

Abstract
The fate of tumor-specific CD4+ T cells is central to the outcome of the host immune response to cancer. We show that tumor antigen recognition by a subset of CD4+ T cells led to their differentiation into cells capable of suppressing naive and Th1 effector cells. Such tumor-induced regulatory T cells (TMTregs) arose both from precommitted “natural” regulatory T cells and CD4+CD25–GITRlow precursors. Once induced, TMTregs were capable of maintaining suppressor activity long after transfer into antigen-free recipients. Suppression was mediated by GITRhigh cells residing within both CD25+ and CD25– subsets. Vaccination of the tumor-bearing host concomitantly expanded TMTregs and effector cells, but suppression was dominant, blunting the expansion of naive tumor-specific T cells and blocking the execution of effector function in vitro and in vivo. These studies illustrate the possibility that therapeutic vaccination could actually worsen host tolerance to tumor antigens and support treatment paradigms that seek to not only increase the frequency of tumor-specific T cells, but to do so in conjunction with strategies that inactivate or remove regulatory T-cell populations.
Introduction
It has become increasingly clear that malignant transformation and cancer progression are immunologically recognizable events in immunocompetent hosts. In the early stages, this recognition may exert selective pressure, influencing the kinetics and character of tumor growth, and altering immunologic features of the emerging cancer.1 Ultimately, however, the immune system is confronted with persistent exposure to tumor antigens, frequently in a noninflammatory context, favoring the establishment of tolerance.2 Much like tolerance to normal self-antigens, tolerance to tumor-associated antigens may arise from a failure to encounter antigen (ignorance) or the deletion or functional inactivation (anergy) of tumor-specific T cells.
A growing body of evidence indicates that dominant forms of tolerance such as T-cell suppression play a particularly important role in preventing reactivity to self-antigens.3,5 The cells that have been implicated as regulatory T cells (Tregs) differ greatly in terms of their origin, differentiation, phenotype, and mode of action. The so-called “natural” CD4+CD25+ Tregs are thought to arise as a distinct lineage from the thymus.6,7 But regulatory function can also be acquired by uncommitted, CD4+ T cells under particular conditions of antigenic stimulation. These so-called “induced” Tregs are likewise heterogeneous, including interleukin 10 (IL-10)–producing type 1 T-regulatory (Tr1) cells8,9 and transforming growth factor β (TGF-β)–producing Th3 cells.10
Although much of the initial attention focused on the role of Tregs in controlling self-reactivity, there is growing evidence that Tregs have a significant impact on the host response to cancer.11,12 In several models, tumor immunosurveillance is augmented when CD4+CD25+ Tregs are depleted.13,16 Removing CD4+CD25+ Tregs has also been shown to enhance tumor immunity elicited by vaccination.17 In humans, CD4+CD25+ T cells have been identified at increased frequency in the peripheral blood and malignant effusions of patients with several types of cancers.18,20 Although the specificities of these populations are largely unknown, a recent study reported isolating CD4+CD25+ T-cell clones specific for a major histocompatibility complex (MHC) class II epitope of the cancer-testis antigen LAGE1 from the tumor-infiltrating lymphocytes of a patient with melanoma.21 Finally, evidence that Tregs may alter the clinical course of cancer progression was recently provided by a detailed analysis of the tumors and malignant ascites of patients with ovarian cancer, where increased densities of CD4+CD25+ T cells were predictive of poor survival.22
Although these studies point to the importance of Tregs in the host response to cancer, to date, the origins and mechanism of action of such cells specific for tumor antigen have yet to be examined. In this report, we directly demonstrate the acquisition of regulatory function by a traceable population of transgenic (Tg) tumor-specific CD4+ T cells arising during tumor progression. Strikingly, tumor-induced regulatory T cells (TMTregs) readily expanded in vivo in response to immunization and efficiently suppressed the expansion and effector function of activated T cells, although they did not impart regulatory function on the cells they suppressed. The ability of “therapeutic vaccination” to significantly expand tumor-induced CD4+ Tregs in vivo raises the possibility that in a setting where tumor antigen–specific Tregs are established, such maneuvers might be counterproductive, further impairing tumor-specific immunity.
Materials and methods
Mice
BALB/c (Thy1.2+/+) mice, 6 to 8 weeks old, were purchased from the National Cancer Institute (Frederick, MD). TCR transgenic mice (6.5 Tg mice) on a BALB/c background expressing an αβ TCR specific for amino acids 110-120 from hemagglutinin (HA) were a gift from H. von Boehmer (Harvard Medical School, Dana-Farber Cancer Institute, Boston, MA). The 6.5 Tg mice on Thy1.1+/+ or Thy1.1+/1.2+ background were used in experiments as specified. The Tg mice expressing HA on pancreatic islet β cells (Ins-HA) were provided by L. Sherman (The Scripps Research Institute, La Jolla, CA) and bred onto a Rag2–/– background. Experiments using mice were conducted in accordance with protocols approved by the Animal Care and Use Committee of the Johns Hopkins University School of Medicine, Baltimore, MD.
Antibodies and flow cytometry
Antibodies for flow cytometry were anti-CD4 (allophycocyanin, peridinin chlorophyll protein [PerCP], and phycoerythrin-Cy5 [Cyc]), Thy1.1 (PerCP and phycoerythrin [PE]), Thy1.2-allophycocyanin, CD25 (allophycocyanin and PE), CD62LPE, CTLA-4-PE, and GITR-PE (R&D Systems, Minneapolis, MN). All antibodies were purchased from BD Biosciences (Mountain View, CA) unless otherwise specified. All fluorescence-activated cell sorting (FACS) analysis was of surface expression except for CTLA-4 and FoxP3, for which cells were permeablized. A total of 30 000 gated events were collected on a FACSCalibur (Becton Dickinson, San Jose, CA) and analyzed using CellQuest software (Becton Dickinson).
Tumor cells and adoptive transfer
The generation and maintenance of A20HA B-cell lymphoma cells was described previously.23 Tumor cells (1 × 106) were injected via tail vein. For adoptive transfer using whole CD4+ T cells, single-cell suspensions obtained from lymph nodes and spleens of 6.5 Tg donors were enriched for CD4+ cells as previously described.24 For experiments using CD4+CD25–GITRlow Tg cells, pre-enriched CD4+ cells were stained with antibodies (Abs) and further fractionated by sorting. The percentage of lymphocytes positive for CD4 and the clonotypic TCR (monoclonal antibody [mAb] 6.5) was determined by flow cytometry. A total of 2.5 × 106 CD4+ 6.5 TCR+ T cells were injected intravenously into each BALB/c recipient. Chloromethylfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR) labeling of purified CD4+ T cells were previously described.24
Cell sorting and quantitative real-time PCR analysis
CD4+ enriched cells were stained with anti-Thy1.1 in combination with specified mAbs and were sorted on FACSAria (Becton Dickinson). Cells were gated on Thy1.1+ population and sorted into CFSEhigh (undivided) and CFSElow (≥ 2 cycles) subpopulations. The purity of the sorted cells was typically greater than 97%.
