

Abstract
Retroviral vectors with long terminal repeats (LTRs), which contain strong enhancer/promoter sequences at both ends of their genome, are widely used for stable gene transfer into hematopoietic cells. However, recent clinical data and mouse models point to insertional activation of cellular proto-oncogenes as a dose-limiting side effect of retroviral gene delivery that potentially induces leukemia. Self-inactivating (SIN) retroviral vectors do not contain the terminal repetition of the enhancer/promoter, theoretically attenuating the interaction with neighboring cellular genes. With a new assay based on in vitro expansion of primary murine hematopoietic cells and selection in limiting dilution, we showed that SIN vectors using a strong internal retroviral enhancer/promoter may also transform cells by insertional mutagenesis. Most transformed clones, including those obtained after dose escalation of SIN vectors, showed insertions upstream of the third exon of Evi1 and in reverse orientation to its transcriptional orientation. Normalizing for the vector copy number, we found the transforming capacity of SIN vectors to be significantly reduced when compared with corresponding LTR vectors. Additional modifications of SIN vectors may further increase safety. Improved cell-culture assays will likely play an important role in the evaluation of insertional mutagenesis.
Introduction
Hematopoietic cells are important targets for somatic gene therapy, considering their availability for in vitro manipulation and their enormous functional capacity. In selected diseases, hematopoietic gene therapy has clearly shown clinical efficacy, creating new perspectives for the entire field.1-4 Because of the high proliferative potential of hematopoietic cells, stable introduction of transgenes into cellular chromosomes is required for successful genetic modification. Retroviral (including lentiviral) vectors confer a predictable efficiency of stable transgene insertion with a controlled copy number.5,6 However, secondary leukemias have been reported, in animal models7-9 and in a clinical trial,10 in which insertional activation of cellular proto-oncogenes by inserted retroviral vectors represented the initiating event. To overcome the present uncertainty in the scientific and regulatory communities, systematic research must be conducted to address the frequency of insertional mutagenesis, the role of contributing factors, and the impact of vector design.6 Indeed, all cases of leukemogenic complications observed to date in clinical trials or animal models of gene therapy involved the use of conventional retroviral vectors with long terminal repeats (LTRs) containing strong enhancer/promoters. This configuration is derived from their strongly leukemogenic parental viruses and may trigger distant enhancer interactions and activation of 3′ located genes by promoter insertion.11 Self-inactivating (SIN) retroviral vectors that contain only one internal enhancer/promoter should reduce the incidence of interactions with nearby cellular genes.6 Experimental evidence supporting this hypothesis would have important implications for the design of future clinical trials.
Considering random vector insertion and a hypothetical “vulnerable region” of 10 kb that might lead to up-regulation of a neighboring proto-oncogene after retroviral vector insertion, the risk of activating insertions in a given proto-oncogene per treated cell might be in the order of 10-5. In line with such predictions, we showed that vector dose escalation can initiate murine leukemias containing combinatorial proto-oncogene activations with a frequency approaching 1 in 1 million treated bone marrow cells.8 Insertional mutagenesis by retroviral vectors may induce a competitive growth advantage to murine bone marrow cells in vivo, allowing the identification of genes regulating stem-cell turnover.12 Accordingly, insertional mutagenesis by retroviral vectors allows the identification of genes that immortalize murine bone marrow cells in vitro.13 In the present study, we took advantage of these findings to develop rapid mutagenesis assays, starting from primary murine bone marrow cells that were transduced with a known multiplicity of infection (MOI). Our data reveal that SIN vectors carrying a strong internal enhancer/promoter may transform primary hematopoietic cells by insertional mutagenesis, though with significantly lower frequency than their LTR-driven counterparts.
Materials and methods
Retroviral vectors and vector production
The retroviral vector SF91-eGFP-wPre (LTRSF; Figure 1B) has been described previously.14 SF91 contains the spleen focus-forming virus LTR (GenBank accession no. AJ224005), the posttranscriptional regulatory element (wPRE) from woodchuck hepatitis virus, and encodes either DsRed2 or eGFP. Retroviral self-inactivating (SinSF) vectors were recently described.15 Briefly, the 3′ U3 region is devoid of all enhancer and promoter elements, leaving the integrase attachment site intact. As an internal promoter, the identical U3 region from the LTR vector was inserted to express DsRed2 or eGFP. Cell-free supernatants were generated by transient transfection of Phoenix-gp packaging cells (kindly provided by G. Nolan, Stanford University, Stanford, CA) with packaging constructs coding for the gag-pol proteins (M57) and the ecotropic envelope.16 Viral titers determined on SC1 fibroblasts were in the range of 106 to 107 infectious U/mL unconcentrated supernatant, depending on the vector backbone and the transgene used. All experiments were performed with thawed vector stocks of known titers (Table 1).
Figure 1.
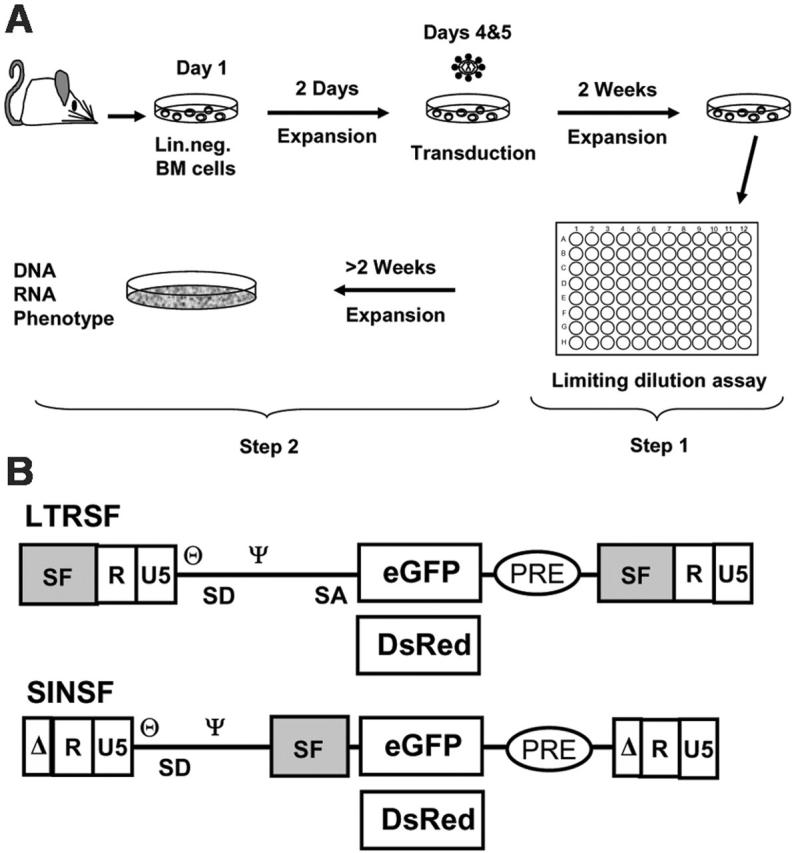
Experimental setup and vectors. (A) Murine Lin- cells were isolated, prestimulated, and transduced with cell-free vector supernatants with the use of MOIs, as indicated in Table 1. The cells were expanded as a mass culture for 2 weeks and subsequently selected on a 96-well plate (step 1). Randomly picked clones were further expanded to numbers exceeding 106 for phenotyping and to harvest DNA and RNA (step 2). (B) Retroviral vectors used for transduction shown as proviruses. LTRSF is an LTR-driven retroviral vector that has been previously described (SF91).14 It contains a splice-competent leader region, including the primer binding site (θ) and the packaging signal (Ψ), and encodes either eGFP or DsRed. The U3 region containing all the enhancer/promoter elements is derived from spleen focus-forming virus (SF). SINSF is a self-inactivating (SIN) retroviral vector.15 The U3 region is almost completely deleted, leaving only the integrase attachment sites intact. eGFP and DsRed are driven by the SF enhancer/promoter, identical to the cis-elements used in the LTR-driven vector.
