

Abstract
Immune mechanisms are involved in the pathophysiology of aplastic anemia (AA) and myelodysplastic syndrome (MDS). Immune inhibition can result from cytotoxic T cell (CTL) attack against normal hematopoiesis or reflect immune surveillance. We used clonally unique T-cell receptor (TCR) variable β-chain (VB) CDR3 regions as markers of pathogenic CTL responses and show that while marrow failure syndromes are characterized by polyclonal expansions, overexpanded clones exist in these diseases and can serve as investigative tools. To test the applicability of clonotypic assays, we developed rational molecular methods for the detection of immunodominant clonotypes in blood and in historic marrow biopsies of 35 AA, 37 MDS, and 21 paroxysmal nocturnal hemoglobinuria (PNH) patients, in whom specific CDR3 sequences and clonal sizes were determined. CTL expansions were detected in 81% and 97% of AA and MDS patients, respectively. In total, 81 immunodominant signature clonotypes were identified. Based on the sequence of immunodominant CDR3 clonotypes, we designed quantitative assays for monitoring corresponding clones, including clonotypic Taqman polymerase chain reaction (PCR) and clonotype-specific sequencing. No correlation was found between clonality and disease severity but in patients treated with immunosuppression, truly pathogenic clones were identified based on the decline that paralleled hematologic response. We conclude that immunodominant clonotypes associated with marrow failure may be used to monitor immunosuppressive therapy.
Introduction
Many clinical similarities, including responsiveness to immunosuppression, suggest that T-cell-mediated immune attack is involved not only in the pathophysiology of aplastic anemia (AA) but also in certain cases of myelodysplastic syndrome (MDS).1-4 Bone marrow failure can result from an autoimmune attack directed against normal hematopoiesis or may reflect an immune surveillance reaction incited by the dysplastic myeloid cells. Large granular lymphocyte leukemia (LGL), which often results in single-lineage cytopenias, can occasionally accompany cases of MDS or even AA. This association may be instructive in many ways.5-7 First, evolution of a LGL clone may occur in the context of a primarily polyclonal immune response. Second, the presence of a LGL clone may suggest a rational target for therapeutic intervention with immunosuppressive agents. LGL clones can be easily functionally characterized and defined based on the presence of a unique T-cell receptor (TCR) rearrangement.8 Finally, in the laboratory, disease-associated clonal expansions of cytotoxic T cells (CTLs) may be used as a model system for polyclonal immune responses.9,10
The principle of molecular clonotypic analysis is based on the unique structure of TCR. Rearrangement of individual V, D, J, and C (variable, diversity, joining, and constant, respectively) regions leads to the assembly of the β (B) chain and creation of the hypervariable complementarity determining region 3 (CDR3), which plays a major role in the recognition of antigenic peptides presented in the context of HLA. CDR1 and CDR2 regions also have an important role in the recognition of peptide major histocompatibility complex (pMHC) but their main function consists of stabilizing the ligated CDR3.11 TCR B chain is the most suitable marker of individual CTLs due to the overall higher diversity of the VB CDR3 (as opposed to variable α [VA] CDR3); in addition, a single T cell can express 2 distinct TCRs with 2 alpha chains but only 1 beta chain.12 Immune responses to antigenic peptides result in expansion of individual reacting T-cell clones. Each T-cell clone carries one unique clonotype defined by the nucleotide sequence of the TCR VB CDR3. Clonotypes can be used as signatures of the individual CTL clones and serve as surrogate markers for their target antigens.13 Current technologies allow for identification and quantitation of immunodominant clonotypes by measuring the frequency of identical sequences within VB CDR3 amplification products. Several TCR-based techniques have been applied to study various immune processes, including graft-versus-host disease.14-16
Various levels of resolution of TCR repertoire analysis have been employed in AA, paroxysmal nocturnal hemoglobinuria (PNH),13,15,17-22 LGL,9,10 and, to a certain degree, also in MDS.17,23,24 In MDS patients undergoing immunosuppressive therapy, a decrease in clonal expansion was reported.3 Recently, we applied molecular analysis of the TCR repertoire to identify and follow immunodominant clones in LGL.9,10 The principles of the clonotypic receptor analysis established in our studies in LGL served as a basis here for more systematic analyses. We hypothesized that if immunodominant clones are involved in the mechanisms of cytopenia and hematopoietic suppression, their size should correlate with the disease course. Thus, determination of individual clones and levels of their representation is a powerful investigative tool that, if further refined and validated, may gain diagnostic significance in the future.
The goal of this work was to isolate and characterize immunodominant, disease-associated T-cell clones in AA, MDS, and PNH; study the clonal kinetics and compare with healthy controls; and determine the changes in the clonal CTL repertoire. To accomplish these tasks, we developed and applied new molecular tools of TCR repertoire analysis including multiplex VB polymerase chain reaction (PCR) and clonotypic Taqman PCR.
Patients, materials, and methods
Patients and controls
Informed consent for sample collection was signed by the individuals in accordance with protocols approved by the Institutional Review Board of the Cleveland Clinic Foundation (Cleveland, OH). Peripheral blood specimens and archived bone marrow biopsies were obtained from 35 patients with aplastic anemia, 37 patients with MDS, 21 patients with PNH, and 20 healthy controls (Tables 1, 2). Patients with AA were diagnosed according to the International Study of Aplastic Anemia and Agranulocytosis criteria. Samples from patients treated with immunosuppressive agents were collected prior to treatment. In selected patients, longitudinal studies were performed after immunosuppressive therapy with horse/rabbit antithymocyte globulin (ATG) and cyclosporin A (CsA) (Table 3). Diagnosis of MDS was established by bone marrow biopsy and peripheral blood counts and classified according to French-American-British (FAB) classification.25 PNH diagnosis and original diagnosis of LGL were based on clinical and laboratory parameters as previously described.26-28
Table 1.
Patient characteristics
| Diagnosis | No. | Mean age, y (range) | Sex, female-male ratio |
|---|---|---|---|
| AA | 35 | 47.1 (4–79) | 18:17 |
| MDS | 37 | 61.8 (25–80) | 13:24 |
| RA and RARS | 19 | 63.5 (41–80) | 8:11 |
| RAEB and RAEB-t | 18 | 60.0 (25–77) | 5:13 |
| PNH | 21 | 46.6 (29–75) | 11:10 |
| Hypoplastic BM* | 42 | 50.4 (10–85) | 17:25 |
| Hyperplastic BM | 24 | 65.1 (25–80) | 9:15 |
AA indicates aplastic anemia; MDS, myelodysplastic syndrome; RA, refractory anemia; RARS, refractory anemia with ring sideroblasts; RAEB, refractory anemia with excess blasts; RAEB-t, RAEB in transformation; PNH, paroxysmal nocturnal hemoglobinuria; and BM, bone marrow
Includes AA and MDS
Table 2.
Laboratory data of AA patients
|
No. patients
|
|||||
|---|---|---|---|---|---|
| Severity | Total | ANC less than or equal to 0.5 × 109/L | PLT less than or equal to 20 × 109/L | HGB less than or equal to 8.5 g/dL | Transfusion dependent |
| mAA | 10* | 2 | 2 | 1 | 1† |
| sAA | 22* | 1 | 18 | 10 | 17† |
| sAA RF | 2 | 1 | 1 | 1 | 1 |
| sAA REL | 1 | 0 | 1 | 0 | 1 |
ANC, PLT, and HGB indicate absolute neutrophil count, platelet count, and hemoglobin count, respectively, at the time of sampling; mAA, moderate aplastic anemia; sAA, severe aplastic anemia; sAA RF, sAA refractory; and sAA REL, sAA relapsed
Laboratory counts were not available for 1 patient with mAA and 1 patient with sAA
The transfusion status was not known in 2 patients with mAA and 3 patients with sAA
Table 3.