RNA was extracted using RNAeasy Kit (Qiagen, Valencia, CA) and was treated with DNaseI. cDNA was synthesized using the SuperScript First-Strand Synthesis System (Invitrogen, Carlsbad, CA). cDNA amounts were analyzed by real-time quantitative polymerase chain reaction (qRT-PCR) with the Taqman system (Applied Biosystems, Foster City, CA). Each sample was assayed in triplicate for target genes with the internal reference, HPRT, using the Taqman Universal PCR Master Mix and the ABI Prism 7700 Sequence Detection System (Applied Biosystems). Primers and probes for FoxP3 were previously reported.25 IFN-γ, IL-2, IL-10, IL-4, TGF-β, and HPRT were purchased as predeveloped assay reagents from Applied Biosystems. The relative mRNA frequencies of target genes were determined by normalization to HPRT. Briefly, each set of samples was normalized using the difference in the average threshold cycles (Ct) between target and HPRT: ΔCt = (CtHPRT – Cttarget). Relative mRNA frequencies of target/HPRT were calculated as 2ΔCt.
In vitro proliferation and cytokine ELISA
Sorted or magnetically enriched CD4+ T cells were incubated with irradiated (3000 rads) BALB/c splenocytes in the presence of HA110-120 peptide. For in vitro suppression assays, CD4+ T cells from 6.5 Rag 2–/– Tg mice were purified (CD4+ Isolation Kit, Miltenyi Biotech, Auburn, CA) and used as responder cells. Responder cells were incubated with the indicated number of sorted cells and irradiated BALB/c splenocytes pulsed with HA peptide. At 72 hours after incubation, 100 μL supernatant from each well was harvested for enzyme-linked immunosorbent assay (ELISA; R&D Systems). Cells were pulsed with [3H]-thymidine (1 μCi/well [0.037 MBq]) and cultured for 12 hours before harvesting and measuring scintillation counts.
In vivo priming with vaccinia-HA
A recombinant vaccinia virus encoding hemagglutinin from the 1934 PR8 strain of influenza (vacHA) was described previously.26 On the days indicated, mice were primed by intraperitoneal inoculation with 1 × 107 plaque-forming units of vacHA suspended in 0.1 mL HBSS.
Monitoring for diabetes
Mice were followed for the development of diabetes by monitoring tail blood glucose levels at the indicated time points using a MediSense Precision QID glucometer (Abbott Labs, North Chicago, IL). A mouse was considered diabetic at the first of 2 consecutive readings of glucose levels greater than 250 mg/dL.
Statistical analysis
The significance of the results was determined using the Student t test. P values less than .05 were considered statistically significant.
Results
Heterogeneity in the cell division profile of anergic, tumor-specific CD4+ T cells reveals intrinsic differences in T-cell function
We have previously described an in vivo system for tracking the fate of a population of CD4+ T cells specific for a model tumor antigen during tumor progression.26,28 In this system, CD4+ T cells bearing receptors specific for an MHC class II restricted epitope of influenza HA are progressively rendered unresponsive after transfer into mice harboring a systemic B-cell lymphoma expressing HA (A20HA). Like many human lymphomas, this tumor disseminates to secondary lymphoid tissues, and at late stages it can be found infiltrating the liver and bone marrow. The low-level expression of HA does not alter the median lethal dose (LD50) or the kinetics of A20HA progression when compared with A20 wild-type tumor in syngeneic, immunocompetent mice also receiving HA-specific CD4+ T cells. However, tumors are not ignored by the immune system because antigen recognition by HA-specific CD4+ T cells is evident from their clonal expansion and loss of naive phenotype. Although these antigen-experienced T cells persist throughout the course of tumor progression, they have a diminished response to antigenic stimulation in vitro and are refractory to priming in vivo, a state we termed “tumor-specific T-cell anergy.” However, the mechanisms of anergy induction, the composition of the anergic population, and the physiologic role played by these cells are not well understood.
To more precisely characterize the composition of the anergic tumor-specific CD4+ T-cell population, purified CD4+ T cells from HA-specific TCR transgenic donors were labeled with CFSE and transferred into A20HA-bearing (TM) or non–tumor-bearing (NT) recipients. Consistent with previous results, the frequency of donor CD4+ T cells increased slightly in the spleens of tumor-bearing mice (Figure 1A; TM/D7 and TM/D21 versus NT/D7). Accompanying this modest expansion, a minor fraction of the donor cells had divided one or more times as revealed by CFSE dilution (Figure 1A; 27% by day 7). Surprisingly however, even 3 weeks after T-cell transfer, when macroscopic tumor nodules were evident throughout the spleen, liver, and mesenteric lymph nodes, fewer than a third of HA-specific CD4+ T cells had divided (Figure 1A; TM/D21). To assess the functional responsiveness of HA-specific T cells in vivo, some mice were challenged with a recombinant vaccinia virus encoding HA (vacHA). The diminished expansion of HA-specific cells in vaccinated TM mice (0.85% in TMvac/D21 versus 4.46% in NTvac/D21) was characteristic of the previously reported tumor-specific T-cell anergy. Despite the limited expansion, the majority of the donor cells (82%) did divide in response to vacHA, although the fraction that underwent multiple rounds of division was substantially less than that observed in vaccinated, NT mice. This difference in cell division profile was reflected in the absolute numbers of divided transgenic T cells that accumulated in the spleens of vacHA-primed TM versus NT mice (Figure 1B).
Figure 1.