Table 1.
Overview of experiments performed in this study
| Vector, MOI | Titer of supernatant, 106/mL | Treated cells | Positive wells on 96-well plate | Frequency of replating cells | No. clones of analyzed samples | Mean copy no., d 7 qPCR |
|---|---|---|---|---|---|---|
| Pilot experiment | ||||||
| LTRSFdsRED, 5 | 3 | 1 × 106 | 56; 81 | 1 in 80 | 1 ; 3 | ND |
| Experiment 1 | ||||||
| LTRSFeGFP, 20 | 15 | 1 × 105 | 50; 70 | 1 in 102 | 2 ; 9 | ND |
| SINSFeGFP, 20 | 5 | 1 × 105 | 2; 3 | 1 in 3790 | ND | ND |
| Mock, 0 | — | 1 × 105 | 4; 5 | < 1 in 2083 | ND | ND |
| Experiment 2 | ||||||
| LTRSFeGFP, 20 | 20 | 1 × 105 | 96; 96 | > 1 in 22 | 2 ; 4 | 11.6 |
| SINSFeGFP, 20 | 5 | 1 × 105 | 0; 6 | 1 in 3150 | ND | 2.9 |
| LTRSFdsRed, 20 | 23 | 1 × 105 | 96; 96 | > 1 in 22 | 2 ; 4 | 13.9 |
| SINSFdsRed, 20 | 5 | 1 × 105 | 0; 0 | < 1 in 9550 | ND | 5.4 |
| Mock, 0 | — | 1 × 105 | 0 | < 1 in 9550 | ND | ND |
| Experiment 3 | ||||||
| LTRSFeGFP, 20 | 8 | 1 × 105 | 52; 40 | 1 in 153 | 1 ; 10 | 3 |
| SINSFeGFP, 20 | 9 | 1 × 105 | 1; 0 | < 1 in 9550 | ND | 3.1 |
| Mock, 0 | — | 1 × 105 | 0; 0 | < 1 in 9550 | ND | ND |
| Experiment 4 | ||||||
| LTRSFeGFP, 20 | 9 | 1 × 105 | 42; 40 | 1 in 180 | 3 ; 3 | 6.4 |
| LTRSFeGFP, 10 | 9 | 1 × 105 | 20 | 1 in 428 | ND | 4.2 |
| LTRSFeGFP, 5 | 9 | 1 × 105 | 15 | 1 in 589 | ND | 2.2 |
| LTRSFeGFP, 2.5 | 9 | 1 × 105 | 15 | 1 in 589 | ND | 0.9 |
| SINSFeGFP, 20 | 10 | 1 × 105 | 32; 34 | 1 in 237 | 2 ; 6 | 4.4 |
| Mock, 0 | — | 1 × 105 | 0 | < 1 in 9550 | ND | ND |
| Experiment 5 | ||||||
| LTRSFeGFP, 20 | 10 | 1 × 105 | 96 | > 1 in 22 | ND | 4 |
| LTRSFeGFP, 5 | 10 | 1 × 105 | 31 | 1 in 256 | ND | 1.5 |
| LTRSFeGFP, 1.25 | 10 | 1 × 105 | 0 | < 1 in 9550 | ND | 0.6 |
| SINSFeGFP, 20 | 10 | 1 × 105 | 13 | 1 in 687 | 1 ; 5 | 1.7 |
| SINSFeGFP, 5 | 10 | 1 × 105 | 0 | < 1 in 9550 | ND | 0.6 |
| SINSFeGFP, 1.25 | 10 | 1 × 105 | 0 | < 1 in 9550 | ND | 0.5 |
| Mock | — | 1 × 105 | 0 | < 1 in 9550 | ND | ND |
Titer was determined by limiting dilution on SC-1 fibroblasts and was adjusted before the transduction ; bone marrow cells. Pairs of numbers separated by semicolons result from duplicate determinations
ND indicates not determined; —, not applicable
Isolation of lineage-negative bone marrow cells and transduction
Lineage-negative (Lin-) bone marrow (BM) cells of untreated C57Bl6/J mice were transduced as previously described.17 Briefly, Lin- cells were isolated from complete BM by magnetic sorting using lineage-specific antibodies (Gr1, CD11b, CD45R/B220, CD3e, TER119; PharMingen, Hamburg, Germany) and were cryopreserved in aliquots. Before retroviral transduction, Lin- BM cells were prestimulated for 2 days (Figure 1A) in StemSpan HS2000 medium (CellSystems, St Katharinen, Germany) containing 50 ng/mL mSCF, 100 ng/mL hFlt-3 ligand, 100 ng/mL hIL-11, 10 ng/mL mIL-3 (PeproTech, London, United Kingdom), 1% penicillin/streptomycin, and 2 mM glutamine at a density of 1-5 × 105 cells/mL. Cells were transduced on day 4 using cell numbers and an MOI as indicated in “Results.” Virus preloading was carried out on RetroNectin-coated (10 μg/cm2; TaKaRa, Otsu, Japan) suspension culture dishes by spinocculation for 30 minutes at 4°C. Typically, 1 × 105 cells were cultured in a single well of a 24-well plate, and the culture volume was 500 μL on day 4 and 1 mL on day 5 to account for increasing cell numbers. On day 5, cells were transferred to freshly prepared plates preloaded with RetroNectin and retroviral vector for a second transduction. If retroviral supernatants used in side-by-side comparisons had different titers, the supernatant with the higher titer was diluted with supernatant harvest medium so that identical volumes were used for preloading. Transgene expression was measured on both days after transduction (days 5 and 6), to control for equal transduction efficiency in all cultures.