Characteristics of AA patients longitudinally studied
| Patient no. | Therapy drug | Response |
|---|---|---|
| 29 | rATG | Partial + |
| 31 | hATG/CsA | + |
| 33 | hATG/CsA | + |
| 39 | CsA | + |
| 40 | h+rATG (2×) | – |
| 41* | Anti-IL2 Ab | + |
| 42 | hATG/CsA | + |
| 45* | rATG | – |
| 46 | hATG | + |
| 47* | rATG/CsA | + |
| 48* | rATG | Partial + |
rATG indicates rabbit antithymocyte globulin; –, no response; hATG, human antithymocyte globulin; +, response to therapy; CsA, cyclosporin A; and partial +, partial response to therapy
These patients were studied only by VB flow cytometry
VB cytometry (VB skewing)
The individual contribution of each of the 19 VB subfamilies identifiable by specific monoclonal antibodies (mAbs) was determined as previously described,12 and results were expressed in percentage of alpha/beta CD4+ or CD8+ cells. VB flow cytometry analysis was performed on fresh peripheral blood according to the manufacturer's instructions (IOTest Beta Mark kit; Beckman Coulter, Fullerton, CA) with the following modifications: 5 μL of phycoerythrincyanine 5.1 (PC5)-conjugated CD4, 5 μL energy-coupled dye (ECD)-conjugated CD8 mAbs, and 20 μL of anti-VB antibody (VB1-5, VB7-9, VB11-14, VB16-18, VB20-23) were added. An additional anti-VB6.7 Ab (not included in the kit) was used. A 4-color protocol was applied and the lymphocyte gate was set according to the size and forward scatter properties. VB overrepresentation was established when contribution of a particular VB family was greater than the mean + 2 standard deviations (SDs) of values found in healthy volunteers.
CD8 lymphocyte separation and cDNA preparation
Mononuclear cells were separated from peripheral blood by density gradient sedimentation (Mediatech, Herndon, VA). CD4+ and CD8+ T cells were isolated by flow cytometric sorting. Samples were stained with a CD8 mAb conjugated with fluorescein isothiocyanate (FITC; PharMingen, San Diego, CA), and CD8+ high fraction was sorted on an Epics Altra high-speed flow cytometer (Beckman Coulter, Miami, FL). Total RNA was extracted from CD8+ T cells using TRIZOL (Invitrogen, Carlsbad, CA) combined with Phase-Lock gel tubes (Eppendorf, Hamburg, Germany) and dissolved in a final volume of 20 μL diethyl pyrocarbonate (DEPC) water. cDNA was generated from 5 to 8 μL RNA by first-strand cDNA synthesis using SuperScript III RT Kit (Invitrogen).
VB-specific PCR and multiplex TCR VB PCR
CDR3 sequences of TCR VB chain were amplified using a VB-specific forward and a CB reverse primer as previously described.9 Alternatively, a 2-tube multiplex PCR (BIOMED2) that covers all VB TCR gene rearrangements was applied as described previously29 on genomic DNA or cDNA samples.
Isolation of genomic DNA from formalin-embedded tissue
DNA was isolated from embedded blocks as previously described.31 Ten formalin-embedded sections were cut (6 μm thick) and dissolved in 400 μL lysis buffer (50 mM Tris; 1 mM EDTA; 0.5% Tween 20, pH8.6) in 105°C for 10 minutes. After addition of 5 μL proteinase K, samples were incubated at 65°C overnight and DNA was extracted using a modified phenol/chloroform method, precipitated in ethanol, and dissolved in nuclease-free water.
CDR3 cloning and sequencing
PCR products were separated on a 1.2% agarose gel, excised, and purified using the Gel Extraction Kit (Eppendorf) following the manufacturer's instructions. Four microliters of the purified PCR product was ligated into the TA cloning vector pCR2.1 (Invitrogen) overnight at 14°C. Ligations were heat-shock transformed into TOP10F Escherichia coli and plated on X-gal-covered agarose plates overnight. Colony PCR and subsequent sequencing of positive colonies were performed as previously described.9 An expanded immunodominant clonotype was defined according to its frequency among sequenced VB CDR3 regions within an expanded VB family. The pathologic expansion was based on the average size of maximal expansions in healthy individuals and was greater than 12% of all clones, as calculated and described by us previously.9 VB CDR3 region sequences were analyzed using the ImMunoGeneTics information system TCR alignment tool31 or manually using Word macros for CDR3 sequence analysis written in our laboratory. In this study, we focused on comparison of CDR3 regions that include invariant portions of VB and JB chains (in accordance with the CDR3 numbering proposed on the ImMunoGeneTics website32).
Clonotype-specific PCR on archived tissues
Clonotypic reverse primers were designed from immunodominant clonotypic sequences derived from peripheral blood to span the CDR3 region and were used with VB family-specific forward primers (for sequences see Table 4). To increase the fidelity, each clonotypic primer covered the variant amino acids (aa's) of each JB chain. A 2-step clonotypic PCR was performed. In the first-step PCR, 2 μL genomic DNA from patients' bone marrow biopsies and a healthy control DNA were added to a master mix as for regular VB PCR. Multiple PCRs were run to establish optimal temperature conditions and the following protocol proved efficient: initial 2.5-minute denaturation at 94°C and 8 amplification cycles of 30 seconds/94°C and 60 seconds/70°C were followed by 8 touchdown cycles (30 seconds/94°C, 30 seconds/67°C/-1°C, 30 seconds/72°C) extended for 20 more cycles with an annealing temperature of 60°C. PCR products (including blank) were diluted 1:100 in water and 6 μL was used in a second-step PCR with temperature as follows: initial denaturation at 94°C followed by 9 touchdown cycles (30 seconds of each 94°C, 68°C/-1°C, and 72°C), and final 11 cycles (30 seconds of each 94°C, 60°C, and 72°C). Eight microliters of PCR product was run on a 2.5% agarose gel. The fidelity of the clonotype PCR was verified by direct sequencing of CDR3 amplicons.
Table 4.
Sequences of primers and probes used for clonotypic assays
| Probes | Primers |
|---|---|
| Quantitative clonotypic assays* | |
| jb1-1FAM | FAM-CAAGGCACCAGACTCACAGT-BHQ-1 |
| jb2-1FAM | FAM-AGCAGTTCTTCGGGCCAG-MGBNFQ |
| jb2-3FAM | FAM-CAGTATTTTGGCCCAGGCA-MGBNFQ |
| vb12jb2.3BM sense, patient no. 31 | 5′-AGTTATGGAGCGGGGGCTAGCAC-3′ |
| vb13jb2.3BM sense, patient no. 37 | 5′-AGTTCCATCGGAAGGACTAGTAC-3′ |
| vb6jb2.3PB sense, patient no. 39 | 5′-TGCGTATCCACCGGAGGAGAG-3′ |
| vb14jb2.3BM sense, patient no. 40 | 5′-AGCAGGGAGACTAGCGGTCACAC-3′ |
| vb23jb2.1PB sense, patient no. 40 | 5′-CATTGATTCCCTGTCCCGGGAAG-3′ |
| vb15jb2.3PB sense, patient no. 41 | 5′-CTGCGTATCTGTGTCCCCTGTCC-3′ |
| vb24jb2.5PB sense, patient no. 41 | 5′-GGTCTCTCCTCCTACTCTGC-3′ |
| vb7jb2.1PB sense, patient no. 42 | 5′-TGTAAGGGTAGGCCCCGGTG-3′ |
| CB antisense | 5′-CTGCTTCTGATGGCTCAAACAC-3′ |
| Clonotypic PCR assays on biopsy tissues† | |
| vb6 sense | 5′-TCTCAGGTGTGATCCAAATTCGGG-3′ |
| vb7 sense | 5′-CCTGAATGCCCCAACAGCTCTCTC-3′ |
| vb15 sense | 5′-CAGGCACAGGCTAAATTCTCCCTG-3′ |
| vb23 sense | 5′-GCAGGGTCCAGGTCAGGACCCCCA-3′ |
| vb24 sense | 5′-CCCAGTTTGGAAAGCCAGTGACCC-3′ |
| vb6jb2.3PB antisense, patient no. 39 | 5′-TGCGTATCCACCGGAGGAGAG-3′ |
| vb23jb2.1PB antisense, patient no. 40 | 5′-CATTGATTCCCTGTCCCGGGAAG-3′ |
| vb15jb2.3PB antisense, patient no. 41 | 5′-CTGCGTATCTGTGTCCCCTGTCC-3′ |
| vb24jb2.5PB antisense, patient no. 41 | 5′-GGTCTCTCCTCCTACTCTGC-3′ |
| vb7jb2.1PB antisense, patient no. 42 | 5′-TGTAAGGGTAGGCCCCGGTG-3′ |
| jb2.1 antisense | 5′-CCTTCTTACCTAGCACGGTGA-3′ |
| jb2.3 antisense | 5′-CCCGCTTACCGAGCACTGTCA-3′ |
| jb2.5 antisense | 5′-CGCGCACACCGAGCAC-3′ |
FAM-labeled and minor groove binder (MGB) or black hole quencher 1 (BHQ1) quenched probes were used for quantitative PCR. CB reverse primer was designed to cover both CB1 and CB2 chains
PB indicates primer designed from CDR3 sequence detected in peripheral blood; and BM, primer designed from BM-derived clonotype
Shown in Figure 4A, C
Shown in Figure 4B
Quantitative clonotype-specific PCR of peripheral blood CD8+ cells
Sequences derived from expanded clones detected in bone marrow biopsies or in peripheral blood were used to design a clonotypic assay employing clonotype-specific forward primers, JB-specific Taqman probes, and a CB reverse primer covering both human CB chains (Table 4). In brief, 15 μLof 2× Taqman universal master mix (Applied Biosystems, Foster City, CA) was used with 0.6 μL (50 μM) forward clonotypic and reverse CB primers, 0.66 μL (10 μM) JB Taqman probe, 6 μL cDNA (1:3 dilution with water), and 1 μL water (for primer and probe sequences, see Table 4). In parallel, GAPDH gene expression was analyzed by using the human GAPDH kit (Applied Biosystems). All samples were run on a 7500 Real-Time PCR system (Applied Biosystems) in duplicates at 95°C for 10 minutes followed by 45 cycles of 15 seconds at 95°C and 60 seconds at 60°C.