Heterogeneity of tumor-specific CD4+ T cells. (A) A total of 2.5 × 106 CFSE-labeled HA-specific CD4+Thy1.1+ T cells were transferred into BALB/c recipients (Thy1.2+/+) either tumor free (NT) or inoculated with 1 × 106 A20HA 10 days earlier (TM). Sixteen days after T-cell transfer, some mice were immunized with vacHA and analyzed 5 days later. Mice were killed at indicated time points. The frequency of transferred Thy1.1+CD4+ cells in the spleen was measured by FACS analysis. Percentage of the gated population is displayed in each dot plot. CFSE profiles of the gated cells are shown. The percentage of the divided cells in the gated population is indicated in each histogram. (B) Absolute number of divided donor cells recovered from spleen (total splenocyte count × percent CD4+Thy1.1+ × percent donor cells that are CFSElow). For unvaccinated NT mice, only CFSEhigh donor cells were counted. Each group had a minimum of 3 mice. Results are shown as mean ± SE. (C) Cells were sorted based on cell division status as indicated in panel A. Sorted cells (2 × 104) were stimulated with 10 μg/mL HA110-120 peptide in the presence of 2 × 105 irradiated BALB/c splenocytes. Cell proliferation was measured by 3H-thymidine incorporation. Supernatants were analyzed by ELISA for detection of IL-2 and IFN-γ. (D) qRT-PCR analysis of sorted cells. Purified CD4+ T cells from 6.5 Tg mice were included as naive controls. Each symbol represents data from one mouse. Each sample was run in triplicate for each gene, with HPRT as internal reference. mRNA abundance of the target gene was normalized to HPRT and represented as relative mRNA frequency.
Given the considerable heterogeneity of HA-specific T cells in TM mice demonstrated by CFSE profiling, we sorted divided (≥ 2 divisions) versus undivided cells within this population and examined whether they differed functionally when pulsed with antigen (Figure 1C). Similar to what had been described previously for unfractionated HA-specific T cells in TM mice, divided “tumor antigen–experienced” cells (TM-CFSElow) were hypoproliferative, made little IL-2, and failed to secrete IFN-γ on peptide stimulation. In contrast, when separated away from the divided cells, undivided HA-specific T cells (TM-CFSEhigh) from the same TM mice proliferated, produced abundant IL-2, but had not differentiated into IFN-γ–producing cells. Although vaccination increased the number of divided HA-specific T cells in the spleens of TM mice by over 7-fold (Figure 1B), these divided cells (TMvac-CFSElow) functioned similarly to the divided cells from unvaccinated TM mice (TM-CFSElow). Importantly, despite vacHA immunization, they failed to produce IFN-γ, indicative of a maintained anergic phenotype. This was in marked contrast to the effector cells generated in vaccinated NT mice (NTvac-CFSElow), which proliferated in response to peptide, were capable of producing some IL-2, and made abundant amounts of IFN-γ, indicative of successful Th1 differentiation following immunization.
We further quantified the expression of a limited set of genes in these fractionated cells. RNA isolated from the sorted cells was subjected to qRT-PCR analysis. As shown in Figure 1D, although IFN-γ mRNA was somewhat more abundant in divided cells from TM mice (TM-CFSElow) than in naive T cells, vaccination did not increase this signal compared to effector cells (TMvac-CFSElow versus NTvac-CFSElow). Interestingly, tumor antigen–experienced cells consistently expressed increased message for IL-10, whether or not vaccination was performed. In contrast, IL-10 transcripts were uniformly low in naive and effector cells. In parallel to the IL-10 transcription profile, the transcripts of Foxp3, a master control gene involved in the development and function of CD4+CD25+ Tregs,25,29 were increased only in the tumor antigen–experienced cells.
These data collectively demonstrate that the HA-specific CD4+ T cells in TM mice, previously defined as anergic cells, are heterogeneous in their cell division status, gene expression profile, and functionality in vitro. Only a minor portion of this population, having divided one or more times on encountering tumor antigen in vivo, is rendered intrinsically unresponsive. When isolated, the undivided cells, which constitute the majority of the population, function like naive cells.
Anergic, tumor antigen–experienced CD4+ T cells have regulatory function
The existence of a small number of anergic cells with elevated expression of Foxp3 and IL-10 (Figure 1; TM-CFSElow) masking the functional competence of many naive-like cells (TM-CFSEhigh) suggested that these anergic cells might have regulatory function. Therefore, in vitro suppression assays were conducted by coculturing naive HA-specific CD4+ T cells with cells sorted by FACS (Figure 2A). Tumor antigen–experienced T cells (TM-CFSElow) suppressed naive cell proliferation in a dosage-dependent manner. In contrast, the undivided cells in TM mice (TM-CFSEhigh) were not suppressive, but instead added to the total proliferation of the responder cells, further supporting the notion that they are functionally competent cells. Although vaccination of the TM mice led to an increase in the percentage and absolute number of divided cells (Figure 1A-B), these cells (TMvac-CFSElow) suppressed similarly to, or possibly even more potently than, TM-CFSElow cells. In contrast, effector cells (NTvac-CFSElow) did not substantially affect the proliferation of naive CD4+ T cells.
Figure 2.

Tumor antigen–experienced CD4+ T cells have regulatory function. (A) Suppression assay using purified CD4+ T cells from Rag2–/– 6.5 Tg mice as responders. Responder cells (2 × 104/well) were mixed with sorted cells at the indicated ratios in the presence of 10 μg/mL HA peptide and 2 × 105 irradiated BALB/c splenocytes. Proliferation of the culture in the absence of peptide was less than 1000 cpm. (B) Suppression of Th1 effector cells. Sorted effector cells and TMTregs (2 × 104/well), either cultured alone or mixed together at a 1:1 ratio, were stimulated with HA peptide and irradiated BALB/c splenocytes. Cell proliferation and IFN-γ production were measured as described in Figure 1C. Results were shown as mean ± SE of triplicate cultures. The data shown are representative of 3 separate experiments with similar results.
Because vaccination increased the number of divided HA-specific T cells available for sorting, but did not appreciably alter the anergic or suppressive properties of these cells compared with their counterparts from unvaccinated TM mice, we termed these TMvac-CFSElow cells TMTregs and used them in subsequent experiments addressing the mechanisms of suppression.
Effective regulation of adaptive immunity might be expected to have an impact on the initiation of de novo responses or the reactivation of previously generated memory/effector cells. In addition to their in vitro suppression of naive cells (Figure 2A), TMTregs were able to suppress the proliferation and IFN-γ production of fully differentiated Th1 effector cells (Figure 2B).
Phenotype of TMTregs
Consistent with their regulatory function, TMTregs had increased expression of antigens that are reportedly expressed at high levels by natural Tregs, including CD25, CD62L (L-selectin), CTLA4, and GITR (Figure 3). However, in contrast to thymus-derived natural Tregs, which display this phenotype in the absence of prior exposure to nominal antigen, only the highly divided cells from vaccinated TM mice acquired the regulatory phenotype, suggesting that development of TMTregs is associated with cell differentiation. In sum, tumor antigen–experienced CD4+ T cells preferentially express a panel of Treg-associated markers, although these antigens fail to define a uniform phenotype for this population.
Figure 3.