Replating assays
After retroviral transduction, BM cells were expanded as mass cultures for 2 weeks in StemSpan medium containing 50 ng/mL mSCF, 100 ng/mL hFlt-3 ligand, 100 ng/mL hIL-11, 10 ng/mL mIL-3, 1% penicillin/streptomycin, and 2 mM glutamine. During this time, cell density was adjusted to 5 × 105 cells/mL every 3 days, and medium was gradually shifted to IMDM, 10% FCS, 1% penicillin/streptomycin, and 2 mM glutamine containing the same cytokines described. After mass culture expansion for 14 days BM cells were plated into 96-well plates at a density of 100 cells/well (also at 10 cells/well in some experiments). Two weeks later the positive wells were counted, and the frequency of replating cells was calculated based on Poisson statistics using L-Calc software (Stem Cell Technologies, Vancouver, BC, Canada). Selected clones were expanded for further characterization.
Phenotypic characterization of clonal cells
Cell surface markers of the cells from clones generated in the mutagenesis assay were characterized by FACS analysis using antibodies for cell-surface markers (antibodies specific for the surface markers CD11b, Gr1, B220, CD3, Ter119, Sca1, and c-Kit). Cell morphology was analyzed on cytospin slides stained with May-Grünwald/Giemsa solution.
Southern blot analysis
Genomic DNA for Southern blot analysis was isolated from cells of expanded clones using the blood DNA separation kit (Qiagen, Hilden, Germany). Ten micrograms genomic DNA was digested with appropriate enzymes, and Southern blot analysis was performed according to standard protocols. Detection was carried out with α32P-labeled DNA probe corresponding to the eGFP or DsRed2 cDNA, the wPRE, or the Flk1 intronic enhancer (accession no. AF061804; 500-bp probe).
PCR
Ligation-mediated polymerase chain reaction (LM-PCR) and linear amplification-mediated (LAM-PCR) used to obtain integration sites of LTR vectors were performed as described8,18,19 with 300 to 500 ng genomic DNA. To amplify integration sites of SIN vectors, 3′ LM-PCR was established using a primer that annealed in the wPRE sequence (SINPRE; 5′-[bio]GCACTGATAATTCCGTGGTGTTGTC-3′). For exponential PCR, the following primers were designed: SIN LTR2, 5′-GATATCGAATTCACAACC-3′; SIN LTR3, 5′-CCAATAAAGCCTCTTGCTGT-3′. Linker and linker primers were used as described,8,18,19 with small modifications: linker, 5′-GACCCGGGAGATCTGAATTCGAGTGGCACAGCAGTTAGGACG-3′; linker primer 1, 5′-GACCCGGGAGATCTGAATTCG-3′; linker primer 2, 5′-AGTGGCACAGCAGTTAGGACG-3′. LM and LAM amplicons were isolated, purified, and directly sequenced or they were shotgun cloned into the TOPO TA vector (Invitrogen, Carlsbad, CA) and sequenced. A 37-bp sequence in the unprimed LTR was used to verify the specificity and correctness of the insertion sites sequenced. Quantitative PCR for the wPRE was performed on an i-Cycler (Bio-Rad, Hercules, CA) using QuantiTect SYBR Green (Qiagen). The wPRE-specific primers (forward, 5′-GAGGAGTTGTGGCCCGTTGT-3′; reverse, 5′-TGACAGGTGGTGGCAATGCC-3′) amplified a 94-bp fragment. The wPRE-specific signal was normalized by the signal of a housekeeping gene (Flk1 intron enhancer, gene ID AF061804, bases 352-459, forward 5′-GTGAATTGCAGAGCTGTGTGTTG-3′ and reverse 5′-ATTCATTGTATAAAGGTGGGATTG-3′). Results were quantified using the comparative CT method. For quantitative RT-PCR, RNA was extracted from expanded mutant clones using Trizol (Invitrogen) and the RNAeasy kit (Qiagen). Reverse transcription was performed with 1 to 2 μg RNA using PowerScript MLV reverse transcriptase, and real-time PCR for deregulation of cellular genes using Assay-on-Demand (Applied Biosystems, Foster City, CA).
Results
Murine bone marrow cells acquire a growth advantage in vitro by retroviral transduction
To evaluate insertional mutagenesis by retroviral gene transfer in a relevant target cell type, we transduced murine Lin- BM cells harvested with a purity of greater than 90% from steady state hematopoiesis of C57Bl6 mice. Using a protocol that allows efficient and dose-controlled retroviral gene transfer in serum-free, cytokine-supplemented expansion cultures,17 cells were treated with ecotropic retroviral particles used at a defined multiplicity of infection (MOI). Two rounds of transduction were performed on days 4 and 5 of the expansion culture, and gene-transfer rates were monitored by flow cytometry. After retroviral gene transfer, cells were grown in cytokine-supplemented media for another 2 weeks before they were replated in 96-well plates. Under these conditions, mock-treated cells barely survived.
In a pilot experiment (Table 1), we transduced 106 Lin- cells using an MOI of 5 with replication-defective retroviral LTR vectors (construct SF91) that expressed the DsRed2 red fluorescent protein.8 After initial bulk culture for 14 days and replating in 96-well plates, we obtained a large number of wells with proliferating cell populations (step 1, replating; Figure 1A). Cells isolated from individual wells of the 96-well plates were further expanded for preparation of DNA, RNA, flow cytometry, and cytospin analyses (step 2, expansion; Figure 1A). In subsequent experiments, we reduced the initial cell numbers to 105 per assay, thus saving experimental animals and expenses for serum-free media and recombinant cytokines and allowing the MOI to increase up to 2 × 10 without vector concentration. Under these conditions, we reproducibly recovered clones when replating cells after transduction with high vector doses (Table 1).
