

Abstract
Growing evidence indicates that the affinity of monoclonal antibodies (mAbs) for CD16 (FcγRIII) plays a central role in the ability of the mAb to mediate antitumor activity. We evaluated how CD16 polymorphisms, and mAb with modified affinity for target antigen and CD16, affect natural killer (NK) cell phenotype when CD20+ malignant B cells were also present. The mAb consisted of rituximab (R), anti-CD20 with enhanced affinity for CD20 (AME-B), and anti-CD20 with enhanced affinity for both CD20 and CD16 (AME-D). Higher concentrations of mAb were needed to induce CD16 modulation, CD54 up-regulation, and antibody-dependent cellular cytotoxicity (ADCC) on NK cells from subjects with the lower affinity CD16 polymorphism. The dose of mAb needed to induce NK activation was lower with AME-D irrespective of CD16 polymorphism. At saturating mAb concentrations, peak NK activation was greater for AME-D. Similar results were found with measurement of CD16 modulation, CD54 up-regulation, and ADCC. These data demonstrate that cells coated with mAb with enhanced affinity for CD16 are more effective at activating NK cells at both low and saturating mAb concentrations irrespective of CD16 polymorphism, and they provide further evidence for the clinical development of such mAbs with the goal of improving clinical response to mAb.
Introduction
Monoclonal antibodies (mAbs) are an integral component of therapy for a number of cancers, but there is still much we do not understand about their mechanisms of action. Laboratory and clinical correlative studies are beginning to shed some light on how mAbs can induce tumor regression. Evidence in both human in vitro systems and animal models suggests mAb-induced apoptotic signaling via CD201,2 and fixation of complement3,4 can play a role in rituximab-mediated elimination of CD20+ cells. In vitro and animal model studies demonstrate that various human effector cell populations, including natural killer (NK) cells,5 monocytes,6 and granulocytes7 can mediate antibody-dependent cellular cytotoxicity (ADCC) under select conditions.
Among the most convincing evidence that ADCC plays a role in mediating the clinically relevant antitumor response of rituximab is the demonstration that polymorphisms in CD16, also known as Fcγ receptor IIIa or the low-affinity Fcγ receptor, affect clinical response to rituximab (R). Two groups have demonstrated that R is more effective in patients with follicular lymphoma homozygous for valine (VV) at CD16 amino acid position 158 compared with subjects who are heterozygotes (VF) or homozygous for phenylalanine (FF) at that position.8,9 Weng et al10 also reported a correlation between the higher-affinity CD16 polymorphism and response to active idiotype immunization. Dall'Ozzo et al11 found that NK cells from subjects with the higher-affinity polymorphism for CD16 mediate ADCC at a lower mAb concentration than do NK cells from subjects with the low-affinity CD16 polymorphism; however, there was considerable intersubject variability. Polymorphisms in CD16 did not correlate with clinical response to R or alemtuzumab in chronic lymphocytic leukemia (CLL),12,13 or in preliminary reports of patients treated with the combination of chemotherapy and R. Nevertheless, these data provide convincing evidence that response to mAb, at least with some clinical scenarios, is dependent on the interaction between CD16 and mAb-coated target cells.
Much of the effort over recent years in the area of mAb engineering has focused on decreasing immunogenicity, or producing mAbs that target different antigens. We have used a directed evolution approach to produce mAb with varying affinity for Fc receptors and for antigen to investigate the functional effect of modifying mAb sequences.
We also recently reported a system using peripheral blood mononuclear cells (PBMCs) and target cells to assess how mAbs affect NK-cell phenotype.14 The mAb of the IgG1 subclass induced modulation of CD16 and up-regulation of CD54 on NK cells when the appropriate target cells were present. Greater concentrations of mAbs were needed to induce these changes on NK cells from subjects with the lower-affinity CD16 polymorphism. Phenotypic changes were greater in NK cells from subjects with the higher-affinity polymorphism even when saturating concentrations of mAb were used, demonstrating that increased concentration of mAb can overcome some, but not all, of the influence CD16 polymorphisms have on NK activation. These studies provide a straightforward and easily reproducible technique to measure the ability of mAb-coated tumor cells to activate NK cells in vitro.
We therefore evaluated anti-CD20 mAbs with modified affinity for target antigen alone, or target antigen and CD16, for their ability to activate NK cells. These data indicate that tumor cells coated with mAb with enhanced affinity for CD16 are more effective at activating NK cells at both low and saturating mAb concentrations irrespective of CD16 polymorphism. These studies provide further support for the clinical development of such mAbs with the goal of improving clinical response to mAb.