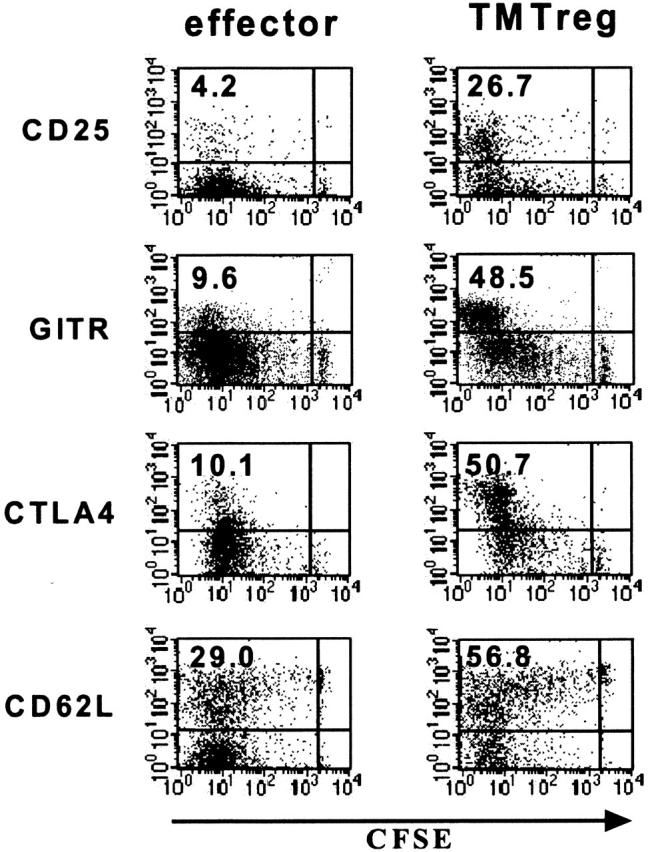
Phenotype of TMTregs. T-cell transfer and vaccination were conducted as described in Figure 1. Spleen cells from vaccinated tumor-free and TM mice were stained with anti-CD4, anti-Thy1.1, and the indicated mAb. Dot plots shown are gated on CD4+Thy1.1+ donor cell population. Value shown in each plot is the percentage of the indicated population. The data shown are representative of 3 separate experiments with similar results. GITR indicates glucocorticoid-induced TNF receptor.
GITR expression, but not CD25, distinguishes concomitantly developed effector and regulatory cells
In naive mice, immunization with vacHA generates potent HA-specific Th1 responses (Figure 1C; NTvac-CFSElow). In vaccinated TM mice, the inability to detect such a response in the total population of HA-specific T cells could either be due to impaired Th1 differentiation in this environment or concomitant induction of Th1 cells and TMTregs, with the latter masking the function of the former. Based on the observation that TMTregs had sustained expression of CD25 (range, 20%-50%) and GITR (40%-70%), we examined whether either marker could account for the suppressive activities of TMTregs and whether the removal of these subsets would unmask the priming of effector T cells in mice receiving therapeutic vaccination. To this end, TMTregs were sorted based on CFSE dilution and CD25 or GITR expression. Interestingly, both CD25+ and CD25– cells from TMTregs were hypoproliferative to peptide stimulation in vitro (Figure 4A). Moreover, both subsets were suppressive to naive cells, though the CD25+ subset was more potent (Figure 4B). These results demonstrated that tumor-induced regulatory CD4+ T cells include both CD25+ and CD25– subsets.
Figure 4.
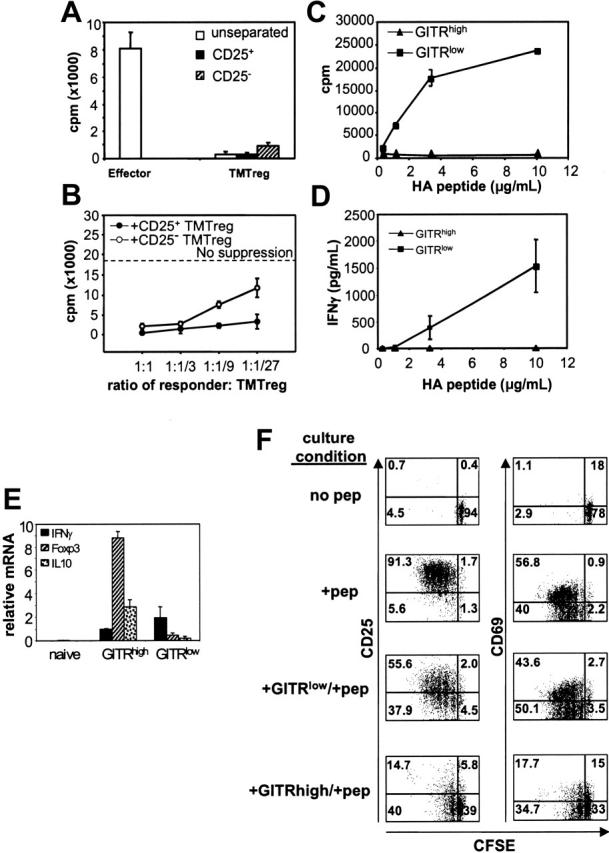
Tumor-induced CD4+ regulatory cells contain both CD25+ and CD25– cells, whereas antigen-specific suppression resides exclusively in a GITR+ subset. Spleen cells were pooled from vaccinated TM mice that had previously received CFSE-labeled Tg CD4+Thy1.1+ T cells as described in Figure 1. CFSElow cells were further separated into CD25+ and CD25– subsets. (A) Proliferation of CD25-separated and unseparated TMTregs. CFSElow effector cells from vaccinated NT mice were included for comparison. (B) In vitro suppression assay. Rag2–/– 6.5CD4+ responder T cells were mixed with CD25-separated TMTregs at the indicated ratios and assayed as in Figure 2A. (C) CFSE CD4+Thy1.1+ low cells from vaccinated TM mice were also sorted into GITRhigh and GITRlow subsets. GITR-fractionated cells were stimulated with irradiated fresh BALB/c splenocytes and varied concentration of HA peptide. Cell proliferation (C) and IFN-γ production (D) were measured as described in Figure 1C. (E) qRT-PCR analysis of GITR-fractionated cells. mRNA frequencies of the indicated genes were normalized to HPRT. (A-E) Results are shown as mean ± SE of triplicate cultures or reactions. (F) GITRhigh subset exclusively suppresses responder cells in vitro. Thy1.2+CD4+ responder cells from Rag2–/– 6.5 Tg mice were CFSE labeled and cultured with irradiated BALB/c (Thy1.1+/+ background) splenocytes and HA peptide, either alone or with equal number of GITR-fractionated cells (Thy1.1+/+). Three days later, cells were stained with anti-Thy1.2 and either anti-CD25 or anti-CD69 mAb. Plots shown are gated on Thy1.2+ responder cells. The data shown are representative of 2 separate experiments with similar results. Numbers indicate the percentage of cells in each quadrant.
