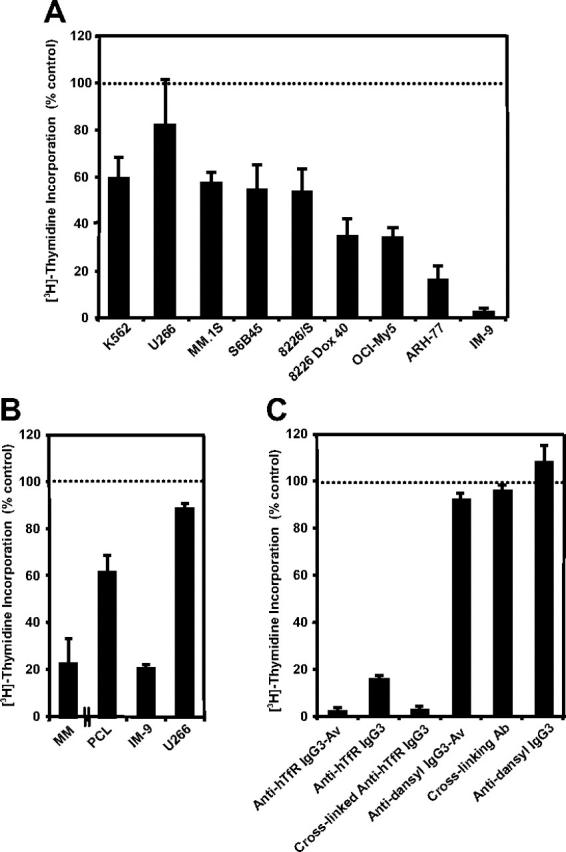
Anti-hTfR IgG3-Av inhibits the growth of malignant hematopoietic cells. (A) Proliferation assay of a panel of hematopoietic malignant cell lines incubated with 34 nM anti-hTfR IgG3-Av for 72 hours. Cells were incubated with 4 μCi (0.148 MBq)/mL [3H]-thymidine for an additional 24 hours prior to harvesting. Data represent the mean of quadruplicate samples of 2 independent determinations of [3H]-thymidine incorporation. (B) Proliferation assay of primary cells isolated from the bone marrow of a patient with MM and a patient with PCL. Myeloma cells from the patients were isolated in 2 independent experiments by positive selection. As a control, 2 cell lines, IM-9 (highly sensitive) and U266 (less sensitive), were tested in parallel. Control data from the experiment with the PCL patient are shown. All cells were treated in triplicate with 100 nM anti-hTfR IgG3-Av for 48 hours and then incubated with 4 μCi (0.148 MBq)/mL [3H]-thymidine for an additional 48 hours prior to harvesting and determination of radioactivity. (C) ARH-77 cells were incubated with 11 nM anti-hTfR IgG3-Av, anti-hTfR IgG3, anti-hTfR IgG3 cross-linked with a 5-fold excess of secondary Ab, secondary Ab alone, the anti-dansyl IgG3 isotype control, or the anti-dansyl IgG3-Av for 72 hours. Cells were incubated with 4 μCi (0.148 MBq)/mL [3H]-thymidine for an additional 24 hours prior to harvesting and determination of radioactivity. Data shown are representative of 3 independent experiments. All data are presented as the percentage [3H]-thymidine incorporation compared with control cells treated with buffer. Error bars indicate the standard deviation.
Anti-hTfR IgG3-Av or anti-hTfR IgG3 cross-linked with a secondary Ab inhibits the growth and induces apoptosis in ARH-77 cells
Previous studies suggest that anti-hTfR IgG3-Av inhibits cell growth by cross-linking the TfR.13 To test this hypothesis, we measured proliferation and apoptosis in ARH-77 cells treated with different anti-hTfR molecules. Anti-hTfR IgG3-Av completely inhibited [3H]-thymidine incorporation (Figure 1C) and resulted in a significant increase in the number of dead (annexin V bright, PI bright) and apoptotic (annexin V bright, PI dim) cells from 48 to 96 hours (Figure 2A). The total percentage of dead cells including apoptotic cells (annexin V bright, PI bright and annexin V bright, PI dim) resulting from anti-hTfR IgG3-Av treatment increased from 63% at 48 hours, to 79% at 72 hours, and to 86% at 96 hours. In another experiment, treatment with anti-hTfR IgG3-Av induced cell death in 85% of the cells compared with 47% in cells treated with a 3-fold higher concentration of anti-hTfR IgG3 (Figure 2B). However, the growth inhibitory and proapoptotic effects of anti-hTfR IgG3 could be enhanced to levels comparable with those of anti-hTfR IgG3-Av by cross-linking with a secondary mAb (Figures 1C and 2B). Similar results were obtained using IM-9 cells (data not shown). These results suggest that the tetravalent (4 antigen-binding sites) anti-hTfR IgG3-Av has a stronger cytotoxic effect than anti-hTfR IgG3 due, at least in part, to its increased cross-linking of cell-surface TfR.
Figure 2.
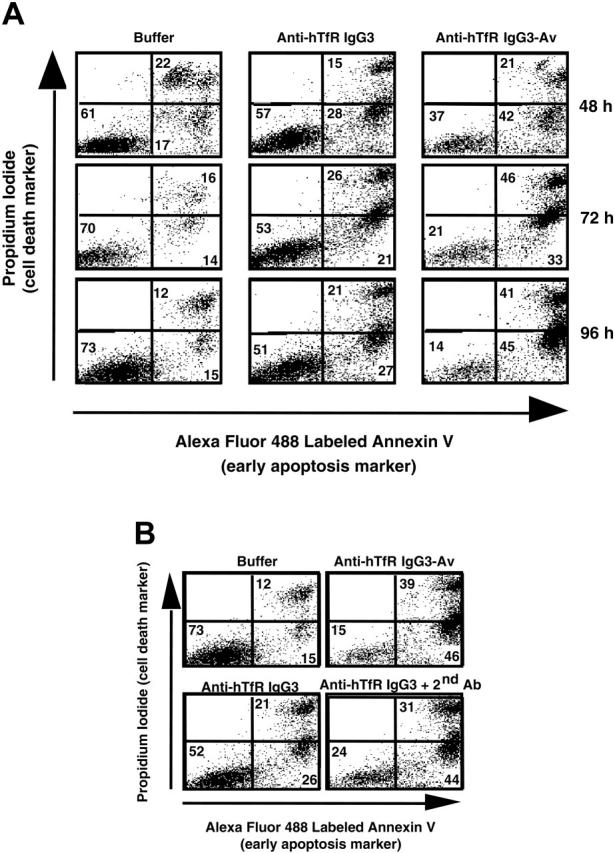
Anti-hTfR IgG3-Av induces apoptosis in ARH-77 cells. (A) Cells were incubated with 11 nM anti-hTfR IgG3 or anti-hTfR IgG3-Av for the indicated times. Cells were then washed, stained with annexin V-Alexa Fluor 488 and PI, and analyzed by flow cytometry. Data shown are representative of 3 independent experiments. (B) ARH-77 cells were treated with 1.2 nM anti-hTfR IgG3-Av, 3.7 nM anti-hTfR IgG3, or 3.7 nM anti-hTfR IgG3 cross-linked with a 5-fold excess of secondary Ab for 96 hours and analyzed by flow cytometry. The percentage of cells located in each quadrant is shown in the corner. Results are representative of 2 independent experiments.