Patients, materials, and methods
Antibodies
Rituximab (R) (Biogen-Idec, Cambridge, MA; Genentech, South San Francisco, CA) was purchased commercially. AME-B and AME-D are anti-CD20 IgG mAbs with human germ line framework regions that were generated using directed evolution technology. Functional analyses using intact cells were used to screen and select mAbs with the most promising characteristics.
For AME-B, libraries for all 6 CDRs were synthesized using a mutagenesis procedure that introduced diversity through the targeted insertion of synthetic oligonucleotide pools. CDR libraries were inserted into human germ line frameworks. The resulting libraries were screened for binding to fixed Ramos human B lymphoma cells in an enzyme-linked immunosorbent assay (ELISA) format. Variants binding to CD20 were sequenced, and selected Fabs were subjected to further rounds of combinatorial library modification and screening. AME-B was identified as a mAb with high affinity for CD20+ cells. It was expressed as a full-length human IgG1, stably expressed in Chinese hamster ovary cells, and purified from tissue culture supernatant by Protein A chromatography followed by anion exchange chromatography.
AME-D was selected by subjecting AME-B to a second round of directed evolution, this time focused on the Fc region. Briefly, diversity was introduced into the constant region of the mAb using synthetic oligonucleotides and screening variants isolated from the resulting libraries in an ADCC assay. AME-D demonstrated approximately 5- to 10-fold enhanced ADCC relative to AME-B. Purification of AME-D was performed as outlined for AME-B. Analysis of glycosylation patterns of AME-B and AME-D demonstrated they were similar to those seen with rituximab.
Thus, AME-B and AME-D share a common variable region and demonstrate high binding to target cells. In addition, AME-D has a modified Fc region with enhanced ADCC activity (Figure 1).
Figure 1.
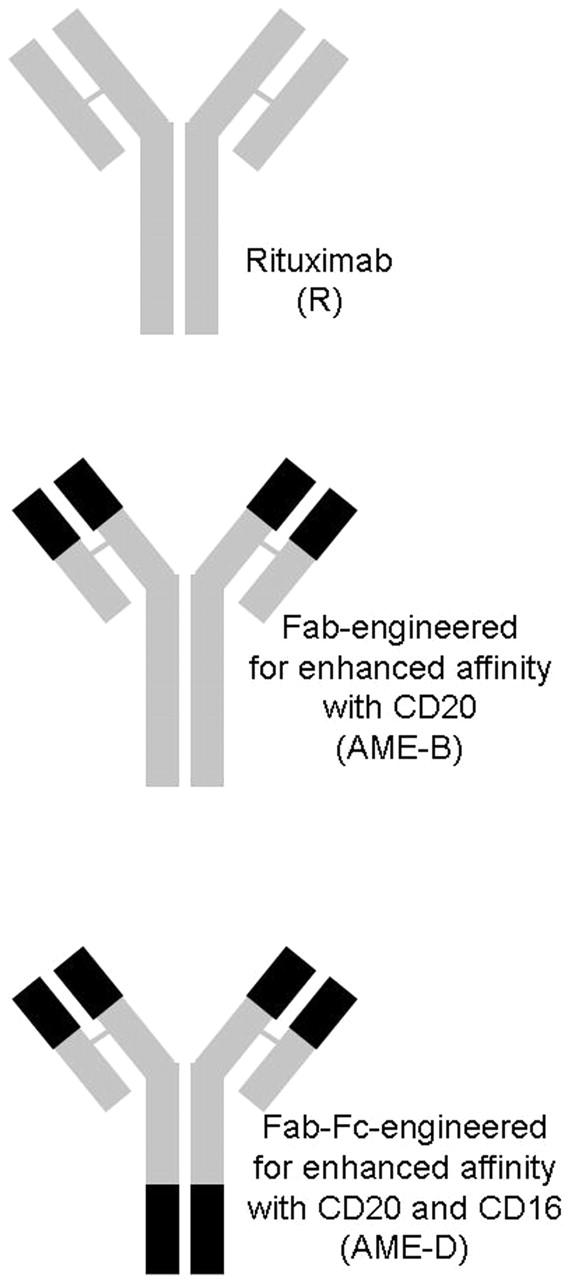
Anti-CD20 mAbs. Schematic illustrating AME-B with modifications to enhance affinity to CD20 and AME-D with modifications to enhance affinity to both CD20 and CD16.
Samples from human subjects
Peripheral blood mononuclear cells were obtained from normal volunteers or subjects with chronic lymphocytic leukemia. Mononuclear cells were isolated, and red blood cells were removed by resuspending the cells in 5 mL red cell lysis buffer according to standard procedures. Informed consent was provided according to the Declaration of Helsinki. The University of Iowa institutional review board (IRB) approved use of healthy donor and CLL patient peripheral blood mononuclear cells for the described studies.