In contrast, when the divided HA-specific T cells from vacHA-primed, TM mice were separated based on GITR expression, the GITRlow subset was competent in proliferation and IFN-γ production, whereas GITRhigh cells were totally unresponsive (Figures 4C-D). Furthermore, qRT-PCR analysis revealed that the molecular signature associated with Tregs resided in the GITRhigh cells, whereas GITRlow cells had an effector signature (Figure 4E). To clearly define the function of these 2 subsets, in vitro suppression assays were conducted using naive HA-specific CD4+ T cells as responders. As shown in Figure 4F, up to 97% of responder cells had gone through 1 to 5 cell divisions after 3 days in culture with cognate peptide. Responder cells in the presence of GITRlow cells divided to a similar extent (93%), although the peak of the divided cells (cycle 2 and 3) was about one cycle delayed compared to that of responder cells cultured alone (cycle 3 and 4). In contrast, in the presence of GITRhigh cells, not only was there a reduction in percentage of responder cells that divided (∼50%), but the extent of division was restricted within 2 cycles. As expected, early activation markers such as CD25 and CD69 were all up-regulated in activated responder cells either cultured alone or together with GITRlow cells (Figure 4F, second and third row, respectively). In sharp contrast, elevation of both activation markers on the responders was severely inhibited in the presence of GITRhigh cells (Figure 4F, bottom row). Taken together, our data demonstrate that tumor-specific regulatory cells and Th1 effector cells develop concomitantly in vaccinated TM hosts. GITR-expressing regulatory cells, however, function dominantly in the in vitro assays.
TMTregs maintain suppressor properties long term in the absence of antigen
Our finding that therapeutic vaccination coamplified TMTregs and effector cells, the latter undergoing Th1 differentiation, suggested that induced Tregs may also represent a distinct T-cell differentiation pathway. We therefore wished to address whether TMTregs, once induced, could maintain their phenotype and suppressive function, in a fashion analogous to committed memory/effector cells. TMTregs were sorted from TM mice and transferred into normal tumor-free mice, where they were rested for 40 days before receiving vacHA challenge. TMTregs expanded in response to vaccination (data not shown). Importantly, the expanded long-term TMTregs still displayed a regulatory cell phenotype, that is, elevated expression of CD25, GITR, and CTLA4 (Figure 5A). Consistent with their phenotype, these cells were unresponsive to restimulation in vitro and suppressive to naive responder cells (Figure 5B). These results strongly suggest that tumor-specific regulatory cells, which are induced by progressing tumor in the periphery, are a stable committed population.
Figure 5.
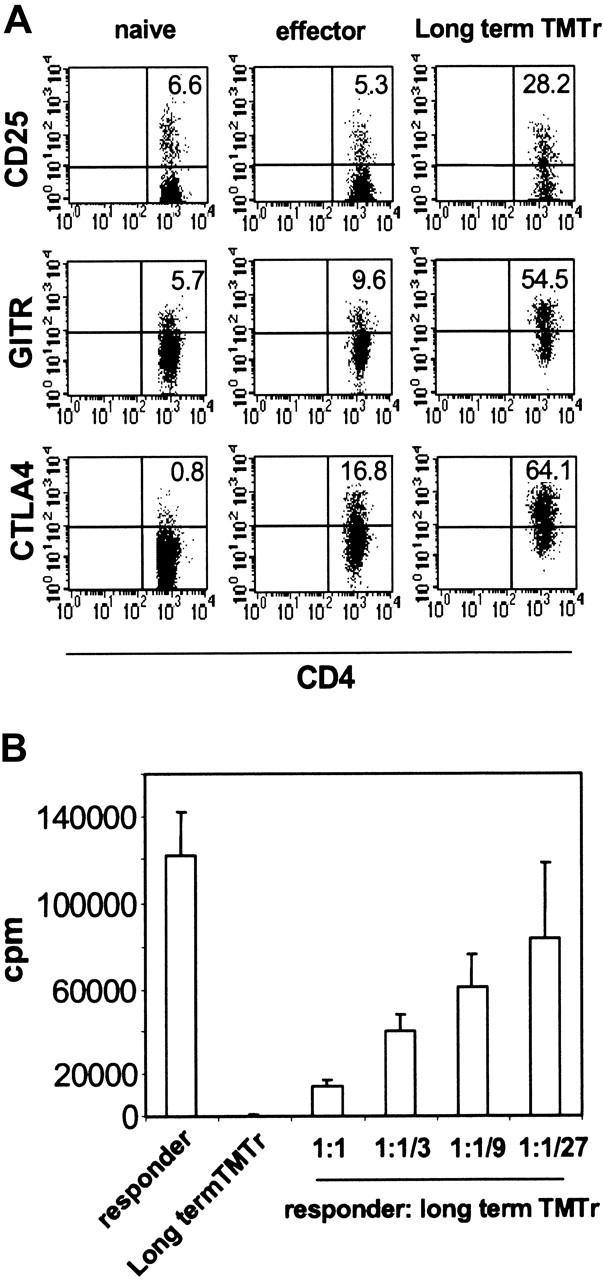
Tumor-specific regulatory cells maintain suppressor activity in the absence of antigen. CFSE-labeled Thy1.1+ TgCD4+ T cells were transferred into A20HA-bearing mice that were immunized with vacHA 2 weeks later. Thy1.1+CFSElow TMTregs were isolated after 5 days and transferred into NT BALB/c mice. Forty days after transfer, the recipient mice were challenged with vacHA and spleen cells were harvested 5 days later. (A) Surface staining of long-term TMTregs. Plots shown are gated on Thy1.1+ donor cells. Cells from NT mice that received naive TgCD4+ T cells without or with vaccination were included as naive or effector controls, respectively. The number in each plot indicates the percentage of positive cells in the gated population. (B) Thy1.1+ cells recovered from those described in panel A were sorted and stimulated with peptide-pulsed BALB/c splenocytes. Purified CD4+ T cells from Rag–/– 6.5 Tg mice were used as responders in the suppression assay. Results are shown as mean ± SE of triplicate cultures.