Normalization of expression levels and transduction efficiency of SIN and LTR vectors
To determine whether the SIN architecture reduces the risk for insertional side effects, we used γ-retroviral SIN vectors in which the internal expression cassette is under control of the same retroviral enhancer/promoter sequences that constitute the U3 region of the LTR-driven constructs.15 Two pairs of ecotropic vectors (Figure 1B) expressing either eGFP or DsRed2 were produced and used with identical MOIs for subsequent experiments. As described earlier,15 the SIN vectors expressed levels of eGFP or DsRed2 similar to those of their LTR counterparts, ruling out that potential differences in transforming capacity would be related to transgene-expression levels. PCR analysis confirmed the deletion of the U3 region in the SIN vector LTRs after retroviral transduction of bone marrow cells (data not shown).
Equal infectivity of the SIN constructs was suggested by flow cytometry of Lin- cells after retroviral gene transfer (Figure 2, Table 1). However, quantitative PCR data obtained on day 7 after transduction revealed that transduction with SIN vectors often resulted in lower average copy numbers compared with LTR vectors used at the same MOI. In contrast, studies in 32D cells revealed equal infectivity of SIN and LTR vectors with consistent data obtained by flow cytometry, Southern blot, and real-time PCR (Table S1 and Figures S1-S2, available on the Blood website; see the Supplemental Materials link at the top of the online article). The mechanism underlying the trend to higher average transgene copy numbers obtained with LTR vectors in primary cells remains to be elucidated. We thus determined transgene copy number on day 7 after the last exposure to vector particles by real-time PCR in all subsequent experiments (Table 1). Day 7 was chosen for the real-time PCR studies to ensure the absence of episomal vector copies and potential plasmid contaminations resulting from the use of supernatants produced by transient transfection of packaging cells.
Figure 2.

FACS analyses of the transduction, selection, and expansion process. eGFP expression and percentage of positive cells of SIN- and LTR-transduced BM cells 1 day after the second transduction (top panel), after 2 weeks (middle panel), and after selection of a single clone (bottom panel). Representative data obtained in experiment 3 and experiment 4 (FACS analysis of the SIN vector-transduced clone).
LTR vector transduction induces significantly higher frequencies of replating cells than SIN vector transduction
After transduction at a high MOI (2 × 10), greater than 90% of the cells were transduced with LTR and SIN vectors (representative data shown in Figure 2). Importantly, when cells were transduced with SIN vectors, replating cells (Figure 1A, step 1) were obtained with reduced frequency. To account for potential differences in infectivity, we calculated the ratio of replating frequency per vector copy. On summarizing all experiments in which this value could be calculated (Table 1), the ratio was 0.00276 ± 0.00310 (n = 10) for LTR vectors and 0.00023 ± 0.00029 for SIN vectors (n = 6). An unpaired 2-sided t test revealed the difference to be statistically significant (P = .037), despite interexperimental variability (Figure 3). This interexperimental variability may be explained by the variable vector copy number found in replating clones (Figure 4) and by experimental parameters related to the culture of primary cells. Of note, the replating frequency reflects a combination of the number of independent mutants and their competitive growth in the initial bulk culture. This implies that the average frequency of replating cells does not directly reflect the number of distinct mutants. Therefore, the 12-fold numeric difference in the average frequency of replating cells per copy number between LTR and SIN vectors must not be overinterpreted. To reduce interexperimental variability, we attempted to split the cultures into 96-well plates at low density immediately after transduction but were unsuccessful. These conditions might have been too stressful to allow for outgrowth of insertional mutants.
Figure 3.

Replating ability of Lin- cells depends on vector design. Correcting the frequency of replating cells for the average vector copy number detected on day 7 after transduction, LTR vectors showed a significantly increased risk for transforming Lin- cells to acquire replating potential. The median is indicated as a thick black line. If no clones were obtained, the frequency of 1 in 9550 was taken for calculation; if all 96 wells contained replating cells, the frequency was estimated as (at least) 1 in 22 cells.
Figure 4.

Genetic analyses of insertional mutants obtained after step 2. (A) Representative Southern blot analysis using 10 μg genomic DNA of expanded clones. Only selected clones of each experiment are shown displaying a different band pattern within each blot. The cDNA for eGFP or DsRed or a fragment spanning the wPRE were used for probing. Clone names are indicated above each lane. (B) Location of insertions into the Evi1 allele. The Mds1 locus lies further upstream in the same transcriptional orientation. LTR and SIN vectors recovered in our in vitro studies show the same pattern, consistent with our previous findings made with LTR vectors in dominant or leukemic clones in vivo.7,8,12 (C) Quantitative real-time PCR was used to determine the transcript level of Evi1 or HoxA7. Bars represent the relative enhancement compared with expression levels in mock-transduced and expanded cells. Clone 6.4 contains a vector insertion upstream of HoxA7, whereas clone C4 does not. Each PCR was performed in triplicate, and bars represent the mean of 3 CT values. Values for clones 1.5 and a1 represent the average of 2 independent determinates performed in triplicate.
The minimal average vector copy number (determined on day 7) capable of triggering replating clones was 0.9 for the LTR vector and 1.7 for the SIN vector (Table 1). For LTR vectors, average copy numbers greater than 1 always gave rise to replating cells (8 of 8 experiments), whereas results achieved with the SIN vector were more variable (2 of 4 experiments). Because we normalized the frequency of replating cells for the vector copy number detected on day 7 after gene transfer, a firm conclusion can be drawn that transduction with LTR vectors significantly increases replating ability of primary murine bone marrow cells when compared with SIN vectors containing the same enhancer/promoter in an internal position. However, our data also revealed that SIN vectors were not free of transforming potential when used at a high MOI in primary murine bone marrow cells.
Insertional mutagenesis is the driving force of in vitro transformation and can also be detected after the use of SIN vectors
To examine whether insertional mutagenesis represented the driving force of the enhanced fitness detected in the replating assays, cells had to be further expanded before sufficient DNA were obtained for Southern blot, LM-PCR, cellular RNA, and live cells for phenotyping studies. Importantly, only a subset (80%) of the cells surviving the replating in 96-well plates could be successfully expanded to numbers exceeding 106 (Figure 1A, step 2). These clones showed high levels of marker-gene expression, irrespective of the vector used (Figure 2). One of these clones (eGFP clone B; Table 2) showed robust growth and could be kept as an immortal culture. This clone also showed the most primitive cytology (Figure 5A) and the highest frequency of cells coexpressing c-Kit and Sca1 (Figure 5B). All clones showed a high contribution of cells expressing myeloid lineage antigens (Figure 5B). More mature cells of granulocytic morphology were not observed. Erythroid or lymphoid markers were not detected by flow cytometry (Ter119, CD3, B220; data not shown).