Anti-hTfR IgG3-Av down-regulates cell-surface expression of TfR
The level of surface TfR on ARH-77 cells incubated with anti-hTfR molecules was measured by flow cytometry using a Tf-FITC conjugate. Based on our findings (data not shown) and published data on 128.1,21 anti-hTfR IgG3 and anti-hTfR IgG3-Av do not inhibit the binding of Tf to the TfR. Therefore, their presence does not affect the binding of Tf-FITC to the receptor. Both anti-TfR Abs caused an extensive and rapid decrease in surface TfR expression, which remained low throughout the experiment (Figure 3; also see Figure S1, available on the Blood website by clicking on the Supplemental Figure link at the top of the online article). Although the differences were small, anti-hTfR IgG3-Av consistently induced greater cell-surface TfR down-regulation than anti-hTfR IgG3. Treatment of IM-9 cells yielded similar results (data not shown). Comparable TfR down-regulation was also observed in cells treated with anti-hTfR IgG3 cross-linked with a secondary mAb (data not shown).
Figure 3.

Anti-hTfR IgG3-Av down-regulates TfR expression on the surface of ARH-77 cells. Cells treated with buffer (•), 11 nM anti-hTfR IgG3 (▪), or 11 nM anti-hTfR IgG3-Av (▴) were harvested at 4, 12, 24, 48, and 72 hours. Cells were then incubated with Tf-FITC, washed, and analyzed by flow cytometry. This experiment was repeated twice with similar results.
Anti-hTfR IgG3-Av or anti-hTfR IgG3 cross-linked with a secondary Ab induces intracellular degradation of the TfR
To determine the fate of down-regulated surface TfR, we analyzed the lysates of treated cells by immunoblotting. Intact TfR vanished between 4 to 12 hours after treatment with anti-hTfR IgG3-Av. In contrast, anti-hTfR IgG3 induced less extensive receptor loss (Figure 4A). However, TfR loss following binding of anti-hTfR IgG3 could be increased by cross-linking with a secondary mAb (Figure 4B). When cells were treated with anti-hTfR IgG3-Av for shorter times, over half of the intact receptors had disappeared by 20 minutes (Figure 4C).
Figure 4.

Anti-hTfR IgG3-Av induces the highest levels of intracellular degradation of the TfR. (A) ARH-77 and IM-9 cells were treated with buffer, 11 nM anti-hTfR IgG3, or 11 nM anti-hTfR IgG3-Av. ARH-77 was treated for 4, 12, and 24 hours, while IM-9 was treated for 4 hours. (B) ARH-77 cells were treated with buffer, 11 nM anti-hTfR IgG3, 11 nM anti-hTfR IgG3 cross-linked with secondary mAb in 1:1 or 1:5 ratios, or 11 nM anti-hTfR IgG3-Av for 8 hours. (C) ARH-77 cells were treated with buffer or 11 nM anti-hTfR IgG3-Av for 5, 10, and 20 minutes. (D) ARH-77 cells were treated with buffer or 11 nM anti-hTfR IgG3-Av for 4 hours. In all cases, cells were harvested, and whole-cell lysates were prepared and analyzed by immunoblotting with a murine mAb against the intracellular N-terminus of the human TfR followed by incubation with goat anti-mouse IgG-HRP conjugate. The blot was then stripped and reblotted with a murine anti-β-actin mAb. In panels A, B, and C, only the regions of the 95-kDa intact TfR and β-actin are shown. In panel D, the blot from 14 to 95 kDa is shown. These experiments were repeated at least twice with similar results.
The rapid disappearance of TfR could be a result of shedding of surface TfR. However, we were unable to detect any TfR fragments in the culture media of treated cells (data not shown). Alternatively, TfR could be degraded intracellularly. In fact, immunoblotting of lysates from cells treated with anti-hTfR IgG3-Av showed small fragments of TfR (approximately 16 and 20 kDa) concomitant with the disappearance of full-length TfR (95 kDa) (Figure 4D). Based on their sizes and the fact that they were recognized by a mAb against the cytoplasmic N-terminus of TfR (a type II transmembrane protein), these fragments appeared to be a result of proteolytic cleavages in the extracellular region of the TfR. The degradation may take place in the lumen of an intracellular compartment since anti-hTfR IgG3-Av is internalized through TfR-mediated endocytosis.13
Iron supplement blocks the antiproliferative and proapoptotic effects of anti-hTfR IgG3-Av
To determine if anti-hTfR IgG3-Av inhibits growth by disrupting TfR-dependent iron uptake, we incubated cells with the fusion protein in the presence of metal supplements. Iron, but not another divalent transition metal, zinc, completely blocked the growth inhibitory and proapoptotic effects of anti-hTfR IgG3-Av (Figure 5A-B). Similar results were obtained in cells treated with anti-hTfR IgG3 alone or cross-linked with a secondary Ab (data not shown). Iron supplement in untreated cells did not promote cell growth (data not shown). In conclusion, these data suggest that the anti-hTfR Abs induce apoptosis, at least in part, by causing iron deprivation.
Figure 5.

Iron supplement blocks the antiproliferative and proapoptotic effects of anti-hTfR IgG3-Av. (A) ARH-77 and IM-9 cells were treated for 72 hours with 11 nM anti-hTfR IgG3-Av alone or in the presence of 30 μM zinc sulfate (ZnSO4) or 25 μM ferric ammonium citrate (FAC). The cells were then cultured in the presence of [3H]-thymidine for an additional 24 hours and harvested, and the incorporation of [3H]-thymidine was determined. Each value is the mean of quadruplicate samples expressed as the percentage of the control mean (controls are cells treated with buffer alone, with or without metal salt supplement; data not shown). (B) ARH-77 cells were treated for 96 hours with buffer or 11 nM anti-hTfR IgG3-Av with or without 50 μM FAC. The cells were then washed, stained with annexin V-Alexa Fluor 488 and PI, and analyzed by flow cytometry. The percentage of cells located in each quadrant is shown at the corner.