Results
TCR VB utilization patterns in bone marrow failure syndromes and related conditions by flow cytometry and CDR3 size distribution analysis
We focused our analysis on CTL responses and analyzed a large cohort of patients with various related BM failure syndromes for the skewing of the VB family patterns (Tables 1, 2, 3, 5; Figure 1). Our theory was that expansion of individual pathogenic CTL clones can lead to overrepresentation of the corresponding TCR VB family as measured by a panel of VB-specific antibodies. Abnormal expansions of VB-expressing CTLs were defined based on the mean values +2× standard deviation, calculated within healthy individuals (due to this definition, 95% of healthy individuals did not harbor any pathologic expansions).9 Our previous analysis included patients with LGL in whom monoclonality and the principles of flow cytometric identification of CTL expansions are well established.9 The results of this study demonstrate a continuum of VB skewing patterns, from expected extreme monoclonal VB overexpansions in LGL (not shown) to multiple nominally smaller, albeit prominent, expansions identified in AA and MDS patients (Table 5; Figure 1). To compare differences in the degree of CTL expansions and account for the size of individual VB families in healthy controls (eg, 1.8% for VB4 or 4% for VB8, etc), we have calculated expansion ratios. As expected, the expansion ratios in AA and MDS patients were nominally lower than in the LGL patient group (Table 5). We expected that using our methodical approach we would identify differences between MDS subgroups that were previously classified based on marrow cellularity. Theoretically, hypoplastic MDS may show a more prominent VB family skewing (likely due to an immune-mediated process) than hypercellular MDS forms but such differences were not found (Table 5). However, using VB flow cytometry we detected several LGL-like “overexpansions” in MDS patients (Figure 1; Table 5). The detection rate for the presence of expansions approached 95% for all groups studied.
Table 5.
Characteristics of clonal repertoire in studied patients
| Diagnosis | No. clones per patient | Average clone, % | SEM | Expansion factor | Detection rate, % |
|---|---|---|---|---|---|
| AA | 2.32 | 6.75 ± 4.59 | 0.54 | 2.72 | 81 |
| MDS | 3.11 | 8.68 ± 7.6 | 0.71 | 3.42 | 97 |
| RA and RARS | 3.11 | 9.54 ± 9.34 | 1.22 | 3.46 | 100 |
| RAEB and RAEB-t | 3.11 | 7.78 ± 5.71 | 0.68 | 3.09 | 94 |
| PNH | 3.95 | 8.45 ± 9.47 | 1.04 | 3.26 | 95 |
| Hypoplastic BM* | 2.62 | 7.09 ± 5.17 | 0.49 | 2.78 | 84 |
| Hyperplastic BM | 3.04 | 9.50 ± 8.79 | 1.03 | 3.54 | 96 |
Expansion factor indicates the average fold expansion of VB families above the greater-than-2 SD values derived from healthy controls
SEM indicates standard error of mean; AA, aplastic anemia; MDS, myelodysplastic syndrome; RA, refractory anemia; RARS, refractory anemia with ring sideroblasts; RAEB, refractory anemia with excess of blasts; RAEB-t, RAEB in transformation; PNH, paroxysmal nocturnal hemoglobinuria; and BM, bone marrow
Includes AA and MDS
Figure 1.
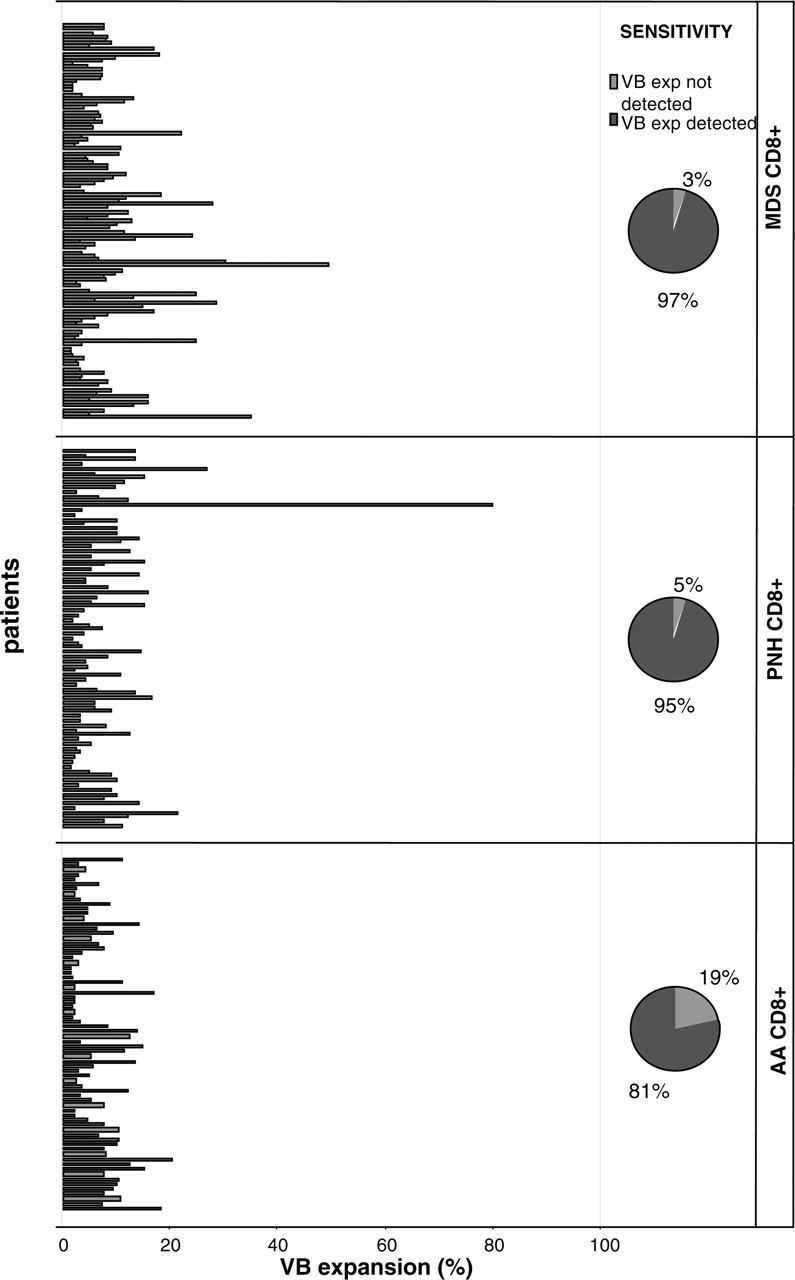
Patients with immunodominant expansions of TCR VB families. The size of pathologic VB expansion was calculated based on the values obtained from healthy controls and was defined as greater than 2 SDs above the normal values. The figure shows significant expansions of single CTL VB families in 4 patient groups. The pie diagrams depict the detection rate of immunodominant clonal expansions.