FcγRIIIa (CD16) genotyping
DNA was extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA) and stored at -20°C until use. The polymorphism of CD16 that predicts a valine (V) versus phenylalanine (F) substitution at amino acid position 158 was determined by performing allele-specific polymerase chain reaction (PCR). Allele-specific amplification of the FcγRIIIa gene was performed as described by Leppers-van de Straat et al15 with slight modification. Briefly, the PCR reaction was optimized using 1.5 μL template DNA, either 17 pmol or 25 pmol of valine-specific or phenylalanine-specific primers, respectively, 0.5 mM dNTPs, 2 mM MgSO4, and 1 U Platinum Taq High-Fidelity polymerase (Invitrogen, Carlsbad, CA) combined and diluted in High-Fidelity PCR buffer (Invitrogen) to a final volume of 50 μL. For PCR amplification, an initial denaturation step of 5 minutes at 95°C was followed by 45 cycles (94°C for 30 seconds, 65.6°C for 30 seconds, 72°C for 30 seconds) and final extension at 72°C for 8 minutes. Samples were run in pairs using either valine-specific (5′-CTG AAG ACA CAT TTT TAC TCC CAA C-3′) or phenylalanine-specific (5′-CTG AAG ACA CAT TTT TAC TCC CAA A-3′) forward primers combined with an FcγRIIIa-specific reverse primer (5′-TCC AAA AGC CAC ACT CAA AGA C-3′). The resulting amplicons of 73 base pairs were separated on 3% agarose gels and visualized under ultraviolet (UV) radiation. Subjects were classified as heterozygous (VF), or homozygous (VV or FF). Ambiguous results were confirmed by nucleotide sequencing.
Alternatively, a protocol modified from that of Cartron et al8 and Leppers-van de Straat et al15 was used for genotyping the allelic polymorphism at position 158 of FcγRIIIa. Briefly, genomic DNA was isolated from peripheral blood from healthy human volunteer donors using the QiaAmp kit. FcγRIIIa was amplified from genomic DNA by PCR using the primers 1 (5′-ATATTTACAGAATGGCA-3′) and 2 (5′-GGTGATGTTCACAGTCTCTGAAGACACATTTTTACTGTCAA-3′). PCR products were purified and digested with HinCII at 37°C for 16 hours. Digested samples were analyzed on 3% NuSieve agarose gels (Cambrex, Baltimore, MD). Primer 2 creates a HinCII site in the PCR product only when the allele encodes a valine at position 158. Under these conditions F/F gives 1 fragment of 148 base pairs (bp); F/V gives 3 fragments of 148, 109, and 39 bp; and V/V gives 2 fragments of 109 and 39 bp. When necessary, genotype was confirmed by DNA sequencing.
Cocultures
Cocultures were performed as previously reported using fresh PBMCs and Raji cells (Burkitt lymphoma cell line) or CLL cells at a 1:1 ratio at a final concentration of 1 × 106 effector cells and 1 × 106 target cells/mL.14 Cultures were treated with the varying concentrations of rituximab, AME-B, AME-D, or no mAb for 20 hours at 37°C in RPMI media supplemented with 10% heat-inactivated fetal calf serum, 2 mM l-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μM 2-mercaptoethanol.
Flow cytometry
Immunofluorescent staining was performed on cocultured cells following 20 hours of incubation with mAbs. For surface-marker staining, cells were washed once, then stained with directly conjugated commercial antibodies (including anti-human CD56 AlexaFluor 647, CD54 PE, from BD Pharmingen, San Diego, CA; CD16 FITC [clone LNK16], from Serotec, Raleigh, NC; and CD3 PE-Cy7 from Caltag Laboratories, Burlingame, CA) per the manufacturer's protocol for 15 minutes on ice. Cells were washed twice, fixed in 2% formaldehyde solution, and stored at 4°C for flow cytometry within 24 hours. Four-color flow cytometry analysis was performed on the LSR II (BD Immunocytometry Systems, San Jose, CA). Data were analyzed using FlowJo software (TreeStar, Ashland, OR). CD16 and CD54 expression of NK cells was determined by gating on CD3-, CD56+ lymphocytes.