Anti-hTfR IgG3-Av directs TfR to an intracellular compartment expressing LAMP-1, but degradation of the receptor is not sensitive to increase of pH in the lysosome
To determine if anti-hTfR IgG3-Av directs TfR to the lysosome for degradation, we analyzed treated cells by confocal microscopy. Increased colocalization of TfR and LAMP-1 was observed beginning 15 minutes after treatment with anti-hTfR IgG3-Av (Figure 6; Table 1). Despite the data indicating that TfR travels to the lysosome, the degradation of the receptor could not be blocked by coincubation with 20 mM ammonium chloride or 200 μM chloroquine (data not shown), chemicals that increase the pH of the lysosome. This suggests that the proteases responsible for the degradation of TfR are still functional at higher pHs.
Figure 6.

Anti-hTfR IgG3-Av directs the TfR to an intracellular compartment containing the lysosomal marker LAMP-1. ARH-77 cells treated with 11 nM anti-hTfR IgG3-Av for 0 (buffer control), 15, 30, or 45 minutes were fixed, permeabilized, and stained as described in “Materials and methods.” Cells were then analyzed by confocal microscopy. The image of a representative cell from each time point is shown immunostained for TfR (green) and LAMP-1 (red) with overlay. The area where colocalization occurs is shown in white. The percent colocalization was calculated as described in “Confocal microscopy” (Table 1).
Table 1.
Colocalization of TfR and LAMP-1 in ARH-77 cells incubated with 11 nM anti–hTfR IgG3-Av
|
Experiment 1
|
Experiment 2
|
Experiment 3
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Incubation time, min | 0 | 15 | 30 | 90 | 0 | 30 | 60 | 0 | 15 | 30 | 45 |
| Average % colocalization | 10.2 | 20.0 | 16.3 | 18.2 | 13.6 | 20.8 | 18.7 | 11.9 | 21.7 | 19.2 | 39.8 |
| SD | 1.5 | 4.0 | 2.6 | 3.4 | 3.8 | 3.0 | 3.9 | 3.5 | 8.1 | 1.4 | 9.8 |
| SE | 0.5 | 1.4 | 0.9 | 1.4 | 1.4 | 1.1 | 1.5 | 1.0 | 2.5 | 0.5 | 3.1 |
| No. of fields | 11 | 8 | 8 | 6 | 7 | 8 | 7 | 12 | 10 | 9 | 10 |
| Average no. of cells/field | 17 | 14 | 16 | 18 | 19 | 21 | 24 | 19 | 11 | 17 | 7 |
| P | — | < .001 | < .001 | < .002 | — | < .002 | < .03 | — | < .005 | < .001 | < .001 |
— indicates not applicable
A papainlike cysteine protease is involved in the degradation of TfR induced by anti-hTfR IgG3-Av
To identify the proteases that degrade the TfR, we used immunoblotting to detect TfR and its cleavage products in the lysates of ARH-77 cells treated with anti-hTfR IgG3-Av and a variety of proteases inhibitors (Figure 7). None of the agents completely inhibited the destruction of the 95-kDa TfR, indicating that the process involves more than one protease family. PMSF, pepstatin A, and bestatin, specific inhibitors of serine, aspartic acid proteases, and aminopeptidases, respectively,28,29 did not inhibit the formation of the 16- and 20-kDa fragments containing the N-terminus. In contrast, antipain and leupeptin, inhibitors of both cysteine and serine proteases, and E64d, which inhibits most cysteine proteases and trypsin,28-30 blocked the cleavage that generated the 16- and 20-kDa fragments. However, they were not able to block cleavage at a more C-terminal site that resulted in an approximately 23-kDa fragment (Figure 7). The fact that only papainlike cysteine proteases are inhibited by antipain, leupeptin, and E64d, but not by PMSF, suggests that one or more of these enzymes are involved in the generation of the 16- and 20-kDa fragments. Inhibition of metalloproteinases using metal chelators such as o-phenanthroline and GM6001 also did not interfere with the production of the 16- and 20-kDa fragments (data not shown). In conclusion, these results indicate that upon treatment with anti-hTfR IgG3-Av, the extracellular portion of the internalized TfR is cleaved at multiple sites by different proteases including a papainlike cysteine protease.
Figure 7.

A papainlike cysteine protease is involved in the degradation of TfR induced by anti-hTfR IgG3-Av. ARH-77 cells preincubated with buffer, 250 μM E64d, 250 μM antipain, 250 μM leupeptin, 250 μM bestatin, 500 μM pepstatin A, or 500 μM PMSF for 1 hour were treated with buffer or 50 nM anti-hTfR IgG3-Av for an additional 4 hours before being harvested. Cell lysates were analyzed by immunoblotting with a murine mAb against the intracellular N-terminus of the human TfR followed by incubation with goat anti-mouse IgG-HRP. The blot was then stripped and reblotted with a murine anti-β-actin mAb. The regions of the 95-kDa intact TfR, 16- to 23-kDa TfR fragments, and β-actin are shown. Except for the PMSF data, these experiments were repeated at least once with similar results.
Anti-hTfR IgG3-Av induces the activation of caspases 9, 8, and 3
To determine if treatment with anti-hTfR IgG3-Av induces caspase activation, we measured the activity of caspases 9, 8, and 3 in ARH-77 cells treated with the fusion protein. Caspases 9 and 8 are key components of the mitochondrial and death receptor-mediated apoptotic pathway, respectively, while caspase 3 is the executioner caspase of both pathways.31 Caspase activity determined by using a specific fluorogenic substrate has been shown to correlate with immunoblots detecting the active form of the protease.32 Activation of all 3 caspases was seen only between 44 hours and 52 hours, with the highest increase observed 48 hours after treatment with anti-hTfR IgG3-Av (Figure 8A). We did not find a significant difference in the timing of activation among the caspases.
Figure 8.

Anti-hTfR IgG3-Av induces caspase activation, mitochondrial depolarization, and a partially caspase-independent cell death in ARH-77 cells. (A) Cells treated with buffer or 50 nM anti-hTfR IgG3-Av for 48 hours were lysed and caspase activities measured by specific fluorogenic substrates. Each value is the mean of quadruplicate samples. (B) Cells preincubated with or without 100 μM Z-VAD-FMK were treated with buffer, 50 μM DFO, or 50 nM anti-hTfR IgG3-Av for 48 or 96 hours. Cells were then washed and stained with DiOC6(3), annexin V-Alexa Fluor 350, and PI, and analyzed by flow cytometry. The annexin V/PI profiles of treated cells at 48 and 96 hours are shown with the percentages of cells located in each quadrant. (C) Nonapoptotic cells (lower left quadrants of panel B) at 48 hours were gated and their mitochondrial membrane potentials shown in log fluorescence intensity. These experiments were repeated in both ARH-77 and IM-9 cells with similar results.