Table 2.
Insertion sites in dominant clones
| Clone (recovered loci) and loci of nearest genes hit | Gene ID | Chromosome | Distance to TSS | Ori | Predicted gene function | RTCGD |
|---|---|---|---|---|---|---|
| dsRED 6.4 (7 of 9) | ||||||
| Lgals1 | 16852 | 15E1 | –3888 | F | Lectin, galactose-binding, soluble 1 | — |
| Itsn1 | 16443 | 16C3+3 | 9059, intron 3 | F | Intersectin 1 | — |
| Mamdc1 | 320772 | 12C1 | 517643, intron 5 | F | Integral membrane protein | — |
| 23100075C12Rik | 71929 | 9A1 | –830 | R | Unknown | — |
| 2900091E11Rik | 67282 | 10C1 | 693 | F | Unknown | — |
| HoxA7 | 15404 | 6B2+3 | –737 | R | Transcription factor | 21 |
| Evi1 | 14013 | 3A3 | –118152 | R | Transcription factor | 21 |
| dsRED 1.5 (3 of 4) | ||||||
| Gstm2 | 14863 | 3F2.3 | –18239 | F | Glutathione-S-transferase | — |
| C80879 | 26374 | 1F | –139200 | R | Unknown | — |
| Cxxc6 | 52463 | 10B4 | 15 079, intron 1 | R | CXXC finger 6 | — |
| dsRED D2 (6 of 7) | ||||||
| Fdps | 110196 | 3F1 | –1274 | F | Farnesyl diphosphate synthetase | — |
| Pcna | 18538 | 2E1 | 4869, downstream | R | Proliferating nuclear cell antigen | — |
| Prkab-1 | 19079 | 5F | 1206, intron 1 | F | Protein kinase, AMP/activated, β1 noncatalytic subunit | — |
| Adora3 | 11542 | 3F2.3 | –6558 | R | Adenosine A3 receptor | — |
| Evi1 | 14013 | 3A3 | –18478 | R | Transcription factor | 21 |
| Recq15 | 170472 | 11E2 | 21755, intron 9 | F | Helicase activity | 1 |
| eGFP A2 (3 of 7) | ||||||
| A2300063L24 | 319555 | 8B3.3 | 12572, intron 2 | F | Unknown | — |
| Evi1 | 14013 | 3A3 | –56381 | R | Transcription factor | 21 |
| Map3k7ip2 | 68652 | 10A1 | –142950 | R | Mitogen-activated protein kinase kinase kinase 7 interacting protein 2 | — |
| eGFP a1 (3 of 3) | ||||||
| 2310015N21Rik | 76438 | 17C | 169113 intron 14 | R | Unknown | — |
| Evi1 | 14013 | 3A3 | –117497 | R | Transcription factor | 21 |
| Hbs1-like | 56422 | 10A3 | –11793 | R | Eukaryotic release factor; Myb is on the other side | — |
| eGFP 1.8 (4 of 7) | ||||||
| Zfpn1a2 | 22779 | 1C3 | 2350, intron 2 | R | Zinc fingers and homeobox protein 1 (Helios, dimerization partner to Ikoras) | — |
| Mtrf1 | 211253 | 14D3 | 27609, downstream | R | Mitochondrial translational release factor 1 | — |
| LOC433776 | 433776 | 4E1 | 4525 | F | Similar to elongation factor-2 | — |
| PSAT1 | 107272 | 19A | 108469, downstream | R | Phosphoserine aminotransferase 1 | — |
| eGFP 2.5 (8 of 8) | ||||||
| Zfpn1a2 | 22779 | 1C3 | 2350, intron 2 | R | Zinc fingers and homeobox protein 1 (Helios, dimerization partner of Ikoras) | — |
| Evi1 | 14013 | 3A3 | –19126 | R | Transcription factor | 21 |
| A130034M23Rik | 320759 | 16B2 | 68750 | F | Unknown | — |
| Timm44 | 21856 | 8A1.1 | 582, intron 1 | R | Translocator of inner mitochondrial membrane 44 | — |
| Zc3h12a | 230738 | 4D2.2 | 4827, intron 2 | F | Zinc finger CCCH type containing 12A | 1 |
| Anln | 68743 | 9A3 | –914 | F | Anillin, actin-binding protein | — |
| Rpl27a | 26451 | 7E3 | –483 | R | Ribosomal protein L27a | — |
| LOC625213 | 625213 | 15B1 | 66012 | F | Unknown | — |
| eGFP cloneB (5 of 7) | ||||||
| Mad1 like1 | 17120 | 5G2 | 169941, intron 13 | F | Negative regulator of cell cycle | — |
| Polg2 | 50776 | 11E1 | 19984, downstream | R | Polymerase γ2, accessory subunit | — |
| Wdr45 | 54636 | XA1.1 | –414 | R | WD repeat domain | — |
| Evi1 | 14013 | 3A3 | –121245 | R | Transcription factor | 21 |
| Akap6 | 238161 | 12B3 | –441 | R | A kinase anchor protein 6 | — |
| SIN7 (6 of 7) | ||||||
| Slc6a6 | 98488 | 6D1 | –5179 | F | Solute carrier 6 | — |
| Lgals1 | 16852 | 15E1 | –855 | F | Lectin, galactose-binding, soluble 1 | 1 |
| BC037703 | 2446273 | 3F2.2 | 35902 | R | Unknown | — |
| Tgfbr3 | 104637 | 5E5 | 89510, intron 4 | F | TGF-β receptor 3 | — |
| Evi1 | 14013 | 3A3 | –117305 | R | Transcription factor | 21 |
| Tmem23 | 2444110 | 19C1 | –13539 | F | Transmembrane protein 23, SMS1 (sphingomyelin synthase 1) | 4 |
| SIN11 (5 of 6) | ||||||
| Slc35b4 | 58246 | 6B1 | 23596, downstream | R | Solute carrier family 35, member B4 | — |
| LOC 626636 | 626638 | 17E4 | –22650 | F | Other side SOCS5, 60331 downstream | — |
| Slfn9 | 237886 | 11 C | 17494, downstream | F | Schlafen 9 | — |
| Nos1ap | 70729 | 1 H2 | 112929, intron 3 | F | Nitric oxide synthase 1 (neuronal) adaptor protein | — |
| Evi1 | 14013 | 3A3 | –16 781 | R | Transcription factor | 21 |
| SIN1.1 (6 of 10) | ||||||
| Rasgrp4 | 233046 | 7A3 | 335, intron 1 | F | RAS guanyl-releasing protein 4 | * |
| Evi1 | 14013 | 3A3 | –116692 | R | Transcription factor | 21 |
| Lrrfip1 | 16978 | 1 D | 10 602, intron 1 | F | Leucine-rich repeat (in Fli1) interacting protein | 1 |
| 9430070013 Rik | 77352 | 1G3 | 11820, intron 5 | R | Unknown | — |
| Gse1 | 382034 | 8E1 | 47048, intron 1 | F | Genetic suppressor element 1 | 6 |
| Tbc1d23 | 67581 | 16C1.1 | 43797, intron 12 | R | TBC1 domain family, member 23 | — |
Gene hits according to NCBI mouse genome database (frozen January 2006). Insertions are defined with respect to the transcriptional start sites (TSS; mRNA start according to NCBI) of neighboring genes
Ori indicates orientation; RTCGD, retroviral tagged cancer gene database20; F, forward (with respect to the gene's transcriptional direction); R, reverse; —, no entry in the RTCGD
RTCGD lists Rasgrpl (24 hits) and Rasgrp2 (2 hits)20
Figure 5.