Development of TMTregs is independent of pre-existing natural Tregs
As with non-Tg CD4+ T cells, roughly 5% to 10% of mature HA-specific CD4+ T cells express CD25, and in the absence of prior exposure to nominal antigen, this subpopulation can suppress CD25– TCR Tg T-cell proliferation in vitro (data not shown). Therefore, it was of interest to determine whether tumor-induced Tregs were derived from pre-existing CD4+CD25+ T cells. To this end, highly purified CD4+CD25–GITRlow cells sorted from 6.5 TCR Tg mice were transferred to A20HA-bearing recipients. In a complementary approach, Rag2–/– 6.5CD4+ T cells were used as donor cells, exploiting the fact that the HA-specific TCR Tg mice lack natural Tregs when bred onto a Rag2–/– background (Figure 6A). Interestingly, even when the input population lacked natural Tregs, GITRhigh cells still emerged in the divided cells from vaccinated TM mice (groups iii and iv in Figure 6A). Furthermore, purified Tg CD4+CD25–GITRlow and Rag2–/– 6.5CD4+ input donor cells, both having very low but detectable levels of Foxp3 transcripts (0.031 ± 0.008 and 0.011 ± 0.002, respectively), gave rise to an output population expressing significantly increased levels of Foxp3 mRNA (1.86 ± 0.13 and 1.72 ± 0.76, respectively) following their isolation from vaccinated, TM recipients. As expected, the expanded subfraction of GITRhigh cells expressed even higher levels of Foxp3 message (Foxp3/HPRT > 3) than the population as a whole. The suppressive function of these GITRhigh cells was verified in vivo, using a mouse diabetes model in which HA is expressed as a self-antigen on pancreatic islet cells (Rag2–/–/Ins-HA mice). Transfer of divided, HA-specific T cells isolated from vaccinated TM mice into Rag2–/–/Ins-HA recipients resulted in diabetes only when GITRhigh cells were removed prior to transfer (fraction R2 in Figure 6A) but not following the transfer of the unfractionated population (fraction R3).
Figure 6.
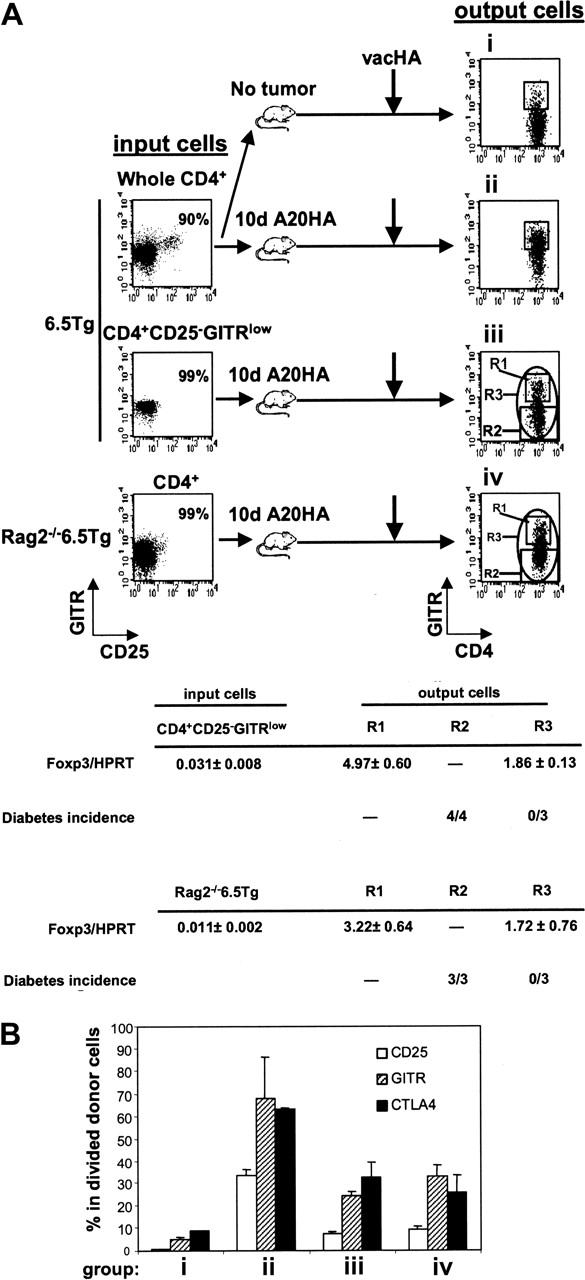
Induction of TMTregs in TM mice is independent of pre-existing natural Tregs. (A) Enriched CD4+ T cells (whole CD4+) from 6.5 Tg mice were labeled with CFSE and transferred into mice without tumor (group i) or with a 10-day A20HA tumor burden (group ii). Alternatively, sorted CD4+CD25– GITRlow cells from 6.5 Tg mice or CD4+ from Rag2–/– 6.5 Tg mice were labeled with CFSE and transferred into TM mice (groups iii and iv, respectively). An aliquot of the sorted donor cells was stained for expression of CD4, CD25, and GITR to evaluate their purity and RNA was isolated for qRT-PCR. Fourteen days after transfer, all recipients received vacHA, and responses were analyzed 5 days later. Spleen cells were stained for expression of CD4, TCR clonotype, and GITR. Plots shown were gated on divided donor cells. Cells were sorted based on the indicated gating regions (R1, R2, or R3). qRT-PCR analysis of Foxp3 mRNA was performed on sorted cells and the relative mRNA frequencies of Foxp3/HPRT are listed. The function of the sorted cells was tested by transferring them into Rag2–/– Ins-HA mice and monitoring diabetes development. Each mouse received either 50 000 cells sorted from the R2 region (CFSElow GITRlow), or cells from the R3 region (CFSElow GITR-unfractionated). Diabetes induction was monitored during a 30-day period. The incidence of diabetes is listed in Table 1. (B) Percentage of donor cells expressing CD25, GITR, and CTLA4 at the time of analysis as determined by FACS analysis. Each group had 3 mice. Results are shown as mean ± SE.
These results directly demonstrate that Tregs in a TM host need not be derived from pre-existing CD4+CD25+GITRhigh natural Tregs. The presence of such natural Tregs, however, did result in an increase in the frequency of the CD25+CTLA4+GITRhigh output population (group ii versus iii and iv in Figure 6B). It is currently unclear whether natural Tregs contribute to this increase by direct expansion or by enhancing Treg induction. It is also unclear what contribution, if any, is made by non-Tg natural Tregs of the recipient. Finally, although these findings support the hypothesis that Tregs may be induced in the periphery from uncommitted progenitors, they do not rule out the possibility that such cells arise from committed Treg precursors residing within the CD25–GITRlow population.