Cell death induced by DFO or anti-hTfR IgG3-Av is partially blocked by a broad-spectrum caspase inhibitor
Although we had evidence of caspase activation in cells treated with anti-hTfR IgG3-Av, it was unclear whether cell death was caspase dependent. To answer this question, we coincubated cells with anti-hTfR IgG3-Av and the broad-spectrum caspase inhibitor Z-VAD-FMK. We also treated cells with DFO, a membrane-permeable iron chelator, as a positive control for iron deprivation. The levels of apoptosis and viability of cells were determined.
In anti-hTfR IgG3-Av-treated cells, inhibition of caspase activity by treatment with Z-VAD-fmk only partially blocked cell death (Figure 8B). The number of dead cells (annexin V bright, PI bright and annexin V bright, PI dim) decreased from 48% to 29% in cells treated with anti-hTfR IgG3-Av alone or pretreated with Z-VAD-fmk, respectively, at 48 hours. Similar results were observed after 96 hours of treatment. These results suggest that anti-hTfR IgG3-Av induced cell death, in part, through a caspase-independent mechanism. However, it is important to note that in several independent experiments we have observed that Z-VAD-FMK was less efficient in blocking apoptosis induced by anti-hTfR IgG3-Av than by DFO, suggesting that the 2 reagents may induce apoptosis through slightly different pathways.
DFO and anti-hTfR IgG3-Av induce mitochondrial depolarization
To determine if the PI-negative, annexin V-negative cells that survived the treatment of DFO or anti-hTfR IgG3-Av in the presence of Z-VAD-FMK were truly healthy, we stained cells treated for 48 hours (Figure 7B) with a third fluorescent reagent, DiOC6(3), that measures mitochondrial potential. A cell with normal mitochondrial potential accumulates DiOC6(3) in the organelle.33 All apoptotic cells lost mitochondrial potential regardless of the treatment or the presence of Z-VAD-FMK (data not shown). In contrast, nonapoptotic (annexin V, PI double negative) cells present at 48 hours varied in their DiOC6(3) staining profiles (Figure 8C). When cells were treated with DFO or anti-hTfR IgG3-Av without Z-VAD-FMK, a majority of the nonapoptotic cells had mitochondrial potentials comparable with those of control cells (Figure 8C, left column). In contrast, when Z-VAD-FMK was added, there was an increase in the number of nonapoptotic cells (Figure 8B, 48 hours), but a large percentage of these cells had sustained significant mitochondrial depolarization (Figure 8C, right column). These results suggest that in the absence of Z-VAD-FMK, cells that were sensitive to DFO or anti-hTfR IgG3-Av underwent apoptosis, leaving mostly bona fide healthy cells in the nonapoptotic population. In contrast, in the presence of Z-VAD-FMK, many cells suffering from mitochondrial damage did not undergo apoptosis by 48 hours. However, with impaired mitochondrial potentials, many of these cells eventually died34 (Figure 8B, 96 hours).
Discussion
We have studied the growth-inhibitory effect of anti-hTfR IgG3-Av using a panel of hematopoietic malignant cell lines and primary cells isolated from MM and PCL patients. We observed a wide range of sensitivities in the cell lines studied, while primary cells from the 2 patients displayed intermediate sensitivity. However, it is important to note that additional studies using a larger cohort of MM patients are required.
Using the 2 most sensitive cell lines, ARH-77 and IM-9, we found that the tetravalent anti-hTfR IgG3-Av induces significantly more apoptosis than does the bivalent anti-hTfR IgG3. Cytotoxicity is associated with cell-surface TfR down-regulation, rapid TfR degradation, and iron deprivation. The fact that these events can be reproduced in cells treated with anti-hTfR IgG3 cross-linked with a secondary Ab suggests that, first, the potent cytotoxicity of anti-hTfR IgG3-Av stems from its ability to increase the cross-linking of cell-surface TfR; and second, increased cross-linking of TfR results in more efficient degradation of the receptor and more severe cytotoxicity. These observations provide new insights into the mechanism of growth inhibition by anti-TfR Abs.
The growth inhibitory property of anti-TfR Abs has been appreciated since the 1980s.35-39 A rat anti-murine TfR IgM (RI7 208) that extensively cross-linked the TfR was reported to block the internalization of the TfR-Tf complex, resulting in iron deprivation and growth inhibition.40,41 In addition, a murine anti-human TfR IgA (42/6) showed potent growth inhibitory activity that could not be attributed completely to its ability to block Tf binding to the receptor.21,42-47 A rat anti-murine TfR IgG (RI7 217) and 2 murine anti-human TfR IgGs (OKT9 and B3/25) did not cause significant growth inhibition unless used in combination or cross-linked with secondary Abs.21,40,41,47,48 Based on these data, it was suggested that polymeric molecules such as anti-TfR IgA and IgM, and anti-TfR IgGs cross-linked with secondary Abs, inhibit growth by blocking internalization of the TfR-Tf complex.18,21,41,47 Whether TfR on cells treated with anti-TfR IgA or cross-linked anti-TfR IgGs is degraded intracellularly was not determined.
In contrast to the model described in the previous paragraph, our data indicate for the first time that some polymeric molecules (anti-hTfR IgG3-Av, cross-linked anti-hTfR IgG3) induce significant levels of apoptosis not by blocking TfR internalization but instead by down-regulating cell-surface TfR and causing its intracellular degradation. Consistent with this hypothesis, previous studies of the anti-human TfR IgA 42/6, a tetravalent Ab, had shown that treated cells readily internalized 42/6 and down-regulated surface TfR.44,47 However, neither the fate of the internalized TfR nor the contribution of surface TfR down-regulation to the overall inhibitory effect of 42/6, which blocks Tf binding to TfR, was known. In addition, the growth of the human leukemic T-cell line CCRF-CEM was strongly inhibited by treatment using pairs of 2 anti-TfR IgG mAbs targeting different epitopes on the TfR; this likely increases the level of TfR cross-linking. It was found that these pairs of anti-TfR IgGs down-regulated cell-surface TfR.21 Overall, these studies suggest that polymeric molecules such as anti-hTfR IgG3-Av, anti-TfR IgA, and anti-TfR IgG cross-linked with a secondary Ab inhibit growth by a mechanism different from what had been reported for the anti-TfR IgM.