Phenotype of clones obtained after step 2. The “immortal” clone B has the lowest frequency of differentiating myeloid cells. (A) Cytospin preparations of mock-expanded cells and one “immortal” clone selected after transduction with LTRSFeGFP (May-Grünwald/Giemsa staining). In no case did we detect mature forms of the granulocytic lineage. Images were visualized using an Olympus BX51 upright microscope (Olympus, Hamburg, Germany) equipped with a 40 ×/0.75 numeric aperture objective lens. Images were processed using the Colorview Soft Imaging System and analySIS Five software (Olympus). (B) Clones were subjected to FACS analysis using antibodies as indicated.
Southern blot data revealed that the expanding clones obtained in a given experiment contained 3 to 10 vector insertions and were often genetically identical or shared many of their insertions (Figure 4A). Thirty-nine of the 51 insertion sites predicted for the set of LTR clones were sequenced from clones by LM-PCR and LAM-PCR18,19 and were compared with the RTCGD database of proto-oncogenes obtained in studies with replication-competent retroviruses.20 As shown in Table 2, in 6 of 8 clones examined after transduction with LTR vectors, an insertion in the Evi1 proto-oncogene was recovered. The insertion pattern (Figure 4B) was reminiscent of our previous studies in which Evi1 insertions led to its up-regulation associated with benign clonal dominance or leukemia induction in vivo.7,8,12 Real-time RT-PCR revealed up-regulation of Evi1 in all 8 clones examined after transformation by LTR vectors (Figure 4C). This included 2 clones (clones 1.5 and 1.8) in which no Evi1 insertion was detected, probably because of the incomplete recovery of insertion sites. Alternatively, high expression of Evi1 in these clones might have represented a direct or an indirect consequence of other insertional hits. These results are in agreement with a recent report13 suggesting that insertional mutagenesis is the driving force of clonal outgrowth in our experimental conditions. The Hoxa7 proto-oncogene, which harbored a promoter-proximal insertion in clone 6.4, was also found to be up-regulated (Figure 4C). Of note, some clones contained more than one insertion in a potential proto-oncogene or another gene encoding proteins involved in cellular signaling pathways (Table 2). Many clones showed additional hits in loci that were less likely to contribute to cell expansion, probably because of the high number of insertions within one clone. Differences in copy number per clone contributed to the interexperimental variability described (Figure 3).
To examine insertion sites in clones obtained after transduction with SIN vectors, we developed a modified LM-PCR, with the first primer initiating the elongation from the wPRE sequence beyond the 3′ LTR. The number of bands obtained by this approach was consistent with the Southern blot data, and sequencing of the PCR products revealed the recovery of authentic SIN vector integration junctions (data not shown). Seventeen of the 23 bands obtained from 3 clones could be sequenced. The SIN clones also showed insertions upstream of the third exon of Evi1, matching the preferred areas of LTR vector insertions (Figure 4B). The third exon contains the translational start codon of a shortened Evi1 protein, which lacks the so-called PR-domain.21 In all cases, the orientation was the reverse of the transcriptional orientation of Evi1, consistent with an enhancer-mediated interaction on Evi1. Two of the 3 clones obtained after transduction with SIN vectors also showed a strong up-regulation of Evi1 (Figure 4C). Only for clone SIN1.1 did the Evi1-expression level increase slightly compared with that of normal cells. This clone could have been transformed by 3 additional hits in potential proto-oncogenes (Table 2). Strikingly, one of the SIN clones (Table 2; SIN7) showed an insertion upstream in forward orientation to Lgals1 (encoding a soluble, galactose-binding lectin) similar to that in LTR clone 6.4 (Table 2). Although more clones must be evaluated, these data reveal that SIN vectors may hit the same genomic loci as their LTR counterparts. The almost default recovery of hits in Evi1 indicates that insertions in this locus might be mandatory to sustain expansion of serially replating clones. It is possible that the cells that did not survive step 2 of the protocol contained other insertions, not necessarily including Evi1.
Within the limitations of our data set, the frequency of sustained clonal outgrowth induced by LTR vectors at high MOI was at least 2 ± 0.7 in an initial pool of 100 000 treated Lin- cells. Given that not all wells containing live cells after step 1 were tested for expansion or were genetically analyzed, the incidence of mutagenesis obtained with LTR vectors might have been higher. To further delineate the mutation frequency, we reduced the number of initially exposed Lin- cells from 105 to 104 but could not recover any clones after transduction with the LTR vector (MOI, 2 × 10; n = 4; Table 1). Under our assay conditions, the incidence of insertional mutants obtained with LTR vectors was at least 2 ± 0.7 in 100 000 treated cells and lower than 1 in 10 000 treated cells. Although fewer clones were obtained with SIN vectors, greater numbers would have to be characterized to obtain reliable statistics on the incidence of insertional mutants, as defined by step 2 of the experiment.