VB family expansion reflects oligoclonal proliferations of individual VB clones
In order to determine whether the VB expansions are due to oligoclonal CTL proliferations, we amplified the VB CDR3 regions using VB and CB primers and, after cloning, sequenced large numbers of CDR3 clonotypes. The frequency of identical VB regions corresponds to the degree of expansion for the individual CTLs. Previously we determined the degree of “physiologic” clonotype expansions within the CTL population derived from healthy blood donors. As examples, we studied VB 3, 9, 13, 18, and 21 CDR3 PCR products derived from CD8 cells using VB-specific and constant primers. Within these exemplary VB families, the identical sequences corresponding to expanded clones are encountered at a similar rate among individual VB families of healthy donors and can involve an average of 10.3% ± 2.3% (on an average of 22 clones we sequenced per VB family).
In both AA and MDS, most of the expanded VB families identified by flow cytometry showed presence of immunodominant clonotypes. As previously shown in LGL, all expanded families are clonally highly polarized9; such a polarity was also occasionally present among patients with MDS, AA, or PNH. No striking differences in the size of the immunodominant clones were found between these 3 diseases. Immunodominant clonotypes identified in blood of patients with AA, MDS, or PNH are shown in Table 6.
Table 6.
Expansion of immunodominant clonotypes in bone marrow failure patients
| Patient no. | Diagnosis | VB | CDR3 | JB | % clonal expansion |
|---|---|---|---|---|---|
| 1 | MDS/LGL | 14 | ASSLLTKTGSYEQE | 2.5 | 100 |
| 2 | MDS/LGL | 9 | ASSLPGTPTE | 1.1 | 100 |
| 3 | MDS | 4 | AALMPPQPRETP | 2.5 | Direct |
| 4 | MDS | 11 | ASSGTGSYE | 2.7 | 63 |
| 5 | MDS | 7 | APAAKMERLT | 2.1 | Direct |
| 5 | MDS | 1 | ASSSELTFRGDT | 2.3 | Direct |
| 6 | MDS | 23 | ATSAGSE | 2.7 | Direct |
| 7 | MDS | 23 | ASSLTDT | 2.2 | 100 |
| 8 | MDS | 8 | ASSFAPMTSGGALDT | 2.3 | 60 |
| 8 | MDS | 21 | ASSPRLAGALETPWET | 2.3 | 50 |
| 9 | MDS | 3 | ASSLGDVNQPQH | 1.5 | 100 |
| 10 | MDS | 3 | ASSKPGSPYQEQ | 2.5 | 50 |
| 10 | MDS | 13 | ASSSLEGGQGN | 2.3 | 36 |
| 11 | MDS | 21 | ASSPGWDRGLE | 2.5 | 100 |
| 12 | MDS | 21 | ASSFRLAGANNE | 2.1 | 67 |
| 13 | MDS | 4 | AALGPPTMSS | 2.5 | Direct |
| 13 | MDS | 1 | ASSLGQGSSYEE | 2.5 | Direct |
| 13 | MDS | 8 | ASSLPSVAN | 2.2 | Direct |
| 14 | MDS | 18 | ASSTGENTG | 2.2 | 75 |
| 30 | AA | 7 | ASSPPGGARMGE | 2.5 | 100 |
| 30 | AA | 16 | ASSQDLAGGPRRET | 2.3 | 56 |
| 30 | AA | 16 | ASSRGRHTDT | 2.3 | 33 |
| 30 | AA | 1 | ASSVGHGVNE | 1.4 | 100 |
| 31 | AA | 18 | ASSPTSAAYG | 1.2 | 70 |
| 31 | AA | 5 | ASSSANYRTDT | 1.1 | 90 |
| 32 | AA | 23 | ASSSHITDT | 2.3 | 80 |
| 33 | AA | 5 | ASSLDRASTPE | 1.1 | 100 |
| 33 | AA | 9 | ASSPANGLADAYE | 2.7 | 50 |
| 35 | AA | 12 | ASSEITEPG | 2.2 | 50 |
| 36 | AA | 15 | ATSDYDREVGDT | 2.3 | 40 |
| 36 | AA | 3 | ASAGTGGNE | 1.4 | 33 |
| 37 | AA | 13 | ASSSIGRTSTDT | 2.3 | 36 |
| 38 | AA | 13 | ASSYYG | 1.2 | 25 |
| 39 | AA | 6 | ASSSPPVDT | 2.3 | 77 |
| 39 | AA | 15 | ATSDLASFNTG | 2.2 | 46 |
| 40 | AA | 22 | ASKWEQGVGNTE | 1.1 | 93 |
| 40 | AA | 23 | ASSFPGQGINE | 2.1 | 100 |
| 41 | AA | 15 | ATGTGDTDT | 2.3 | 33 |
| 41 | AA | 24 | ATSRVGGE | 2.5 | 50 |
| 42 | AA | 7 | ASSTGAYPYNE | 2.1 | 70 |
| 43 | AA | 3 | ASSFRDREETNQPQH | 1.5 | 27 |
| 44 | AA | 2 | CSASFRVE | 2.5 | 57 |
| 20 | PNH | 20 | ASSWKPPPIPYE | 2.7 | 33 |
| 20 | PNH | 8 | ASSLAVAQE | 2.5 | 67 |
| 21 | PNH | 8 | PLTGLTLSE | 2.5 | 36 |
| 22 | PNH | 13 | ASSRRANTG | 2.2 | 30 |
| 22 | PNH | 1 | ASSAAGVKE | 2.5 | 100 |
| 23 | PNH | 13 | ASSSSGSYNE | 2.1 | 60 |
| 24 | PNH | 3 | ASGQRGGA | 2.4 | 61 |
| 25 | PNH | 3 | ASSWTGYE | 2.7 | 76 |
| 25 | PNH | 13 | ASSYGGGQPQFH | 1.5 | 55 |
| 16 | MDS | 4 | ASSPRGTF | 1.5 | 67 |
| 29 | AA | 12 | AISLGGELFF | 2.2 | 50 |
| 29 | AA | 17 | ASSRGLADTDTQYF | 2.3 | 67 |
VB indicates variable region of the T-cell receptor beta chain (TCR); CDR3, complementarity determining region 3 of the TCR beta chain; JB, joining region of the TCR; AA, aplastic anemia; and direct, direct sequencing of the amplification product
We hypothesized that pathogenic clonotypes identified in patients will not be detectable in healthy controls if amplified under fixed conditions. For a meaningful comparison, individuals matched for HLA were used. We took advantage of HLA-matched siblings who were available to some of the patients. Interestingly, when 20 CDR3 regions were sequenced for VB1 and VB5 (originally expanded in the corresponding AA patient) in an HLA-matched sibling donor, we did not find any sequences shared between patients and their sibling donors. Similar results were obtained for a pair of twins discordant for the presence of MDS; when 20 sequences for VB21 and VB8 (each) were sequenced and compared with the counterpart MDS twin, no shared sequences were identified. Overall, TCR variability was higher in HLA-matched healthy individuals compared with bone marrow failure patients, consistent with the presence of expanded VB families and the corresponding CTL clones (data not shown).
Expanded clonal CTLs can be detected in bone marrow biopsies of patients with bone marrow failure
Immunodominant clones detected in blood could represent a variety of immune-mediated processes. Therefore, we determined whether a VB multiplex PCR-based technology can be used for the identification of immunodominant clones directly in the target tissue (bone marrow). For that purpose we used paraffin-embedded bone marrow (BM) biopsies (Figure 2). As flow cytometric VB detection is not possible on the biopsies, we determined the immunodominant clones by amplifying the entire TCR VB repertoire (by multiplex VB PCR) and calculated the frequency of expanded (redundant) CDR3 sequences within a large number of clones sequenced. Using this technique, we retrospectively demonstrated that the marrow of AA patients contained a skewed representation pattern of CTL clones and were able to identify immunodominant clones in most of the biopsies studied. Examples of such analysis and CDR3 immunodominant clonotypes are shown in Table 7. Conversely, biopsy-derived clonotypes were also detected in blood; 2 of the shown clonotypes are immunodominant (for examples, see Figure 3A).