Although anti-hTfR IgG3, anti-hTfR IgG3 cross-linked with a secondary Ab, and anti-hTfR IgG3-Av all down-regulate cell-surface TfR, the latter 2 are far more efficient in directing the receptor for degradation. This may explain the difference in the proapoptotic activities of the different treatments. Therefore, iron uptake may not be completely blocked in cells treated with anti-hTfR IgG3. This hypothesis is supported by the finding that TfR bound by the anti-TfR IgG OKT9 continued to cycle and mediate iron uptake.48 In addition, cells that were not sensitive to the growth inhibitory activity of anti-TfR IgG B3/25 continued to take up a small amount of Tf despite down-regulation of the TfR. In contrast, in the same experiment cells inhibited by the IgA 42/6 did not internalize any Tf.47 Therefore, these studies suggest that polymeric anti-TfR Abs have higher growth inhibitory activities than bivalent anti-TfR Abs due to their ability to induce more efficient degradation of the TfR, resulting in more severe iron deprivation.
Collectively, the results discussed so far indicate that all anti-TfR Abs inhibit cell growth through iron deprivation but the underlying mechanism may differ and depend on the extent of TfR cross-linking. Increase in valence from an anti-TfR IgG (v = 2) to anti-hTfR IgG3-Av and anti-hTfR IgA 42/6 (v = 4) may increase cell-surface TfR down-regulation with an accompanying enhancement of TfR degradation, and more severe iron deprivation. On the other hand, rat anti-murine TfR IgMs (v = 10, 12) extensively cross-linked cell-surface TfR, thereby blocking TfR-Tf complex internalization resulting in iron deprivation but through a different mechanism. It is important to mention that the studies done to date have used different anti-TfR Abs binding to different epitopes. To confirm the contribution of valence to TfR down-regulation and degradation, it would be important to compare IgG, IgA, and IgM containing the same variable regions. For anti-hTfR IgG3-Av, it is also possible that avidin, a positively charged molecule with heparin-binding ability,49 the extended hinge region of human IgG3,50 and/or the binding of the Fc fragment of IgG3 to Fc receptors may contribute to TfR degradation.
Studies conducted in our laboratory and elsewhere indicate that the ability of anti-TfR IgGs to inhibit growth depends on the cell type. Anti-human TfR IgGs B3/25 and 43/31 inhibited the growth of normal granulocyte/macrophage progenitor cells45 but not mitogen-stimulated mononuclear47 and CCRF-CEM cells.21 The murine IgG1 128.1, from which we cloned the variable regions to construct anti-hTfR IgG3 and anti-hTfR IgG3-Av, did not inhibit the growth of CCRF-CEM21 or K562 cells.13 However, 128.1 inhibited the growth of ARH-77 cells (data not shown). Consistent with these observations, we show that there are also significant differences among malignant plasma-cell lines and primary cells in sensitivity to anti-hTfR IgG3-Av. Further studies are needed to determine if the proposed mechanism of growth inhibition operates in cell lines other than ARH-77 and IM-9, and to identify additional factors that contribute to the overall cellular sensitivity to anti-hTfR IgG3-Av. A possibility is that TfR cross-linking initiates a proapoptotic signal independent of iron deprivation. It has been reported that treatment with an anti-TfR Ab results in tyrosine phosphorylation of the ζ-chain of T-cell receptors (TCRs), which participate in intracellular signaling.51 Furthermore, stimulation of either receptor induces tyrosine phosphorylation on the other. Receptors in B cells may also interact with the TfR. However, the fact that the inhibitory effect of anti-hTfR IgG3-Av can be reversed by iron supplement suggests that this signaling event, if it exists, does not play a major role in determining the sensitivity of ARH-77 and IM-9 to anti-hTfR IgG3-Av.
Earlier studies showed that the degradation of an internalized anti-TfR IgG could be inhibited by leupeptin and chloroquine, and suggested that the TfR-Ab complex was degraded in the lysosome.48 Electron microscopy by Hopkins and Trowbridge indicated that the internalized TfR-Ab complex trafficked to a compartment that is morphologically similar to the lysosome.52 In this report, we provide direct evidence that TfR travels to the lysosome after treatment with anti-hTfR IgG3-Av based on its colocalization with LAMP-1. However, an unexpected result is that we could not inhibit TfR degradation by adding chloroquine or ammonium chloride to the cell culture. This result suggests that the proteases responsible for the degradation of TfR are functional at higher pHs. Thus, even though the TfR trafficks to the lysosome, it may be degraded elsewhere such as the proteasome through the ubiquitin pathway similar to the degradation reported for HER/neu targeted by the recombinant humanized Ab trastuzumab.53 However, preliminary studies using antiubiquitin in Western blots and proteosomal inhibitors such as MG-132 (10 μM), lactacystin (20 μM), and PS-341 (500 nM) found no evidence of TfR degradation through this pathway (P.P.N. and M.L.P., unpublished results, June 2004).
Several studies have shown that iron deprivation leads to mitochondrial damage and subsequent activation of caspases of the mitochondrial apoptosis pathway.32,54-59 Leukemic T cells have been shown to undergo mitochondrial depolarization after incubation with a murine Ab that competes with Tf for binding to TfR,60 and the human promyelocytic leukemic cell line HL-60 showed increased release of cytochrome c from the mitochondria, activation of caspases, and apoptosis after treatment with DFO.58,61 ARH-77 cells treated with either anti-hTfR IgG3-Av or DFO exhibit similar mitochondrial depolarization and apoptosis, with iron supplement completely blocking the mitochondrial depolarization and cell death induced by anti-hTfR IgG3-Av (data not shown). Anti-hTfR IgG3-Av induced activation of caspases 9, 8, and 3 in ARH-77 cells, but we could not find a significant difference in the timing of activation among the different caspases. Of interest, we found that inhibition of caspase activity could not prevent mitochondrial depolarization and only partially blocked cell death induced by DFO and anti-hTfR IgG3-Av. It has been reported that damaged mitochondria release a variety of molecules, such as apoptosis-inducing factor (AIF), that induce cell death independent of caspases.62 Further studies are needed to determine if anti-hTfR IgG3-Av induces cell death through this mechanism.
The anti-hTfR IgG3-Av fusion protein, alone or in combination with biotinylated or nonbiotinylated therapeutics, can potentially be used both in vivo and ex vivo in the efficient purging of hematopoietic malignant cells during the expansion of progenitors for use in autologous transplantation. A concern regarding this antibody fusion protein is its potential cytotoxicity to normal hematopoietic stem cells (HSCs). However, malignant cells including myeloma cells express much higher levels of TfR than normal tissues such as HSCs.16,63,64 It is important to note that the TfR is expressed at very low levels in early, noncommitted HSCs,65-69 and there is the general agreement that the noncommitted human adult bone marrow HSC has a CD34+/CD71lo phenotype.66-68 In addition, there are subsets of bone marrow and peripheral blood HSCs that are negative for TfR expression (CD34+/CD71- phenotype).65-68 Therefore, since the level of TfR is minimal in these HSCs, the anti-hTfR IgG3-Av fusion protein is not expected to have a significant effect on these cells. The HSCs should be able to self-renew and further differentiate to give rise to the various subpopulations. In fact, it has even been shown that an anti-murine TfR antibody conjugated to the potent plant toxin ricin is not cytotoxic to early mouse progenitors and HSCs in vitro.17 This issue needs to be explored in future studies to be certain that the anti-hTfR IgG3-Av fusion protein does not affect the viability of pluripotent HSCs.