ADCC
ADCC assays were performed using a standard approach with a set effector-target ratio and varying mAb concentrations. Heparinized peripheral blood was obtained from 4 donors of each FcγRIIIa genotype. PBMCs were isolated on Histopaque and incubated overnight in tissue culture flasks to deplete plastic-adherent cells. Effector cells were incubated for 4 hours together with mAb and CD20-expressing SKW 6.4 target B lymphoma cells (ATCC, Manassas, VA) using an effector-target ratio of 20:1. Target cell lysis was measured by detecting the release of lactate dehydrogenase from the cytoplasm of damaged cells into the culture supernatant using a commercial colorimetric-based detection kit (Roche, Indianapolis, IN).
Statistical analysis
To facilitate comparison between donors tested on different occasions, data were normalized to the maximal response for mAb in each test. Dose response curves were generated using Prism software (GraphPad Software, San Diego, CA) by plotting the log of the antibody concentration against the normalized CD16 median fluorescence, normalized percentage of CD54bright+, or ADCC percentage of maximal specific response for each subject. The EC50 value (antibody concentration inducing a response halfway between baseline and maximum) and log EC50 value were calculated for each curve. Mean comparisons of the log EC50 between genotypes and treatment groups were performed with repeated measures ANOVA followed by Tukey tests for multiple comparisons. All statistical tests were 2-sided and carried out at the 5% level of significance using the SAS statistical software (Cary, NC).
In vivo evaluation
A CD20 tumor model was used to assess the relative in vivo efficacy of AME-D and rituximab. Male 6- to 8-week-old CB17-SCID (severe combined immunodeficient) mice (Taconic, Germantown, NY) were injected subcutaneously in the right and left flanks with 5 × 106 Raji cells. Three days later, animals received a single injection of antibody intraperitoneally at 0.42 to 5 mg/kg (AME-D) or 0.5 mg/kg (rituximab). Control animals received saline. Each treatment group contained 10 mice with 2 tumors each for a total of 20 tumors/group. Once palpable tumors were evident in the control group (approximately day 17), tumor length, width, and height were measured by caliper 3 times per week, and tumor volume was calculated (l × w × h × 0.52 mm3). The experiment was terminated 34 days after initial injection of tumors.
Results
CD16 modulation
Initial studies evaluated the effect of mAb-coated target cells on NK-cell expression of CD16. In these assays, PBMCs were combined in a 1:1 effector-target ratio with Raji lymphoma cells. The mAb was added at the indicated concentration, and surface expression of CD16 on NK cells was determined using flow cytometry. Three anti-CD20 mAbs with varying affinity for target antigen or CD16 were evaluated. The mAbs studied included R, AME-B with enhanced affinity for CD20, and AME-D with enhanced affinity for both CD20 and CD16. As in our prior studies, a change in NK-cell phenotype was seen only when both mAb and target cells were present. Curves were normalized for each person, with 100% being defined as baseline expression of CD16 in the absence of mAb. As illustrated in Figure 2A, all 3 mAbs induced down-modulation of CD16 on NK cells. Loss of surface CD16 was seen at a lower mAb concentration with AME-D when compared with AME-B or R. We defined the effective concentration 50 (EC50) as the concentration of mAb necessary to induce 50% modulation of CD16. As illustrated in Figure 2B, the EC50 of AME-D was lower than that of AME-B or R.
Figure 2.

Modulation of NK cell CD16. CD16 expression of NK cells as measured by CD16 median fluorescence (n = 12). (A) Dose response curves for different mAbs. Error bars indicate SEM. (B) EC50 values for different mAbs. Loss of surface CD16 occurred at lower mAb concentrations with AME-D. Horizontal lines represent the mean.
The EC50 for each individual mAb was also evaluated based on the polymorphism at position 158 of CD16 of the PBMC donor. A larger dose of each mAb was needed to induce CD16 modulation on NK cells from individuals with FF when compared with individuals with VV at that position, with heterozygotes having intermediate values. The difference between VV and FF individuals was statistically significant with AME-B and R, but not AME-D (Figure 3). Heterozygotes had intermediate values with all 3 mAbs. For each polymorphism, a lower concentration of AME-D was needed to modulate CD16 when compared with AME-B or R.
Figure 3.

Effect of mAb and CD16 polymorphism on modulation of CD16. The mAb EC50 as measured by decrease in CD16 expression was determined on NK cells from 12 subjects with various CD16 polymorphisms (n = 4 VV, 4 VF, 4 FF). (A) EC50 values for AME-B, AME-D, and R grouped by polymorphism. (B) The same data with EC50 values for FF, VF, and VV grouped by mAb. Horizontal lines represent the mean.