Figure 2.

Examples of the application of TCR multiplex PCR for detection of immunodominant clonotypes in BM biopsies. Multiplex VB PCR combined with clonotype sequencing allowed for the detection of immunodominant clonotypic expansions in archived bone marrow biopsies. VB indicates T-cell receptor VB chain; CDR3, complementarity determining region 3; JB, joining region of the TCR B chain; and CB, constant B chain. All immunodominant BM-derived clonotypes are shown in Table 7. Images were obtained via digital microscopy using an Olympus BX41 microscope (Olympus America, Melville, NY) equipped with a 40×/0.75 NA UPlan objective lens. Slides were stained with hematoxylin and eosin. Images were captured using a Digital Camera Model no. 11.2 Color Mosaic (Diagnostic Instruments, Sterling Heights, MI), and imaging software included Spot Basic 4.1.3 (Diagnostic Instruments).
Table 7.
Expanded clones detected in marrow biopsies of bone marrow failure patients
| Patient no. and CDR3 | VB | JB | % expanded |
|---|---|---|---|
| 15 | |||
| ASSYSGAPTDT | 13 | 2.3 | 26 |
| ASSIRLGTDT | 14 | 2.3 | 14.8 |
| ASRPSLTSGIYTDT | 14 | 2.3 | 11.1 |
| AISDNNVLTF | 12 | 2.6 | 7.4 |
| ASSVHRGQSYNS | 13 | 1.6 | 7.4 |
| ASSAWGWAASSNE | 17 | 2.1 | 7.4 |
| 30 | |||
| ASSAPQGTDT | 13 | 2.3 | 17.2 |
| 26 | |||
| AIRRVVTDT | 14 | 2.3 | 12 |
| 27 | |||
| ASSQGLAGLTDT | 14 | 2.3 | 26 |
| ASSFSGASDT | 14 | 2.3 | 18.5 |
| ASSPLSRLATDT | 21 | 2.3 | 11.5 |
| ASSYSSHLAGSSYNE | 3 | 2.1 | 7.4 |
| 34 | |||
| ASSEEGWPYG | 13 | 1.2 | 16.6 |
| ASSYSPSYE | 13 | 2.7 | 16.6 |
| 37 | |||
| ASSSIGRTSTDT | 13 | 2.3 | 13 |
| ATSREPYRGPDT | 24 | 2.3 | 8.7 |
| 31 | |||
| ASSYGAGASTDT | 12 | 2.3 | 8 |
| 40 | |||
| ASRETSGHTDT | 14 | 2.3 | 8.7 |
| 16 | |||
| ASSIGDNS | 13 | 1.6 | 10.5 |
| ASSILSEGQP | 4 | 1.3 | 10.5 |
| 18 | |||
| ASSGGTR | 9 | 2.1 | 18.2 |
| 19 | |||
| ASSGGGY | 17 | 2.2 | 25 |
| 39 | |||
| ATRLAGAGDT | 12 | 2.3 | 25 |
| ASSSPPVDT | 6 | 2.3 | 10 |
| ATRRRRKHNE | 15 | 2.1 | 10 |
| 17 | |||
| ASSAGATSTDT | 13 | 2.3 | 14.3 |
| 41 | |||
| ASSKD*REPPRTDT | 10 | 2.3 | 35.7 |
Stop codon (nonproductive CDR3)
Figure 3.
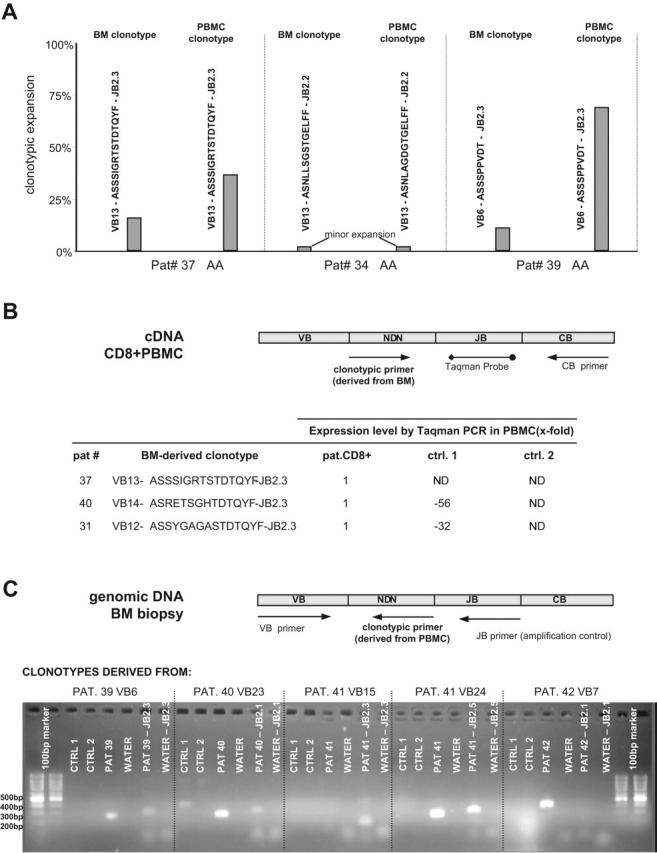
Molecular tracking of immunodominant clonotypes in BM biopsies and peripheral blood CD8+ cells from AA/MDS patients. (A) Independent sequencing of CDR3 regions derived from BM biopsy DNA and peripheral blood CD8+ RNA revealed identical or highly homologous clonotypes. Three examples are shown: in patients no. 37 and no. 39, identical expanded clonotypes were detected in both archived tissue and peripheral blood; patient no. 34 harbors 2 highly homologous minor clonotypes. PBMC indicates peripheral blood mononuclear cell. (B) The presence of clonotypes identified in archival BM tissue was tested on RNA from peripheral blood CD8+ cells using a quantitative Taqman assay. The presence and quantity of each shown clonotype was tested in the original patient and 2 healthy control samples obtained from our laboratory. Expression level in controls is shown as fold decrease in comparison to patient's values, which served as calibrator. ND indicates not detected. (C) Detection of blood-derived clonotypes in BM biopsies using nonquantitative clonotypic PCR. We were able to amplify peripheral blood-specific clonotypes in patient BM biopsies. Each clonotype was tested on genomic DNA from patient BM and genomic DNA from healthy controls. The primer set VB forward - JB reverse was used as an endogenous amplification control. The clonotypic primer derived from patient no. 40 also amplifies a nonspecific product of unexpected length in Ctrl1, possibly suggesting a partially rearranged TCR with high homology to patient's TCR sequence.
Clonotypes originally found in archival BM tissue were also detected in peripheral blood CD8 cells using a quantitative Taqman assay instead of clonotypic sequencing (Figure 3B). Amplification was performed with a clonotype-specific sense primer, a Taqman probe corresponding to the appropriate JB region, and a constant antisense primer. To achieve high specificity of clonotypic amplification, we designed the clonotypic primers to span the terminal portion of the VB chain, the entire NDN region, and the variable portion of the JB region. This would exclude a nonspecific amplification of nearly identical clonotypes with the same JB family restriction yet different variable JB portion. The presence and quantity of each clonotype shown was tested in blood samples from the corresponding patient and 2 healthy controls. When a patient's blood sample was used as a calibrator, the pathogenic clonotype was found at a much lower frequency in blood of one healthy control and was absent in the other control (Figure 3B). Similarly, blood-derived immunodominant clonotypes can also be detected in bone marrow using clonotype-specific PCR (Figure 3C). Immunodominant clonotypes initially found in patients' peripheral blood CD8+ cells were tested on genomic DNA from patients' BM biopsies and from healthy BM. The clonotype-specific primer from patient no. 40 also amplified a nonspecific product of unexpected length in a healthy volunteer, which suggests a partially rearranged clonotype similar to the patient's TCR CDR3. As a positive amplification control, each DNA sample was amplified using a VB forward and JB reverse primer set. Sequencing of PCR products confirmed the fidelity of the PCR assay.