In this report, we have described several key aspects of the mechanism of cell death induced by anti-hTfR IgG3-Av in ARH-77 cells, a widely used model for studying MM.23,24,70,71 This information sheds light on the mechanism of inhibition by anti-TfR Abs in general, and will be useful for designing therapeutics for MM and other hematopoietic malignancies based on anti-hTfR IgG3-Av. These mechanistic studies were restricted to the 2 most sensitive cell lines and further studies are needed to confirm the mechanism of cell death in the other cell lines and primary cells from patients. Further studies are also needed to explain the differences in sensitivity among the panel of hematopoietic malignant cell lines, since we have seen that the sensitivity is not correlated to the levels of TfR expression (P.P.N., G.H., and M.L.P., unpublished data, June 2005). In addition to its proapoptotic activity, anti-hTfR IgG3-Av is the first anti-TfR to contain the human IgG3 constant regions, which are known to bind to Fc receptors and fix complement. These properties have been shown to contribute to the antitumor activity of Abs in patients,72 and may enhance the in vivo effectiveness of anti-hTfR IgG3-Av. The tumoricidal activity of anti-hTfR IgG3-Av can be further increased by combining it with a biotinylated toxin as suggested by preliminary data using malignant hematopoietic cell lines (P.P.N., G.H., and M.L.P., unpublished results, July 2005). Further development of this multifunctional fusion protein may lead to effective therapeutics for hematopoietic malignancies such as multiple myeloma.
Supplementary Material
Acknowledgments
We thank Dr Jay Dela Cruz (UCLA) for his assistance with confocal microscopy. The MM.1S, S6B45, and OCI-My5 cell lines were kindly provided by Drs Kenneth Anderson and Darminder Chauhan (Harvard University), and Alan Lichtenstein (UCLA) generously provided the 8226/DOX40 cell line. The human erythroleukemia-cell line K562 was kindly provided by J. Larrick (Palo Alto Institute of Molecular Medicine, Mountain View, CA). We also thank Drs Thomas Ganz, Elizabeth Neufeld, Koteswara Chintalacharuvu, Juran Kato-Stankiewicz, Milena Pervan, and Lea Guo (UCLA) for their helpful suggestions.
Prepublished online as Blood First Edition Paper, June 27, 2006; DOI 10.1182/blood-2006-04-020263.
Supported in part by grants CA86915 and CA107023 from the National Institutes of Health (NIH), the 2003 Jonsson Cancer Center Foundation Interdisciplinary Grant, and the 2004 Brian D. Novis International Myeloma Foundation Senior Grant Award.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Ponka P, Lok CN. The transferrin receptor: role in health and disease. Int J Biochem Cell Biol. 1999; 31: 1111-1137. [DOI] [PubMed] [Google Scholar]
- 2.Omary MB, Trowbridge IS, Minowada J. Human cell-surface glycoprotein with unusual properties. Nature. 1980;286: 888-891. [DOI] [PubMed] [Google Scholar]
- 3.Faulk WP, Hsi BL, Stevens PJ. Transferrin and transferrin receptors in carcinoma of the breast. Lancet. 1980;2: 390-392. [DOI] [PubMed] [Google Scholar]
- 4.Shindelman JE, Ortmeyer AE, Sussman HH. Demonstration of the transferrin receptor in human breast cancer tissue: potential marker for identifying dividing cells. Int J Cancer. 1981;27: 329-334. [DOI] [PubMed] [Google Scholar]
- 5.Trowbridge IS, Lesley J, Schulte R. Murine cell surface transferrin receptor: studies with an anti-receptor monoclonal antibody. J Cell Physiol. 1982;112: 403-410. [DOI] [PubMed] [Google Scholar]
- 6.Prost AC, Menegaux F, Langlois P, et al. Differential transferrin receptor density in human colorectal cancer: a potential probe for diagnosis and therapy. Int J Oncol. 1998;13: 871-875. [DOI] [PubMed] [Google Scholar]
- 7.Shinohara H, Fan D, Ozawa S, et al. Site-specific expression of transferrin receptor by human colon cancer cells directly correlates with eradication by antitransferrin recombinant immunotoxin. Int J Oncol. 2000;17: 643-651. [DOI] [PubMed] [Google Scholar]
- 8.Brooks D, Taylor C, Dos Santos B, et al. Phase Ia trial of murine immunoglobulin A antitransferrin receptor antibody 42/6. Clin Cancer Res. 1995;1: 1259-1265. [PubMed] [Google Scholar]
- 9.Mayers GL, Raghavan D, Hitt S, Glaves D. Transferrin-gemcitabine conjugates: application to chemotherapy. Proc 89th Annual Meeting Am Assoc Cancer Res. 1998;39: 63-64. [Google Scholar]
- 10.Mayers GL, Razeq J, Abu-Hadid MM. Cytotoxic drug conjugates for treatment of neoplastic disease. US patent no. 5,393,737. February 28, 1995.
- 11.Laske DW, Muraszko KM, Oldfield EH, et al. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery. 1997;41: 1039-1049; discussion: 1049-1051. [DOI] [PubMed] [Google Scholar]
- 12.Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J Neurooncol. 2003;65: 3-13. [DOI] [PubMed] [Google Scholar]
- 13.Ng PP, Dela Cruz JS, Sorour DN, et al. An anti-transferrin receptor-avidin fusion protein exhibits both strong proapoptotic activity and the ability to deliver various molecules into cancer cells. Proc Natl Acad Sci U S A. 2002;99: 10706-10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Habelshaw HA, Lister TA, Stansfeld AG. Correlation of transferrin receptor expression with histological class and outcome in non-Hodgkin lymphoma. Lancet. 1983;1: 498-500. [DOI] [PubMed] [Google Scholar]
- 15.Beguin Y, Lampertz S, Degroote D, Igot D, Malaise M, Fillet G. Soluble Cd23 and other receptors (Cd4, Cd8, Cd25, Cd71) in serum of patients with chronic lymphocytic leukemia. Leukemia. 1993;7: 2019-2025. [PubMed] [Google Scholar]
- 16.Jefferies WA, Brandon MR, Williams AF, Hunt SV. Analysis of lymphopoietic stem cells with a monoclonal antibody to the rattransferrin receptor. Immunology. 1985;54: 333-341. [PMC free article] [PubMed] [Google Scholar]
- 17.Lesley J, Domingo DL, Schulte R, Trowbridge IS. Effect of an anti-murine transferrin receptor-ricin A conjugate on bone marrow stem and progenitor cells treated in vitro. Exp Cell Res. 1984;150: 400-407. [DOI] [PubMed] [Google Scholar]
- 18.Trowbridge IS. Transferrin receptor as a potential therapeutic target. Prog Allergy. 1988;45: 121-146. [DOI] [PubMed] [Google Scholar]
- 19.Coloma MJ. Production and Characterization of Novel Recombinant Antibodies [PhD thesis]. Los Angeles: Department of Microbiology and Molecular Genetics, University of California at Los Angeles; 1997.