TMTregs impair naive T-cell responses in vivo but do not propagate suppression
Given that pre-existing natural Tregs influenced the frequency of TMTregs in a TM host (Figure 6), we wished to evaluate the impact of an established TMTreg population in TM mice on the function of freshly transferred naive tumor-specific T cells. However, cells from the second T-cell transfer rapidly differentiated into TMTregs whether or not a pre-established TMTreg population was present, precluding our ability to assess the influence of a differentiated TMTreg population on the response of naive cells in a TM host (data not shown). Therefore, we isolated TMTregs or effector cells (from vaccinated TM or NT mice, respectively) and transferred these together with naive HA-specific responder cells into secondary, NT recipients, which were vaccinated the next day (schema in Figure 7). Whereas vaccination increased the percentage of responder cells in peripheral blood (Figure 7A), the presence of TMTregs significantly diminished this expansion when compared to responders alone or to responders plus effectors (0.59% versus 1.27% or 1.61%, respectively). Analysis of the absolute number of responder cells in the spleen (Figure 7B) and lymph nodes (data not shown) under each condition confirmed the inhibitory effect of TMTregs on the clonal expansion of responder cells to vaccination. Importantly, vaccination expanded the numbers of TMTregs in the spleen over 30-fold (0.01-0.36 million/spleen), similar to what was observed in blood and lymph nodes, underscoring the capacity of this population to respond to vaccination in vivo. The impact of TMTreg-mediated suppression on the functional differentiation of the expanding responder cells was examined by sorting the relevant populations from the vaccinated, secondary recipients. The expanded responders and TMTregs in the spleens of vaccinated mice were either sorted out together or separately. Whereas TMTregs expanded in response to vacHA in vivo (Figure 7A; 0.02%-0.3%), when sorted and analyzed in vitro, these cells remained hypoproliferative, failed to produce IFN-γ, and were potent suppressors of naive HA-specific T cells (Figure 7C-D). When sorted together, the combined responses mirrored that of TMTregs alone, except that the mixed populations were less potent at suppressing naive T cells in vitro. Surprisingly, however, when separated away from the TMTregs, responder cells proliferated and produced IFN-γ equivalently to the responders that were primed after being transferred alone (Figure 7C). Whereas their function was masked when sorted together with TMTregs, separation revealed that these cells had clearly been primed by vacHA and differentiated into Th1 cells, despite the presence of TMTregs in vivo. Consistent with this interpretation, separated responder cells showed no evidence of suppressive function in vitro (Figure 7D).
Figure 7.
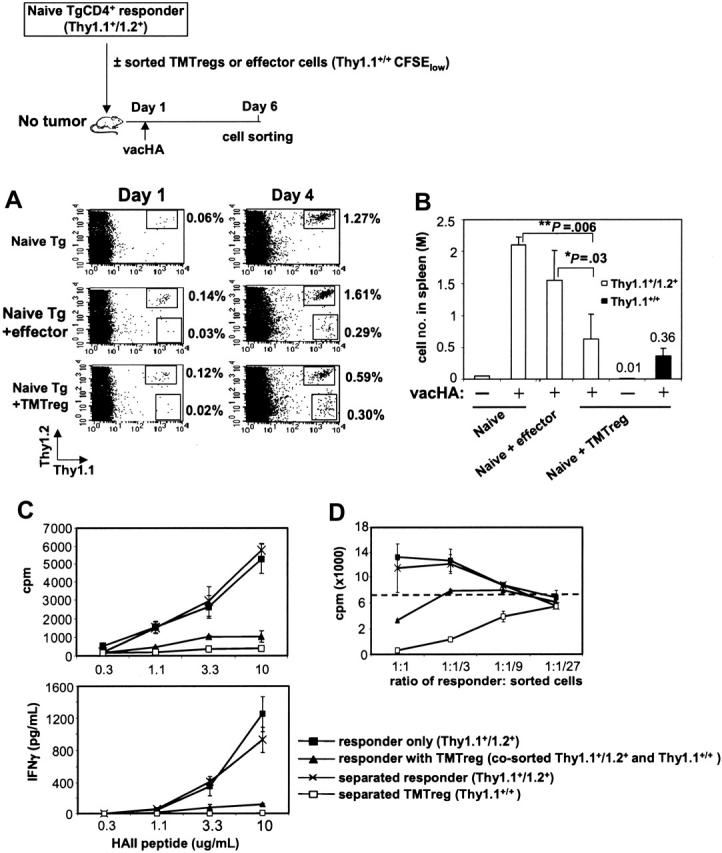
TMTregs inhibit responder cell expansion and effector function but not Th1 differentiation. CD4+ T cells purified from 6.5 Tg mice (Thy1.1+/Thy1.2+) were labeled with CFSE and transferred as responder cells, either alone or together with an equal number of sorted TMTregs or effector cells (Thy1.1+/+), into BALB/c recipients. All mice received vacHA immunization the next day. (A) Cells from tail blood collected at the indicated time points were stained with anti-Thy1.1–PE and anti-Thy1.2–APC mAbs. Percentages of the gated populations are indicated. (B) Spleen cells were harvested, counted, and the absolute numbers of Thy1.1+/Thy1.2+ and Thy1.1+/+ cells were determined 5 days after vaccination. *P < .05; **P < .01. (C) The divided fractions of the Thy1.1+/Thy1.2+ and Thy1.1+/+ populations were sorted by FACS either separately or together and analyzed for proliferation and IFN-γ production in the presence of varied amounts of peptide. (D) In vitro suppression assay. Responder cells (Rag2–/– 6.5CD4+) were mixed with sorted cells at the indicated ratios in the presence of HA peptide and irradiated BALB/c splenocytes. The dotted line represents the proliferation of responders cultured alone with peptide and splenocytes; ▪, sorted responder cells when transferred alone; ▴, cotransferred responders and TMTregs sorted together; ×, separated responders when cotransferred with TMTregs; and □, separated TMTregs when cotransferred with responders. Results were shown as mean ± SE of triplicate cultures. The data shown are representative of 2 separate experiments. Similar results were obtained when TMTregs were transferred in excess of naive responder cells by a 2:1 ratio.