Application of clonotypic TCR VB CDR3 sequences for the design of clonotype-specific assays
Clonotypic sequences obtained from individual patients were used to test their applicability for the design of clonotype-based PCR assays. Expanded clonotypes can be amplified using a simple clonotypic PCR employing a VB forward and clonotypic reverse primer. Simple clonotypic PCR is not quantitative and sequencing a large number of clones is very labor intensive. Therefore we tested possible application of clonotypic Taqman PCR for monitoring of potentially pathogenic clonotypic expansions in blood CD8+ cells (Figure 4A). Correlation with disease activity could serve as surrogate evidence for the involvement of corresponding clones in the pathophysiologic process. For patient no. 40, 2 different clonotypes were studied. VB23-JB2.1 clonotype was not detectable retrospectively in cryopreserved samples (at time points 0 and 1 month) and decreased drastically with the initiation of therapy. The same trend of clonotypic contraction was reproduced in the same patient for another immunodominant clonotype (VB22-JB1). In patient no. 42 with severe AA, ATG therapy resulted in drastic decrease in the frequency of the clonotype in blood. This patient showed an excellent hematologic response with full normalization of hematologic parameters. In patient no. 39 (initially diagnosed with AA), clonotypic Taqman PCR was performed after reinitiation of CsA therapy in part due to worsening blood counts. At first, the expansion of pathogenic clonotype dropped but it subsequently increased after stopping of CsA prompted by discovery of a new cytogenetic abnormality (20q-).
Figure 4.
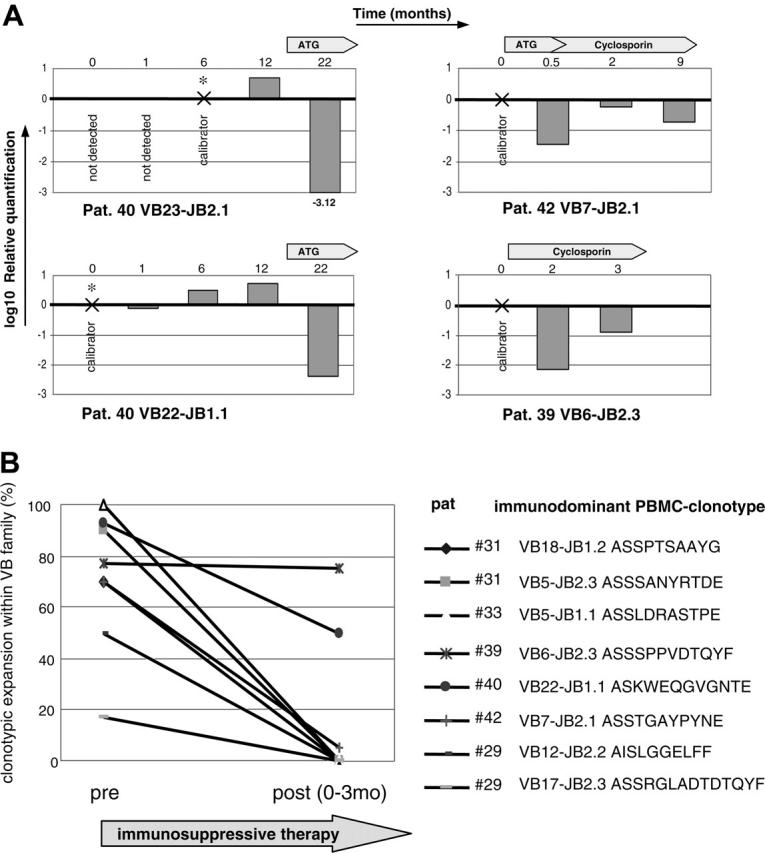
Effect of immunosuppressive therapy on TCR repertoire in AA patient. (A) For the tracking of potentially pathogenic clones, a clonotypic Taqman PCR was performed using patient CDR3-specific forward primers, CDR3-specific JB probe, and CB reverse primer. Four immunodominant clones were tracked during the course of disease in 3 AA patients. RNA was extracted from sorted CD8+ cells. For the calculation of clonotypic expression, samples in which the original immunodominant clonotype was identified were used as calibrators for subsequent or retrospective measurements. GAPDH levels were used for the normalization of RNA amounts. (B) TCR repertoire analysis was performed on 7 patients before and after application of immunosuppressive therapy that is shown in Table 3. AA patient no. 31 was analyzed before and after therapy, and 2 dominant clonotypes were found for VB5 and VB18 with the frequencies of 90% and 70%, respectively. One and 3 months after ATG therapy, normal lymphocyte count was restored and the diversity of obtained CDR3 sequences increased; furthermore, the immunodominant clonotype was not detectable by sequencing. Similar trend was seen in other patients. Patient no. 46 did not harbor any immunodominant expansions and is not shown in the figure.
Quantitative sequencing analysis of immunodominant clones during the course of disease
The TCR VB repertoire was monitored by VB flow cytometry in 11 AA patients before and after administration of immunosuppressive therapy. In 7 of them, the frequency of dominant TCR VB CDR3 clonotypes and the overall TCR repertoire were measured by sequencing. VB flow cytometry identified expansion of VB families, and CDR3 sequencing lead to identification of immunodominant clonotypes within the affected VB family.
In AA patient no. 33, 2 highly expanded clonotypes were detected before immunosuppression: VB5-JB1.1 and VB9-JB2.7, with expansions of 100% and 50%, respectively. The VB5 family immunodominant clone was not present after treatment; the second expanded clonotype (VB9), however, remained stable at 60% before and after therapy (data not shown). This may suggest that only the first of these clones was disease specific. In other patients, a similar trend could be observed: immunosuppressive therapy was associated with disappearance of both dominant clonotypes in patients no. 29 and no. 31. An initial clinical improvement was seen in patient no. 29; however, a new significantly expanded clone emerged within VB12 (VB12-AISEPGANT-JB1.1, 68%; not shown) and the patient recovery stagnated.
Parallel to hematopoietic recovery, the overall TCR variability increased (total CTL repertoire as measured by VB flow cytometry and clonotypic repertoire within a VB family) to values that are observed in healthy controls (data not shown). A decrease in expansion from immunodominant to minor characterized clonotype in patient no. 42, and in patients no. 39 and no. 40 the expanded clonotype remained constant or only slightly decreased after immunosuppressive therapy.
Discussion
We conducted a systematic analysis of the TCR repertoire in BM failure syndromes, focusing on patients with MDS or AA. Our analysis was based on the theory that cytopenias in bone marrow failure patients can be associated with exaggerated CTL-driven immune responses that, if detected, can be applied for the design of clonotypic assays. These assays may allow for monitoring of disease activity. Our study also included patients with LGL (some of whom were previously reported9), serving as a reference for monoclonalities and PNH. We demonstrated that, despite their complexity in AA and MDS, pathologic CTL responses can be successfully detected and characterized. The current study focused on the development of efficient and precise tools for clonotypic monitoring in MDS and AA rather than on the functional characterization of individual clonotypes.
In the first portion of our report, we used flow cytometry to study VB family skewing as a basis for a more stringent molecular clonality analysis. In a large cohort of patients, we detected VB expansions within the CTL populations; no difference was found in the average size of clonal expansions between AA and MDS, and the hypocellularity (irrespective of the diagnosis) was not associated with more pronounced VB skewing. Genotyping of the CDR3 region demonstrated oligoclonality in approximately 2 of 3 of these expanded VB families. Even by this rather crude analysis a continuum of responses was observed, from multiple VB families expanded in AA, PNH, and MDS to extreme monoclonal expansions in typical LGL.