- 20.Morrison SL, Wims L, Wallick S, Tan L, Oi VT. Genetically engineered antibody molecules and their application. Ann N Y Acad Sci. 1987;507: 187-198. [DOI] [PubMed] [Google Scholar]
- 21.White S, Taetle R, Seligman PA, Rutherford M, Trowbridge IS. Combinations of anti-transferrin receptor monoclonal antibodies inhibit human tumor cell growth in vitro and in vivo: evidence for synergistic antiproliferative effects. Cancer Res. 1990;50: 6295-6301. [PubMed] [Google Scholar]
- 22.Shin SU, Wu D, Ramanathan R, Pardridge WM, Morrison SL. Functional and pharmacokinetic properties of antibody-avidin fusion proteins. J Immunol. 1997;158: 4797-4804. [PubMed] [Google Scholar]
- 23.Gado K, Silva S, Paloczi K, Domjan G, Falus A. Mouse plasmacytoma: an experimental model of human multiple myeloma. Haematologica. 2001; 86: 227-236. [PubMed] [Google Scholar]
- 24.Cruz JC, Alsina M, Craig F, et al. Ibandronate decreases bone disease development and osteoclast stimulatory activity in an in vivo model of human myeloma. Exp Hematol. 2001;29: 441-447. [DOI] [PubMed] [Google Scholar]
- 25.Hayman SR, Fonseca R. Plasma cell leukemia. Curr Treat Options Oncol. 2001;2: 205-216. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Calvo M, Peterson EP, Rasper DM, et al. Purification and catalytic properties of human caspase family members. Cell Death Differ. 1999; 6: 362-369. [DOI] [PubMed] [Google Scholar]
- 27.Tomita Y, Bilim V, Hara N, Kasahara T, Takahashi K. Role of IRF-1 and caspase-7 in IFN-gamma enhancement of Fas-mediated apoptosis in ACHN renal cell carcinoma cells. Int J Cancer. 2003;104: 400-408. [DOI] [PubMed] [Google Scholar]
- 28.Beynon RJ, Bond JS. Proteolytic Enzymes: A Practical Approach. New York: Oxford University Press; 2001.
- 29.Zollner H. Handbook of Enzyme Inhibitors. Weinheim, Germany: VCH Verlagsgesellschaft; 1989.
- 30.Sreedharan SK, Verma C, Caves LS, et al. Demonstration that 1-trans-epoxysuccinyl-L-leucylamido-(4-guanidino) butane (E-64) is one of the most effective low Mr inhibitors of trypsin-catalysed hydrolysis: characterization by kinetic analysis and by energy minimization and molecular dynamics simulation of the E-64-beta-trypsin complex. Biochem J. 1996;316(pt 3): 777-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Creagh EM, Conroy H, Martin SJ. Caspase-activation pathways in apoptosis and immunity. Immunol Rev. 2003;193: 10-21. [DOI] [PubMed] [Google Scholar]
- 32.Greene BT, Thorburn J, Willingham MC, et al. Activation of caspase pathways during iron chelator-mediated apoptosis. J Biol Chem. 2002;277: 25568-25575. [DOI] [PubMed] [Google Scholar]
- 33.Korchak HM, Rich AM, Wilkenfeld C, Rutherford LE, Weissmann G. A carbocyanine dye, DiOC6(3), acts as a mitochondrial probe in human neutrophils. Biochem Biophys Res Commun. 1982;108: 1495-1501. [DOI] [PubMed] [Google Scholar]
- 34.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305: 626-629. [DOI] [PubMed] [Google Scholar]
- 35.Trowbridge IS, Domingo DL. Anti-transferrin receptor monoclonal antibody and toxin-antibody conjugates affect growth of human tumour cells. Nature. 1981;294: 171-173. [DOI] [PubMed] [Google Scholar]
- 36.Lesley JF, Schulte RJ. Selection of cell lines resistant to anti-transferrin receptor antibody: evidence for a mutation in transferrin receptor. Mol Cell Biol. 1984;4: 1675-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaickus L, Levy R. Antiproliferative monoclonal antibodies: detection and initial characterization. J Immunol. 1985;135: 1987-1997. [PubMed] [Google Scholar]
- 38.Lesley J, Schulte R. Selection and characterization of transferrin receptor mutants using receptor-specific antibodies. Immunogenetics. 1986; 24: 163-170. [DOI] [PubMed] [Google Scholar]
- 39.Kemp JD, Thorson JA, McAlmont TH, Horowitz M, Cowdery JS, Ballas ZK. Role of the transferrin receptor in lymphocyte growth: a rat IgG monoclonal antibody against the murine transferrin receptor produces highly selective inhibition of T and B cell activation protocols. J Immunol. 1987;138: 2422-2426. [PubMed] [Google Scholar]
- 40.Lesley JF, Schulte RJ. Inhibition of cell growth by monoclonal anti-transferrin receptor antibodies. Mol Cell Biol. 1985;5: 1814-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lesley J, Schulte R, Woods J. Modulation of transferrin receptor expression and function by anti-transferrin receptor antibodies and antibody fragments. Exp Cell Res. 1989;182: 215-233. [DOI] [PubMed] [Google Scholar]
- 42.Trowbridge IS, Lopez F. Monoclonal antibody to transferrin receptor blocks transferrin binding and inhibits human tumor cell growth in vitro. Proc Natl Acad Sci U S A. 1982;79: 1175-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mendelsohn J, Trowbridge I, Castagnola J. Inhibition of human lymphocyte proliferation by monoclonal antibody to transferrin receptor. Blood. 1983;62: 821-826. [PubMed] [Google Scholar]
- 44.Neckers LM, Cossman J. Transferrin receptor induction in mitogen-stimulated human T lymphocytes is required for DNA synthesis and cell division and is regulated by interleukin 2. Proc Natl Acad Sci U S A. 1983;80: 3494-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taetle R, Honeysett JM, Trowbridge I. Effects of anti-transferrin receptor antibodies on growth of normal and malignant myeloid cells. Int J Cancer. 1983;32: 343-349. [DOI] [PubMed] [Google Scholar]