CD54 up-regulation
It is possible that modifying the Fc altered the ability of the mAb to block the anti-CD16 mAb we used to detect surface CD16. It was therefore important that we also evaluate NK activation which would not be affected by this potential artifact. As we reported previously, quantification of the number of NK cells with bright expression of CD54 is a reproducible marker for NK activation induced by mAb-coated tumor cells.14 We therefore evaluated the number of CD54bright NK cells as a measure of NK-cell activation. The NK activation data paralleled that seen with CD16 modulation. AME-D activated NK cells at a lower mAb concentration than did AME-B or R (Figure 4A). CD54bright EC50 for AME-D was lower than for AME-B, which was lower than for R (Figure 4B). This suggests that affinity for CD16 has the major effect on the ability of mAb-coated target cells to activate NK cells and that affinity for target antigen (CD20) has less of an effect in this model that involves target cells with a high density of target antigen. The EC50 of CD54bright expression was lower for NK cells from individuals who were homozygous for valine at position 158 of CD16 (VV) when compared with individuals who were FF, with heterozygotes (VF) having intermediate values for each of the 3 mAbs (Figure 5). The mean EC50 of CD54bright with AME-D in FF subjects was 2.78 ng/mL, whereas that of R in VV subjects was 6.20 ng/mL and of AME-B in VV subjects was 3.87 ng/mL. In other words, AME-D was more effective at inducing activation of NK cells expressing lower-affinity CD16 than R or AME-B were at inducing activation of NK cells expressing higher-affinity CD16. NK activation occurred at lower mAb concentrations with AME-D irrespective of polymorphism. These data indicate that enhanced mAb affinity for CD16 can overcome the decreased affinity seen with CD16 (FF).
Figure 4.

Up-regulation of NK cell CD54. Up-regulation of CD54 expression as measured by the percentage of CD54bright NK cells (n = 12). (A) Dose response curves for different mAbs. Error bars indicate SEM. (B) EC50 values for different mAbs. Up-regulation of surface CD54 occurred at lower mAb concentrations with AME-D. Horizontal lines represent the mean.
Figure 5.

Effect of mAb and CD16 polymorphism on up-regulation of CD54. The mAb EC50 as measured by an increase in the number of CD54bright NK cells was determined on cells from 12 subjects with various CD16 polymorphisms (n = 4 VV, 4 VF, 4 FF). (A) EC50 values for AME-B, AME-D, and R grouped by polymorphism. (B) The same data with EC50 values for FF, VF, and VV grouped by mAb. Horizontal lines represent the mean.
We next determined whether the effect of modified mAb was on EC50 only. If so, more mAb might be able to overcome lower affinity between mAb and CD16. Alternatively, the optimal degree of NK activation might be greater with the modified mAbs. To address this question, we identified the percentage of NK cells activated (as indicated by CD54bright expression) for each mAb at peak activation irrespective of mAb concentration. We then normalized this value by comparing peak CD54 activation for AME-B or AME-D with that of R. As illustrated in Figure 6, the peak number of NK cells activated by mAb-coated target cells was greater with AME-D than for R in all individuals studied (all AME-D/R values are > 1). AME-B-coated target cells also appeared to be more effective at activating NK cells than were R-coated target cells, although this value did not reach statistical significance.
Figure 6.

Peak activation of NK cells. The percentage of NK cells activated (as indicated by CD54bright expression) was identified for each mAb at peak activation irrespective of mAb concentration. Peak CD54 activation for AME-B or AME-D is compared with that of R. NK cell activation by mAb-coated target cells was greater for AME-D than for AME-B or R in each sample tested. (Top) AME-B versus rituximab. (Bottom) AME-D versus rituximab.
Comparison of ADCC to CD16 modulation and CD54 up-regulation
The traditional approach to assessing the ability of mAbs to activate effector cells is to measure ADCC using traditional cytolytic assays. We therefore compared traditional ADCC with CD16 modulation and NK activation as indicated by CD54 up-regulation. As illustrated in Figure 7, the log EC50 values of CD16 modulation, NK activation, and ADCC were consistent. All confirmed the known effect of CD16 polymorphisms on NK action and demonstrated that AME-D is effective at a lower mAb concentration than R.
Figure 7.

Comparison of CD16 modulation, CD54 activation, and ADCC. The EC50 for AME-D and R was evaluated for CD16 modulation (A), CD54 up-regulation (B), and ADCC (C) and plotted on a log scale and grouped by polymorphism (n = 4 VV, 4 VF, and 4 FF for each assay). Similar results were found whether the readout was loss of surface CD16, NK activation as determined by CD54 up-regulation, or lysis of target cells as determined by ADCC. Error bars indicate SEM.