Based on these results, we proceeded with more intricate molecular analysis using CDR3-specific amplification and sequencing of the clonotypic repertoire. We applied the previously used VB CDR3 PCR for patients with known VB family expansion as well as multiplex PCR amplifying total human VB TCR repertoire when VB expansion was not known. In agreement with previous reports involving smaller groups of MDS patients,23,24 we found evidence for oligoclonal T cell-mediated immune responses not only in AA but also in MDS patients. Skewing of CDR3 amplification products in both MDS and AA patients was also previously reported.4,13,17,18,23,24,33 However, in the current study, recently developed and more efficient methodology allowed for detection and characterization of immunodominant clonotypes not only in freshly isolated blood and BM, but also in archived paraffin-embedded specimens from the initial diagnostic biopsy. A multiplex PCR assay proved a very efficient means for identification of the most expanded clonotypes. Following multiplex PCR, VB-specific amplification using VB primers was performed with primers allowing for the amplification of VB family corresponding to the redundant clone detected in the multiplex PCR. This approach allowed for quantitation of clonal expansion within the VB family. Such a rational method lead to identification of oligoclonally expanded clones even when VB flow cytometry was not possible (biopsy material) or not informative. Previously, this method was applied to the analysis of LGL9,10 and has now been adopted to study MDS and AA.
The size of expanded clones identified in our study in MDS and AA patients was comparable, ranging from 25% to 100% of a given VB family. This finding has to be viewed in the context of physiologic variability of the TCR, which was previously estimated from a large number of control sequences derived from healthy individuals.9 Interestingly, the MDS patient group included 3 patients in whom traditional flow cytometric assays detected LGL T-cell clones and confirmed a diagnosis of concomitant LGL. Furthermore, using our strategy, LGL-like VB expansions (> 30% of total CD8 cells) were found in additional MDS patients. This suggests that LGL-like subclinical CTL responses may be more frequently encountered than it would be extrapolated based on traditional clinical testing. Subsequent studies of TCRγ rearrangement confirmed the presence of oligoclonality in these patients. Molecular assays with TCR analysis could be useful to further refine the diagnostic criteria for some MDS patients who share overlapping features with LGL patients not detectable by routine techniques. In addition to the sensitive detection of clonal dominance in blood, identification of expanded clonotypes in biopsy specimens is very instructive and would be difficult to reconcile with the argument that expanded clones are due to infection or alloimmunization.
Globally, our analysis showed that immunodominant CTL clonotypes could be detected at comparable rates in AA and MDS patients irrespective of the morphologic subtype. So far, isolation and characterization of dominant clones have been reported for 4 MDS patients,24 in a smaller series of PNH patients,22 in LGL patients,9,10,34-36 and in AA patients.19,33 In most other studies dealing with CTL responses in BM failure,4,9,10,13,20,23 oligoclonality within a VB family was determined solely by genotyping. Genotyping, in contrast to the Taqman PCR and CDR3-sequencing method, does not provide precise information about the actual clonal size and lacks consistent reproducibility.
Immunodominant clonotypes derived from blood or BM can be used as markers of the disease activity, assuming that these clones are related to the pathogenic process rather than to infections or alloimmunization. The disease specificity of the clonotypes detected in our study was supported by the correlation of their frequency with hematologic response to immunosuppression. Similarly, isolation of immunodominant clonotypes in BM biopsy specimens as well as in blood of patients at presentation indirectly supports the notion of specificity. Most of the patients did have low transfusion burden and none were pathologically infected at the time of sampling. Previously, a clinical correlation for the expanded clonotypes derived from blood of patients with AA was demonstrated.33 Clearly, the most stringent proof would be provided by functional analysis, but such an analysis was not possible due to the lack of putative antigens needed for functional assays. It is worth noting that if a clinical correlation between a clonotype and clinical events should be established, the clonotypic sequence itself could serve as a surrogate marker of the corresponding unknown antigen. Based on this premise, in our study, sequences derived from immunodominant clones were used to test the principle of clonotype-based monitoring using new molecular assays such as clonotype-specific PCR. The sequence analysis of the clonotypic repertoire can be used to estimate TCR variability and the presence of dominant clones during the course of disease.23 Similarly, as reported for 12 patients treated with ATG,23 we demonstrated a decrease in the frequency of the immunodominant clone in correlation with the hematopoietic response to therapy. This finding indirectly suggests a pathologic role of expanded T-cell clones. Previously, the effects of immunosuppression on VB repertoire in AA were mainly based on the determination of skewing patterns of individual VB families17 and on sequencing.33 When we applied a serial sequencing of VB CDR amplicons as a method to estimate clonal size, in addition to the decline in the frequency of the immunodominant clone, we observed a restoration of TCR variability (as a possible effect of immunosuppressive treatment). Moreover, our study can serve as a demonstration for the applicability of quantitative, PCR-based clonotypic testing in MDS and AA. We show that the CDR3 sequences of immunodominant clones can be exploited for the design of quantitative clonotype-specific molecular assays. In 2 of 3 cases that were studied concomitantly by both Taqman PCR and sequencing, excellent correlation was found in 2 (patient no. 40 and no. 42). Of note is that in one of the patients studied serially, no immunodominant clone was detected either prior to or after the treatment (patient no. 46).
This is the first application of clonotypic quantitative PCR to the study of BM failure syndromes. Previously, clonotypic PCR with CDR3-specific probes was used to study expanded clonotypes in graft-versus-host disease (GVHD) and autoimmune neurologic diseases37,38; we have redesigned the technology and use a combination of CDR3-specific primers and JB probes. This technique offers many advantages to the previously described and very labor-intensive direct sequencing method. For example, patients can be screened for the presence of signature clonotypes associated with specific clinical features. Subsequently, clonotype-specific assays may be further tested for their applicability in individualized monitoring of therapeutic responses.
TCR repertoire analysis using molecular techniques indirectly supports the presence of pathologically expanded T-cell clones that might be involved in the immune-mediated inhibition of hematopoiesis. So far, direct functional assays were not successful in proving the pathogenic nature of expanded CD8+ T-cell clones or determining the disease specificity. Conceptually, a differential recognition or high killing efficiency would be expected if the expanded clones directly target hematopoietic cells. However, due to the lack of appropriate HLA-matched controls and the inherent difficulty of obtaining sufficient numbers of autologous target cells, such experiments are extremely challenging and have not been successful so far. In the future, the analysis of CDR3 amino acid sequence patterns may be used for structural comparisons of CTL clones in bone marrow failure syndromes and may find broad applicability in measuring general CTL responses in other conditions, including autoimmune diseases and bone marrow transplantation settings. TCR repertoire assays can facilitate the design of T-cell-specific tests and therapies for some MDS and AA patients.
Prepublished online as Blood First Edition Paper, April 13, 2006; DOI 10.1182/blood-2005-09-3902.