- 46.Taetle R, Rhyner K, Castagnola J, To D, Mendelsohn J. Role of transferrin, Fe, and transferrin receptors in myeloid leukemia cell growth: studies with an antitransferrin receptor monoclonal antibody. J Clin Invest. 1985;75: 1061-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taetle R, Castagnola J, Mendelsohn J. Mechanisms of growth inhibition by anti-transferrin receptor monoclonal antibodies. Cancer Res. 1986; 46: 1759-1763. [PubMed] [Google Scholar]
- 48.Weissman AM, Klausner RD, Rao K, Harford JB. Exposure of K562 cells to anti-receptor monoclonal antibody OKT9 results in rapid redistribution and enhanced degradation of the transferrin receptor. J Cell Biol. 1986;102: 951-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kett WC, Osmond RI, Moe L, Skett SE, Kinnear BF, Coombe DR. Avidin is a heparin-binding protein: affinity, specificity and structural analysis. Biochim Biophys Acta. 2003;1620: 225-234. [DOI] [PubMed] [Google Scholar]
- 50.Phillips ML, Tao MH, Morrison SL, Schumaker VN. Human/mouse chimeric monoclonal antibodies with human IgG1, IgG2, IgG3 and IgG4 constant domains: electron microscopic and hydro-dynamic characterization. Mol Immunol. 1994;31: 1201-1210. [DOI] [PubMed] [Google Scholar]
- 51.Salmeron A, Borroto A, Fresno M, Crumpton MJ, Ley SC, Alarcon B. Transferrin receptor induces tyrosine phosphorylation in T cells and is physically associated with the TCR zeta-chain. J Immunol. 1995;154: 1675-1683. [PubMed] [Google Scholar]
- 52.Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J Cell Biol. 1983;97: 508-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klapper LN, Waterman H, Sela M, Yarden Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res. 2000;60: 3384-3388. [PubMed] [Google Scholar]
- 54.Ackrell BA, Maguire JJ, Dallman PR, Kearney EB. Effect of iron deficiency on succinate- and NADH-ubiquinone oxidoreductases in skeletal muscle mitochondria. J Biol Chem. 1984;259: 10053-10059. [PubMed] [Google Scholar]
- 55.Ranasinghe AW, Johnson NW, Scragg MA, Williams RA. Iron deficiency reduces cytochrome concentrations of mitochondria isolated from hamster cheek pouch epithelium. J Oral Pathol Med. 1989;18: 582-585. [DOI] [PubMed] [Google Scholar]
- 56.Maclean K, Yang H, Cleveland JL. Serum suppresses myeloid progenitor apoptosis by regulating iron homeostasis. J Cell Biochem. 2001;82: 171-186. [DOI] [PubMed] [Google Scholar]
- 57.Walter PB, Knutson MD, Paler-Martinez A, et al. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc Natl Acad Sci U S A. 2002;99: 2264-2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim BS, Yoon KH, Oh HM, et al. Involvement of p38 MAP kinase during iron chelator-mediated apoptotic cell death. Cell Immunol. 2002;220: 96-106. [DOI] [PubMed] [Google Scholar]
- 59.Buss JL, Neuzil J, Gellert N, Weber C, Ponka P. Pyridoxal isonicotinoyl hydrazone analogs induce apoptosis in hematopoietic cells due to their iron-chelating properties. Biochem Pharmacol. 2003; 65: 161-172. [DOI] [PubMed] [Google Scholar]
- 60.Moura IC, Lepelletier Y, Arnulf B, et al. A neutralizing monoclonal antibody (mAb A24) directed against the transferrin receptor induces apoptosis of tumor T lymphocytes from ATL patients. Blood. 2004;103: 1838-1845. [DOI] [PubMed] [Google Scholar]
- 61.Choi SC, Kim BS, Song MY, et al. Downregulation of p38 kinase pathway by cAMP response element-binding protein protects HL-60 cells from iron chelator-induced apoptosis. Free Radic Biol Med. 2003;35: 1171-1184. [DOI] [PubMed] [Google Scholar]
- 62.Lorenzo HK, Susin SA, Penninger J, Kroemer G. Apoptosis inducing factor (AIF): a phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999;6: 516-524. [DOI] [PubMed] [Google Scholar]
- 63.Yeh CJ, Taylor CG, Faulk WP. Transferrin binding by peripheral blood mononuclear cells in human lymphomas, myelomas and leukemias. Vox Sang. 1984;46: 217-223. [DOI] [PubMed] [Google Scholar]
- 64.Lesley J, Hyman R, Schulte R, Trotter J. Expression of transferrin receptor on murine hematopoietic progenitors. Cell Immunol. 1984;83: 14-25. [DOI] [PubMed] [Google Scholar]
- 65.Peschle C, Botta R, Muller R, Valtieri M, Ziegler BL. Purification and functional assay of pluripotent hematopoietic stem cells. Rev Clin Exp Hematol. 2001;5: 3-14. [DOI] [PubMed] [Google Scholar]
- 66.Gross S, Helm K, Gruntmeir JJ, Stillman WS, Pyatt DW, Irons RD. Characterization and phenotypic analysis of differentiating CD34+ human bone marrow cells in liquid culture. Eur J Haematol. 1997;59: 318-326. [DOI] [PubMed] [Google Scholar]
- 67.Bender JG, Unverzagt K, Walker DE, et al. Phenotypic analysis and characterization of CD34+ cells from normal human bone marrow, cord blood, peripheral blood, and mobilized peripheral blood from patients undergoing autologous stem cell transplantation. Clin Immunol Immunopathol. 1994;70: 10-18. [DOI] [PubMed] [Google Scholar]
- 68.Lansdorp PM, Dragowska W. Long-term erythropoiesis from constant numbers of CD34+ cells in serum-free cultures initiated with highly purified progenitor cells from human bone marrow. J Exp Med. 1992;175: 1501-1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sieff C, Bicknell D, Caine G, Robinson J, Lam G, Greaves MF. Changes in cell surface antigen expression during hemopoietic differentiation. Blood. 1982;60: 703-713. [PubMed] [Google Scholar]
- 70.Urashima M, Chen BP, Chen S, et al. The development of a model for the homing of multiple myeloma cells to human bone marrow. Blood. 1997; 90: 754-765. [PubMed] [Google Scholar]
- 71.Mitsiades CS, Treon SP, Mitsiades N, et al. TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: therapeutic applications. Blood. 2001; 98: 795-804. [DOI] [PubMed] [Google Scholar]
- 72.Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1: 118-129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.