Similar results are seen in an autologous system using EBV-transformed B cells as target cells and autologous PBMCs as effector cells (data not shown).
Chronic lymphocytic leukemia as target cells
R and AME-D were evaluated for their ability to activate NK cells in the presence of primary CLL cells. Minimal NK activation was observed with rituximab. In contrast, AME-D was able to activate a subset of NK cells as indicated by CD54 up-regulation (Table 1). AME-D was superior to R at activating NK cells in the presence of CLL cells. Nevertheless, CLL cells coated with AME-D induced less NK-cell activation than did Raji cells (with higher CD20 expression) coated with AME-D.
Table 1.
AME-D activates more NK cells than does R in the presence of fresh CLL cells
| Sample no. | AME-D, % activated NK | R, % activated NK | Activated NK, AME-D/R ratio |
|---|---|---|---|
| 1 | 23 | 3 | 8 |
| 2 | 9 | 1 | 9 |
| 3 | 6 | 1 | 6 |
Values indicate the percentage of NK cells activated (as indicated by CD54bright expression) by 5 μg/mL mAb
In vivo comparison of AME-D to rituximab
There are significant differences between human and murine Fc structure, as well as between human and murine CD16 structure. AME-D has a human constant region and was selected based on its enhanced affinity for human CD16. Data from mouse models are of limited utility in assessing the ability of mAbs with modified Fc, such as AME-D, to induce lysis mediated by CD16. However, murine studies can provide information related to whether modifications might affect other mechanisms that could influence the efficacy of therapy in the murine system. We therefore compared AME-D with R for its ability to inhibit the growth of Raji xenografts in immunodeficient mice. As illustrated in Figure 8, AME-D was able to inhibit Raji xenograft growth in a dose-dependent manner. AME-D and R inhibited growth of Raji cells to a similar degree. This confirmed that the changes made in AME-D did not inadvertently result in a decrease in the ability of AME-D to mediate the antitumor activity responsible for growth inhibition in a xenograft model that might be mediated by other mechanisms.
Figure 8.

Inhibition of Raji xenograft growth by AME-D and R. AME-D and R were compared for their ability to inhibit growth of Raji xenografts in SCID mice. Mice were inoculated with tumor cells on both flanks and treated 3 days later with the indicated dose of AME-D or R. The size of tumors 31 days after tumor inoculation was measured. Horizontal lines represent the mean.
Discussion
Studies demonstrating a correlation between CD16 polymorphisms and mAb efficacy suggest that the strength of the interaction between target cell and CD16, mediated by the mAb, is a key determinant of the clinical efficacy of the mAb. Multiple complex factors related to the interaction between target cell and effector mechanism could affect the efficacy of mAb-based therapy, including target antigen density,16 epitope specificity for target antigen,17 mAb affinity for target antigen,18 mAb concentration,19 mAb affinity for CD16, and CD16 structure.8,9 In addition, for target cell lysis to be observed in a standard ADCC assay (and presumably in patients), the immune effector cells need to be activated by the mAb-coated target cell, and the activated effector cells need to kill the target cells. Various factors, including the percentage of various effector cell populations within the PBMC population (such as NK cells), baseline state of activation of the effector cells, and variability in the lytic mechanisms of the effector cells, can all affect target cell lysis.20
One approach to enhancing mAb efficacy is to alter mAb affinity with either target antigen or IgG receptor. The first step in producing such modified mAb is to produce mAb with a panel of changes. Although our understanding of the relationship between structure and function of mAb is growing,21,22 it is still not possible to predict, with precision, how a given change in sequence will affect the affinity of an mAb or, more importantly, its function. We therefore used a directed evolution approach to produce AME-B with enhanced affinity for CD20 and AME-D with enhanced affinity for both CD20 and CD16. This approach involved sequential evaluation of modified anti-CD20 mAbs for their affinity for target antigen or receptor and the ability to mediate ADCC. ADCC measurements are challenging to perform, are not highly reproducible, and can vary from day to day even when the effector cells are obtained from the same donor. In addition, it is hard to perform ADCC assays on a large number of mAbs. Thus, there is a need for a functional assay that falls between affinity measurements and ADCC assays. Comparison of the ability of mAbs to activate NK cells allows for evaluation of how affinity affects the interaction between target cell and NK cell using an assay that has the advantages of simplicity and reproducibility.