We conclude that SIN vectors might have hit the same genomic loci as their LTR counterparts (Table 2; Figure 4B). Nevertheless, SIN vectors significantly reduced the frequency of insertional “side effects” compared with LTR vectors that contained the same enhancer/promoter sequences in their U3 regions. This was revealed by the lower frequency of replating cells obtained per vector copy number detected on day 7 after gene transfer (Table 1; Figure 3).
Discussion
Through the use of a novel and convenient cell-culture assay that reflected the transforming potential of insertional mutagenesis in primary murine bone marrow cells, the present study revealed that the genotoxic risk of integrating gene-transfer vectors depends on vector architecture. The vectors' transforming potential could be significantly reduced, though not eliminated, with a comparatively simple maneuver: removing the strong retroviral enhancer/promoter sequences from the LTR and placing the same sequences as a monomer into an internal position of a SIN vector. SIN vectors compensate the loss of the enhancer repetition in the LTR with improved RNA processing, thus maintaining sufficient levels of transgene expression from a single vector copy.15 The reduced number of enhancer sequences capable of long distance interactions is expected to be a major reason for reduced insertional transformation of target cells. The SIN design also prevents direct activation of downstream alleles by residual activity of the 3′ LTR promoter or read-through combined with splice interference from the 5′ LTR.
Some gene-therapy strategies need relatively high levels of transgene expression, to name only bone marrow chemoprotection,22 and antagonism of HIV infection by intracellular immunization.23,24 Our study revealed that insertional transformation remains a concern in these cases, given that SIN vectors with strong internal enhancers are used. Insulator sequences incorporated into the residual U3 region of the LTR may attenuate the genotoxic impact of such vectors.25 For those applications that do not require very high levels of transgene expression (eg, correction of metabolic disorders such as Gaucher disease), weaker internal enhancer/promoters (eg, the cellular phosphoglycerate kinase gene promoter, which is more than 5-fold less active in hematopoietic cells) would be expected to further reduce the incidence and severity of insertional side effects.
Hope for reduced insertional side effects of modified gene vectors resulted from earlier findings that lentiviruses and derived vectors integrate more frequently than γ-retroviral vectors into transcribed regions of active genes but less frequently into promoter-proximal regions.26,27 This is underscored by a large study of vector insertion sites recovered from long-term repopulating hematopoietic cells of nonhuman primates. Calmels et al28 were unable to recover EVI1 hits when using a lentiviral SIN vector harboring a strong internal retroviral enhancer/promoter. In contrast, they found a strong overrepresentation of EVI1 insertions following transduction with γ-retroviral LTR vectors,28 similar to observations in murine models.8,12,13 Replating assays as introduced by Du et al13 and modified in the present report could be used to address whether and which lentiviral vectors are capable of activating this allele. Moreover, the assays could be modified to test the impact of culture conditions triggering cell-cycle progression for gene transfer. Hematopoietic cells need significantly more stimulation for γ-retroviral than for lentiviral gene transfer.29
The present assay conditions obviously introduce a bias for clones that up-regulate Evi1 by insertional mutagenesis, at least when focusing on those clones that can be further expanded after the first replating. The selection for clones with Evi1 insertions was more pronounced than in an earlier study of Du et al,13 who used repetitive replating to establish immortal cultures of primary murine bone marrow cells after coculture with retroviral producer cells releasing a gene-marking vector. In contrast to this study, we avoided the pretreatment of donor animals with 5-fluorouracil to ensure that the assays only reflected the impact of insertional mutations, and we developed culture conditions that reported the selective advantage induced by insertional mutagenesis in a relatively short period of time (4-5 weeks). We also demonstrated that it is of great importance to work with cell-free vector supernatants of similar infectivity and to normalize the frequency of transformed cells for the average vector copy number to compare the transforming potential of different vectors.
The insertional genotoxicity assay presented here is relatively convenient, uses an appropriate readout (selective advantage under limiting-dilution conditions) and target cell population (primary hematopoietic cells), and does not require leukemia induction, thus reducing the need for prolonged animal experiments. Importantly, the sensitivity of the assay is 2 orders of magnitude higher than that reported for cell lines in which induction of growth factor independence was used as an indicator of insertional mutagenesis.30 This sensitivity may result from the fact that our assay conditions determined a combined effect of mutation frequency and fitness of transformed cells. A more precise quantification should be possible on the basis of exposed cell numbers, vector dose, and a more comprehensive determination of the number of genetically distinct clones. The sensitivity of the cell-culture assays and the spectrum of “productive hits” might be further increased when using a cell population that is even more prone to immortalization. Candidates are primary hematopoietic cells from genetically defined mouse strains that harbor transforming lesions. Preleukemic genes could also be engineered into the vector used for insertional immortalization. However, depending on promoter and copy number, this might lead to a substantial variability of vector-driven oncogene expression, potentially biasing the results.31 Another interesting outlook is the adaptation of the present assays to disease-specific settings, such as those underlying X-linked severe combined immunodeficiency. All these modifications might result in conditions that are less biased for insertions into Evi1.
Evi1 activation, which apparently was required to sustain the growth of the insertional mutants in the second step of our assay conditions, represents a clinically relevant readout: vector integrations into the human EVI1 allele have been associated with a selective advantage of gene-modified cells in patients receiving retroviral vector-mediated gene therapy for chronic granulomatous disease.4 EVI1 transcripts lacking the first 2 exons are associated with myelodysplastic syndrome and acute myeloid leukemia in humans.21 With its large size and the unusual transcriptional regulation also involving the upstream MDS1 gene,21 this locus may be a suitable target for all varieties of vectors that show a bias for expressed genes. The transforming potential of Evi1 depends on the level of up-regulation,32 underscoring the relevance of this allele to determine the impact of vector enhancer modifications. Using LTR vectors at a high MOI, our assays revealed an incidence of immortalized clones with Evi1 insertions between 10-4 and 10-5 per initially exposed Lin- cell. Considering the high MOI and the size of the vulnerable region of the Evi1 allele (greater than 100 kb), this incidence would still be consistent with a stringent selection based on random vector insertion into this allele.
In summary, improved cell-culture assays will likely play an important role in the evaluation of the functional consequences of insertional mutagenesis and the safety validation of novel vectors designed for genetic therapies. Our study suggests that optimizations of vector design are likely to significantly reduce the toxicity of gene transfer into hematopoietic stem cells.
Supplementary Material
Acknowledgments
We thank Boris Fehse (University Hospital Eppendorf, Hamburg, Germany) and Olga Kustikova (Hannover Medical School) for helpful comments and experimental advice and Anja Weigmann (Institute of Cellular and Molecular Pathology, Hannover Medical School) for consultation in statistical analyses.