Supported in part by a grant from Aplastic Anemia and Myelodysplastic Syndrome (AA & MDS) Foundation and National Institutes of Health (NIH) RO1 HL073429-01A1 (J.P.M.).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Molldrem JJ, Caples M, Mavroudis D, Plante M, Young NS, Barrett AJ. Antithymocyte globulin for patients with myelodysplastic syndrome. Br J Haematol. 1997;99: 699-705. [DOI] [PubMed] [Google Scholar]
- 2.Barrett J, Saunthararajah Y, Molldrem J. Myelodysplastic syndrome and aplastic anemia: distinct entities or diseases linked by a common pathophysiology? Semin Hematol. 2000;37: 15-29. [DOI] [PubMed] [Google Scholar]
- 3.Molldrem JJ, Jiang YZ, Stetler-Stevenson M, Mavroudis D, Hensel N, Barrett AJ. Haematological response of patients with myelodysplastic syndrome to antithymocyte globulin is associated with a loss of lymphocyte-mediated inhibition of CFU-GM and alterations in T-cell receptor Vbeta profiles. Br J Haematol. 1998;102: 1314-1322. [DOI] [PubMed] [Google Scholar]
- 4.Epperson DE, Nakamura R, Saunthararajah Y, Melenhorst J, Barrett AJ. Oligoclonal T cell expansion in myelodysplastic syndrome: evidence for an autoimmune process. Leuk Res. 2001;25: 1075-1083. [DOI] [PubMed] [Google Scholar]
- 5.Lamy T, Loughran TP Jr. Current concepts: large granular lymphocyte leukemia. Blood Rev. 1999; 13: 230-240. [DOI] [PubMed] [Google Scholar]
- 6.Go RS, Lust JA, Phyliky RL. Aplastic anemia and pure red cell aplasia associated with large granular lymphocyte leukemia. Semin Hematol. 2003; 40: 196-200. [DOI] [PubMed] [Google Scholar]
- 7.Saunthararajah Y, Molldrem JL, Rivera M, et al. Coincident myelodysplastic syndrome and T-cell large granular lymphocytic disease: clinical and pathophysiological features. Br J Haematol. 2001;112: 195-200. [DOI] [PubMed] [Google Scholar]
- 8.Plasilova M, Risitano A, Maciejewski JP. Application of the molecular analysis of the T cell receptor repertoire in the study of immune-mediated hematologic disease. Hematol J. 2003;8: 173-181. [DOI] [PubMed] [Google Scholar]
- 9.Wlodarski M, O'Keefe CL, Washawski I, Loughran TP, Rodriguez A, Maciejewski JP. Pathologic clonal cytotoxic T cell responsesnon random nature of the T cell receptor in large granular lymphocytic leukemia. Blood. 2005;106: 2769-2780. [DOI] [PubMed] [Google Scholar]
- 10.O'Keefe CL, Plasilova M, Wlodarski M, et al. Molecular analysis of TCR clonotypes in LGL: a clonal model for polyclonal responses. J Immunol. 2004;172: 1960-1969. [DOI] [PubMed] [Google Scholar]
- 11.Borg NA, Ely LK, Beddoe T, et al. The CDR3 regions of an immunodominant T cell receptor dictate the `energetic landscape' of peptide-MHC recognition. Nat Immunol. 2005;6: 171-180. [DOI] [PubMed] [Google Scholar]
- 12.Padovan E, Casorati G, Dellabona P, Meyer S, Brockhaus M, Lanzavecchia A. Expression of two T cell receptor alpha chains: dual receptor T cells. Science. 1993;262: 422-424. [DOI] [PubMed] [Google Scholar]
- 13.Risitano AM, Kook H, Zeng W, Chen G, Young NS, Maciejewski JP. Oligoclonal and polyclonal CD4 and CD8 lymphocytes in aplastic anemia and paroxysmal nocturnal hemoglobinuria measured by V beta CDR3 spectratyping and flow cytometry. Blood. 2002;100: 178-183. [DOI] [PubMed] [Google Scholar]
- 14.Kondo Y, Shiobara S, Nakao S. Identification of T-cell clones showing expansion associated with graft-vs-leukemia effect on chronic myelogenous leukemia in vivo and in vitro. Exp Hematol. 2001; 29: 471-476. [DOI] [PubMed] [Google Scholar]
- 15.Peggs K, Verfuerth S, Pizzey A, Ainsworth J, Moss P, Mackinnon S. Characterization of human cytomegalovirus peptide-specific CD8(+) T-cell repertoire diversity following in vitro restimulation by antigen-pulsed dendritic cells. Blood. 2002;99: 213-223. [DOI] [PubMed] [Google Scholar]
- 16.Nakao S, Takami A, Takamatsu H, et al. Isolation of a T-cell clone showing HLA-DRB1*0405-restricted cytotoxicity for hematopoietic cells in a patient with aplastic anemia. Blood. 1997;89: 3691-3699. [PubMed] [Google Scholar]
- 17.Kook H, Zeng W, Guibin C, Kirby M, Young NS, Maciejewski JP. Increased cytotoxic T cells with effector phenotype in aplastic anemia and myelodysplasia. Exp Hematol. 2001;29: 1270-1277. [DOI] [PubMed] [Google Scholar]
- 18.Zeng W, Maciejewski JP, Chen G, Young NS. Limited heterogeneity of T cell receptor BV usage in aplastic anemia. J Clin Invest. 2001;108: 765-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karadimitris A, Manavalan JS, Thaler HT, et al. Abnormal T-cell repertoire is consistent with immune process underlying the pathogenesis of paroxysmal nocturnal hemoglobinuria. Blood. 2000;96: 2613-2620. [PubMed] [Google Scholar]
- 20.Plasilova M, Risitano AM, O'Keefe CL, et al. Shared and individual specificities of immunodominant cytotoxic T-cell clones in paroxysmal nocturnal hemoglobinuria as determined by molecular analysis. Exp Hematol. 2004;32: 261-269. [DOI] [PubMed] [Google Scholar]
- 21.Kook H, Risitano AM, Zeng W, et al. Changes in T-cell receptor VB repertoire in aplastic anemia: effects of different immunosuppressive regimens. Blood. 2002;99: 3668-3675. [DOI] [PubMed] [Google Scholar]
- 22.Risitano AM, Maciejewski JP, Muranski P, et al. Large granular lymphocyte (LGL)-like clonal expansions in paroxysmal nocturnal hemoglobinuria (PNH) patients. Leukemia. 2005;19: 217-222. [DOI] [PubMed] [Google Scholar]
- 23.Kochenderfer JN, Kobayashi S, Wieder ED, Su C, Molldrem JJ. Loss of T-lymphocyte clonal dominance in patients with myelodysplastic syndrome responsive to immunosuppression. Blood. 2002;100: 3639-3645. [DOI] [PubMed] [Google Scholar]
- 24.Matsutani T, Yoshioka T, Tsuruta Y, et al. Determination of T-cell receptors of clonal CD8-positive T-cells in myelodysplastic syndrome with erythroid hypoplasia. Leuk Res. 2003;27: 305-312. [DOI] [PubMed] [Google Scholar]
- 25.Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51: 189-199. [PubMed] [Google Scholar]
- 26.Berliner N, Duby AD, Linch DC, et al. T cell receptor gene rearrangements define a monoclonal T cell proliferation in patients with T cell lymphocytosis and cytopenia. Blood. 1986;67: 914-918. [PubMed] [Google Scholar]
- 27.Semenzato G, Zambello R, Starkebaum G, Oshimi K, Loughran TP Jr. The lymphoproliferative disease of granular lymphocytes: updated criteria for diagnosis. Blood. 1997;89: 256-260. [PubMed] [Google Scholar]
- 28.Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. A systematic approach to diagnosis of mature T-cell leukemias reveals heterogeneity among WHO categories. Blood. 2004; 104: 328-335. [DOI] [PubMed] [Google Scholar]
- 29.van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17: 2257-2317. [DOI] [PubMed] [Google Scholar]
- 30.Tbakhi A, Totos G, Pettay JD, Myles J, Tubbs RR. The effect of fixation on detection of B-cell clonality by polymerase chain reaction. Mod Pathol. 1999;12: 272-278. [PubMed] [Google Scholar]
- 31.International Immunogenetics Information System. IMGT/V-QUEST. http://imgt.cines.fr/cgi-bin/IMGTdnap.jv?livret = 0&Option = humanTcR. Accessed September 2005.
- 32.International Immunogenetics Information System. IMGT. http://imgt.cines.fr. Accessed September 2005.
- 33.Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet. 2004;364: 355-364. [DOI] [PubMed] [Google Scholar]
- 34.Akashi K, Shibuya T, Taniguchi S, et al. Multiple autoimmune haemopoietic disorders and insidious clonal proliferation of large granular lymphocytes. Br J Haematol. 1999;107: 670-673. [DOI] [PubMed] [Google Scholar]
- 35.Bowman SJ, Bhavnani M, Geddes GC, et al. Large granular lymphocyte expansions in patients with Felty's syndrome: analysis using anti-T cell receptor V beta-specific monoclonal antibodies. Clin Exp Immunol. 1995;101: 18-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lima M, Almeida J, Santos AH, et al. Immunophenotypic analysis of the TCR-Vbeta repertoire in 98 persistent expansions of CD3(+)/TCR-alpha-beta(+) large granular lymphocytes: utility in assessing clonality and insights into the pathogenesis of the disease. Am J Pathol. 2001;159: 1861-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muraro PA, Wandinger KP, Bielekova B, et al. Molecular tracking of antigen-specific T cell clones in neurological immune-mediated disorders. Brain. 2003;126(Pt 1): 20-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michalek J, Collins RH, Hill BJ, Brenchley JM, Douek DC. Identification and monitoring of graft-versus-host specific T-cell clone in stem cell transplantation. Lancet. 2003;361: 1183-1185. [DOI] [PubMed] [Google Scholar]