The mAbs with enhanced Fc affinity for CD16 would be expected to be better at crosslinking CD16 in the presence of target cells. Therefore, we first compared mAbs that had varying affinity for target antigen and CD16 for their ability to modulate CD16. Second, we evaluated whether this results in activating of the NK cells. Third, we assessed whether measurement of CD16 modulation and NK activation as determined by CD54 expression correlated with the gold standard measurement of ADCC. Several conclusions can be reached from these studies. Enhancing mAb affinity for target antigen (as observed with AME-B) had a minimal effect on the concentration of mAb needed to activate NK cells. In contrast, also changing affinity for CD16 (as observed with AME-D) had a much greater effect. Less mAb was needed to activate NK cells when mAb with enhanced affinity for CD16 was added. This was found across all CD16 polymorphisms but was most notable on NK cells bearing the low-affinity polymorphism (FF at position 158). AME-D activated NK cells from subjects with low-affinity CD16 more effectively than R activated NK cells from subjects with high-affinity CD16. The effect of enhanced affinity is not limited to the EC50. The mAb with enhanced affinity for CD16 was more effective at activating NK cells even when saturating concentrations of mAb were used and peak NK activation was determined. This suggests that the use of mAb with altered affinity for CD16 has an effect that goes beyond effective dose. Although the mechanisms responsible for the observed enhanced NK activation induced by AME-D is still being assessed, this effect is likely due to more stable crosslinking of CD16 by AME-D as a result of a lower AME-D/CD16 rate of dissociation, with activation resulting from enhanced signaling via CD16. The pattern of EC50 values was similar whether the measurements were CD16 modulation, NK activation as indicated by CD54bright expression or ADCC. This supports use of indirect measures such as loss of CD16 expression or enhanced expression of CD54 as surrogate measures of ADCC as additional mAbs with modified Fc are developed.
AME-D was able to activate NK cells in the presence of CLL cells, whereas R was not. The degree of NK activation with AME-D was considerably less than that seen when cells that express larger amounts of CD20 (Raji cells or EBV-transformed lymphoblasts) were present. This finding is not surprising given the low expression of CD20 on these cells. This is consistent with the dose-response curves that demonstrate that mAb concentration affects the ability of cells coated with that mAb to activate NK cells, and that low concentrations of AME-D activate NK cells more effectively than do low concentrations of R. It also demonstrates that AME-D can activate NK cells in the presence of primary malignant B cells.
One approach to comparing the relative potency of mAbs is to assess their activity against human xenografts in immunodeficient mice.23 Because of the differences between human and murine antibody structure, and human and murine IgG receptors, as well as the lack of comparable CD16 polymorphisms in the mouse system, studies in murine models would not be expected to reflect the ability of AME-D to activate human NK cells via CD16 or to mediate ADCC by human effector cells. Therefore, our observation that AME-D and R are similar in a xenograft mouse model is not surprising and tells us little about whether AME-D will be more effective clinically. These results do demonstrate that the changes made to enhance binding to human CD16 did not inadvertently alter the ability to mediate antitumor activity based on other mechanisms that might be active in the xenograft model.
Although these studies suggest that mAbs with enhanced affinity for CD16 could be more effective clinically, many questions related to the clinical potential of these and similar mAbs can be addressed only in clinical trials. Some studies suggest CD16 polymorphisms affect efficacy of mAb therapy,8,9 whereas others do not.13 At this point it is not clear whether this apparent discrepancy is a result of patient heterogeneity and small sample size in some of the studies, or whether different mechanisms of action are responsible for the antitumor effect of mAbs in different clinical settings. We do not know whether mAbs with enhanced affinity for CD16 will have altered pharmacokinetics or immunogenicity. It is also possible that mAbs with modified affinity for CD16 will have an altered toxicity profile because of factors such as nonspecific NK activation, or will alter effector cell trafficking in a way that will affect efficacy. Most importantly, we do not know whether such changes will, indeed, enhance efficacy. Nevertheless, the studies described provide further support for the clinical evaluation of mAb with enhanced affinity for CD16. Such a clinical trial has recently opened.
Prepublished online as Blood First Edition Paper, July 6, 2006; DOI 10.1182/blood-2006-04-020057.
Supported by the National Institutes of Health (grant P50 CA97274).