Prepublished online as Blood First Edition Paper, July 6, 2006; DOI 10.1182/blood-2005-08-024976.
Supported by grants from the German Ministry for Research and Education (Programs Bioprofile and TreatID) (U.M., J.B.), the Integrated Project Concerted safety and efficiency evaluation of retroviral transgenesis (CONSERT) of the European Union (LSHB-CT-2004-005242) (S.K., A.S., M.S., C.V.K.), and the National Cancer Institute (R01-CA107492-01A2) (C.B.).
U.M. and J.B. designed and performed experiments (cell culture, clone phenotyping, and molecular biology [including Southern blot], and real-time PCR and cloning of several insertion sites by LM-PCR), performed statistical analyses, and contributed to writing the paper. M.S. and C.V.K. contributed to the cloning of retroviral insertion sites of LTR clones and database searches. S.K. performed cell-culture assays, flow cytometry and real-time PCR. A.S. cloned and produced retroviral vectors. C.B. initiated the work, designed experiments, and wrote the paper together with the above colleagues.
U.M. and J.B. contributed equally to this study.
The online version of this article contains a data supplement.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346: 1185-1193. [DOI] [PubMed] [Google Scholar]
- 2.Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002; 296: 2410-2413. [DOI] [PubMed] [Google Scholar]
- 3.Gaspar HB, Parsley KL, Howe S, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364: 2181-2187. [DOI] [PubMed] [Google Scholar]
- 4.Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12: 401-409. [DOI] [PubMed] [Google Scholar]
- 5.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4: 346-358. [DOI] [PubMed] [Google Scholar]
- 6.Baum C, Dullmann J, Li Z, et al. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood. 2003;101: 2099-2114. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Dullmann J, Schiedlmeier B, et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296: 497. [DOI] [PubMed] [Google Scholar]
- 8.Modlich U, Kustikova O, Schmidt M, et al. Leukemias following retroviral transfer of multidrug resistance 1 are driven by combinatorial insertional mutagenesis. Blood. 2005;105: 4235-4246. [DOI] [PubMed] [Google Scholar]
- 9.Seggewiss R, Pittaluga S, Adler RL, et al. Acute myeloid leukemia associated with retroviral gene transfer to hematopoietic progenitor cells of a rhesus macaque. Blood. 2006;107: 3865-3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302: 415-419. [DOI] [PubMed] [Google Scholar]
- 11.Mikkers H, Berns A. Retroviral insertional mutagenesis: tagging cancer pathways. Adv Cancer Res. 2003;88: 53-99. [DOI] [PubMed] [Google Scholar]
- 12.Kustikova OS, Fehse B, Modlich U, et al. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science. 2005;308: 1171-1174. [DOI] [PubMed] [Google Scholar]
- 13.Du Y, Jenkins NA, Copeland NG. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood. 2005;106: 3932-3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schambach A, Wodrich H, Hildinger M, Bohne J, Krausslich HG, Baum C. Context dependence of different modules for posttranscriptional enhancement of gene expression from retroviral vectors. Mol Ther. 2000;2: 435-445. [DOI] [PubMed] [Google Scholar]
- 15.Schambach A, Bohne J, Chandra S, et al. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol Ther. 2006;13: 391-400. [DOI] [PubMed] [Google Scholar]
- 16.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7: 1063-1070. [DOI] [PubMed] [Google Scholar]
- 17.Li Z, Schwieger M, Lange C, et al. Predictable and efficient retroviral gene transfer into murine bone marrow repopulating cells using a defined vector dose. Exp Hematol. 2003;31: 1206-1214. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt M, Hoffmann G, Wissler M, et al. Detection and direct genomic sequencing of multiple rare unknown flanking DNA in highly complex samples. Hum Gene Ther. 2001;12: 743-749. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt M, Zickler P, Hoffmann G, et al. Polyclonal long-term repopulating stem cell clones in a primate model. Blood. 2002;100: 2737-2743. [DOI] [PubMed] [Google Scholar]
- 20.Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32: D523-D527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nucifora G, Laricchia-Robbio L, Senyuk V. EVI1 and hematopoietic disorders: history and perspectives. Gene. 2006;368: 1-11. [DOI] [PubMed] [Google Scholar]
- 22.Milsom MD, Fairbairn LJ. Protection and selection for gene therapy in the hematopoietic system. J Gene Med. 2004;6: 133-146. [DOI] [PubMed] [Google Scholar]
- 23.Egelhofer M, Brandenburg G, Martinius H, et al. Inhibition of HIV-1 entry in cells expressing Gp41-derived peptides. J Virol. 2004;78: 568-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolkowicz R, Nolan GP. Gene therapy progress and prospects: novel gene therapy approaches for AIDS. Gene Ther. 2005;12: 467-476. [DOI] [PubMed] [Google Scholar]
- 25.Emery DW, Yannaki E, Tubb J, Stamatoyannopoulos G. A chromatin insulator protects retrovirus vectors from chromosomal position effects. Proc Natl Acad Sci U S A. 2000;97: 9150-9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300: 1749-1751. [DOI] [PubMed] [Google Scholar]
- 27.Hematti P, Hong BK, Ferguson C, et al. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004;2: e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calmels B, Ferguson C, Laukkanen MO, et al. Recurrent retroviral vector integration at the Mds1/Evi1 locus in nonhuman primate hematopoietic cells. Blood. 2005;106: 2530-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ailles LE, Naldini L. HIV-1-derived lentiviral vectors. Curr Top Microbiol Immunol. 2002;261: 31-52. [DOI] [PubMed] [Google Scholar]
- 30.Stocking C, Bergholz U, Friel J, et al. Distinct classes of factor-independent mutants can be isolated after retroviral mutagenesis of a human myeloid stem cell line. Growth Factors. 1993;8: 197-209. [DOI] [PubMed] [Google Scholar]
- 31.Schiedlmeier B, Klump H, Will E, et al. High-level ectopic HOXB4 expression confers a profound in vivo competitive growth advantage on human cord blood CD34+ cells, but impairs lymphomyeloid differentiation. Blood. 2003;101: 1759-1768. [DOI] [PubMed] [Google Scholar]
- 32.Boyd KE, Xiao YY, Fan K, et al. Sox4 cooperates with Evi1 in AKXD-23 myeloid tumors via transactivation of proviral LTR. Blood. 2006;107: 733-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}