Several of the authors (B.A., G.B., M.-A.C., D.M., B.O., and J.B.B.) are employed by Applied Molecular Evolution, whose product was studied in the present work.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Byrd JC, Kitada S, Flinn IW, et al. The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood. 2002;99: 1038-1043. [DOI] [PubMed] [Google Scholar]
- 2.Jazirehi AR, Gan XH, De Vos S, Emmanouilides C, Bonavida B. Rituximab (anti-CD20) selectively modifies Bcl-xL and apoptosis protease activating factor-1 (Apaf-1) expression and sensitizes human non-Hodgkin's lymphoma B cell lines to paclitaxel-induced apoptosis. Mol Cancer Ther. 2003; 2: 1183-1193. [PubMed] [Google Scholar]
- 3.Golay J, Zaffaroni L, Vaccari T, et al. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000;95: 3900-3908. [PubMed] [Google Scholar]
- 4.Di Gaetano N, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171: 1581-1587. [DOI] [PubMed] [Google Scholar]
- 5.Farag SS, Fehniger TA, Becknell B, Blaser BW, Caligiuri MA. New directions in natural killer cell-based immunotherapy of human cancer. Expert Opin Biol Ther. 2003;3: 237-250. [DOI] [PubMed] [Google Scholar]
- 6.Koolwijk P, Van de Winkel JG, Otten I, Bast BJ. Human monocyte-mediated cytotoxicity towards erythrocytes induced by hybrid mouse monoclonal antibodies: effect of antibody binding valency on IgG-Fc gamma R interaction. Immunology. 1992;75: 336-342. [PMC free article] [PubMed] [Google Scholar]
- 7.Valerius T, Repp R, de Wit TP, et al. Involvement of the high-affinity receptor for IgG (Fc gamma RI; CD64) in enhanced tumor cell cytotoxicity of neutrophils during granulocyte colony-stimulating factor therapy. Blood. 1993;82: 931-939. [PubMed] [Google Scholar]
- 8.Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99: 754-758. [DOI] [PubMed] [Google Scholar]
- 9.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21: 3940-3947. [DOI] [PubMed] [Google Scholar]
- 10.Weng WK, Czerwinski D, Timmerman J, Hsu FJ, Levy R. Clinical outcome of lymphoma patients after idiotype vaccination is correlated with humoral immune response and immunoglobulin G Fc receptor genotype. J Clin Oncol. 2004;22: 4717-4724. [DOI] [PubMed] [Google Scholar]
- 11.Dall'Ozzo S, Tartas S, Paintaud G, et al. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004; 64: 4664-4669. [DOI] [PubMed] [Google Scholar]
- 12.Lin TS, Flinn IW, Modali R, et al. FCGR3A and FCGR2A polymorphisms may not correlate with response to alemtuzumab in chronic lymphocytic leukemia. Blood. 2005;105: 289-291. [DOI] [PubMed] [Google Scholar]
- 13.Farag SS, Flinn IW, Modali R, Lehman TA, Young D, Byrd JC. Fc gamma RIIIa and Fc gamma RIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood. 2004;103: 1472-1474. [DOI] [PubMed] [Google Scholar]
- 14.Bowles JA, Weiner GJ. CD16 polymorphisms and NK activation induced by monoclonal antibody-coated target cells. J Immunol Methods. 2005; 304: 88-99. [DOI] [PubMed] [Google Scholar]
- 15.Leppers-van de Straat FG, van der Pol WL, Jansen MD, et al. A novel PCR-based method for direct Fc gamma receptor IIIa (CD16) allotyping. J Immunol Methods. 2000;242: 127-132. [DOI] [PubMed] [Google Scholar]
- 16.Keating MJ, O'Brien S, Albitar M. Emerging information on the use of rituximab in chronic lymphocytic leukemia. Semin Oncol. 2002;29: 70-74. [DOI] [PubMed] [Google Scholar]
- 17.Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103: 2738-2743. [DOI] [PubMed] [Google Scholar]
- 18.Adams GP, Schier R, Marshall K, et al. Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res. 1998; 58: 485-490. [PubMed] [Google Scholar]
- 19.Byrd JC, Murphy T, Howard RS, et al. Rituximab using a thrice weekly dosing schedule in B-cell chronic lymphocytic leukemia and small lymphocytic lymphoma demonstrates clinical activity and acceptable toxicity. J Clin Oncol. 2001;19: 2153-2164. [DOI] [PubMed] [Google Scholar]
- 20.Wisecarver J, Bechtold T, Lipscomb H, et al. Comparison of ADCC and NK activities of peripheral blood mononuclear cells from patients at risk for acquired immune deficiency syndrome (AIDS). AIDS Res. 1983;1: 347-352. [DOI] [PubMed] [Google Scholar]
- 21.Shields RL, Namenuk AK, Hong K, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276: 6591-6604. [DOI] [PubMed] [Google Scholar]
- 22.Presta LG, Shields RL, Namenuk AK, Hong K, Meng YG. Engineering therapeutic antibodies for improved function. Biochem Soc Trans. 2002;30: 487-490. [DOI] [PubMed] [Google Scholar]
- 23.Adams GP, Schier R, McCall AM, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61: 4750-4755. [PubMed] [Google Scholar]