Discussion
These studies provide the first detailed examination of the events accompanying tumor antigen recognition by a subset of tumor-specific CD4+ T cells that differentiate into cells capable of suppressing the host antitumor immune response. Although the phenomenon of tumor-induced T-cell suppression was described nearly 25 years ago,30 the difficulties in tracking specific populations that mediated this effect precluded a full analysis of the underlying mechanisms. Using an experimental protocol favoring the development of immune tolerance, here we show that tumor antigen recognition by CD4+ T cells having a single specificity resulted in the emergence of a population that was heterogeneous with respect to its history of cell division and functional differentiation. Despite widely disseminated cancer, divided tumor antigen–experienced CD4+ T cells represented only a minority of the total pool of tumor-specific T cells, even though the population as a whole was anergic. Cell sorting demonstrated that the features of anergy as measured in vitro resided exclusively in this subset of divided cells (Figure 1C). The majority of tumor-specific CD4+ T cells remained undivided, and when separated from divided cells, functioned comparably to naive cells. Indeed, these studies demonstrate that the global readout of anergy previously observed in unfractionated tumor-specific T cells is a direct reflection of the differentiation of a subset of this population into TMTregs capable of potent suppression of both naive and effector cells in vitro (Figure 2A-B). Several mechanisms may be responsible for the majority of HA-specific T cells remaining undivided. Previous studies have established that for both lymphoid malignancies as well as epithelial cancers, induction of tumor-specific T-cell anergy requires the capture and presentation of tumor antigen by bone marrow-derived antigen-presenting cells (APCs) rather than direct presentation by tumor cells.28,31 It is possible that the number of APCs able to process and present tumor antigen is limited. Alternatively, the developing population of Tregs may block access to antigen by sequestering APCs from naive cells or releasing chemokines that direct naive cells away from APCs that have captured tumor antigen. Finally, it is possible that TMTreg-mediated suppression of naive T-cell proliferation, which is readily demonstrated in vitro (Figure 2), also occurs in vivo (as in Figure 7), especially when antigen is encountered on APCs that have not been activated by infection. The ability of previously undivided cells to enter the cell cycle in response to vacHA (Figure 1), to expand (Figure 1B), and to differentiate into Th1 cells may partly be attributable to Toll-like receptor (TLR)–dependent signals generated in response to vaccinia infection, which have been shown to block the suppressive effects of Tregs.32,33 Nevertheless, suppression is dominant when tumor progresses, whereas effector responses predominate when vaccination occurs in the absence of tumor (Figure 1), or with prophylactic vaccination that results in tumor rejection (data not shown).
The tumor-induced Tregs characterized in this study bear a number of similarities to CD4+CD25+ natural Tregs. They both suppress responder cells in a contact-dependent, IL-10–, and TGF-β–independent manner (data not shown), suggesting that they may use the same mechanisms of suppression in vitro. However, TMTregs are identified based on their cell division status instead of CD25 expression, and the majority of TMTregs are CD25–, with a sizable portion of these CD25– cells simultaneously positive for CTLA4 (Figure 3 and data not shown). Moreover, the CD25– subset of the divided cells from TM mice is both anergic and suppressive (Figure 4A-B) and expresses Foxp3 and IL-10 in a similar pattern to unfractionated TMTregs (data not shown). Interestingly, the CD25+ subset did show greater suppression potency than the CD25– cells (Figure 4B), suggesting that the latter likely contain both regulatory and effector cells. The high CTLA4 expression by CD25– TMTregs has implications for the mechanism by which CTLA4 blockade may augment antitumor immunity. In a study of vaccine-induced tumor rejection, Sutmuller and colleagues demonstrated that antibody to CTLA4 increased antitumor responses in mice depleted of CD4+CD25+ T cells,17 suggesting that in this setting, CTLA4 blockade acts by augmenting the responsiveness of CD25– T cells, rather than by inhibiting T-cell regulation. In the current study, however, the majority of TMTregs are CD25–, and a significant fraction of them overexpress CTLA4, leaving open the possibility that this pathway may be a major contributor to how CTLA4 blockade promotes antitumor immune responses.
Apostolou et al34 reported that transgenic expression of antigen by thymic stroma generated antigen-specific CD4+CD25+ Tregs, whereas CD4+CD25– Tregs were induced de novo from mature monospecific T cells in the periphery when antigen expression was under control of the immunoglobulin κ promoter. Although it is conceivable that A20HA tumor cells, being of B-cell origin, might directly induce TMTregs in an analogous fashion, tumor-specific T-cell anergy also develops in response to antigen expressed by renal cell carcinoma, melanoma, and breast cancer,31,35 and chimera studies show that even in the case of lymphoma, antigen presentation by bone marrow–derived APCs (not the lymphoma itself) is required for tumor-specific anergy.28 Whereas the phenotypic subsets of TMTregs may arise from different developmental pathways, our data demonstrate that at least some GITRhigh TMTregs can be induced from CD25– donor cells. Furthermore, TMTregs can arise in TM mice independent of pre-existing natural Tregs in the donor population (Figure 6), although the involvement of natural Tregs in the host has not been ruled out. Taken together, our data suggest that although targeting CD4+CD25+ Tregs using CD25-depleting antibody as immunotherapy may augment tumor-specific immune responses, residual CD25– TMTregs capable of mediating suppression would still remain.
Despite their impaired proliferation in vitro, TMTregs readily expanded in vivo in response to systemic vaccination, and the expanded TMTregs maintained their suppressive properties (Figure 7). The discrepancy between the in vitro and in vivo proliferative capacity of TMTregs has also been reported for CD4+CD25+ regulatory cells bearing transgenic TCRs.36,37 Klein et al demonstrated that CD4+CD25+ Tregs outgrow naive CD4+CD25– cells with the same antigenic specificity when the 2 populations are cotransferred, leading to the predominance of Tregs in the end.37 Under the conditions studied here, we did not observe an obvious proliferative advantage of TMTregs over cotransferred naive cells, although the former clearly impair the expansion of the latter, and effectively block the execution of their effector function without interfering with their Th1 differentiation (Figures 4 and 7). These data suggest that even if TMTregs do not outcompete their target cells, they can still control the outcome of a response downstream, blocking effector function by localizing to the site of antigen. In this regard, it is interesting that in patients with ovarian cancer, Tregs were shown to be enriched in tumor masses, but were actually less frequent in tumor-draining lymph nodes than in lymph nodes from controls.22
Recent studies indicate that the generation of effective antitumor cytotoxic T-lymphocyte (CTL) responses requires effector CD4+ help38 and can be largely affected by the presence of regulatory CD4+ cells.16,17,39 Regulatory CD4+ cells may promote CD8+ tolerization by preventing “licensing” of APCs by Th1 effector cells, or “educating” tolerogenic APCs,40 or directly modulating CD8+ T cells.41 It has been shown that “helpless” CTLs may have normal primary response, that is, initial CD8+ activation and expansion, but CTL memory response is severely impaired.42 It is noteworthy that in several studies CTLs generated in the presence of CD4+ Tregs resemble the phenotypes of helpless CD8+.43,45 In line with these reports, we found that TMTregs were unable to up-regulate CD40L on rechallenge, although primary CD8 response to vacHA was not affected in the presence of TMTregs (data not shown), implying that TMTregs may largely influence the effector/memory phase of CTLs.
Since the resurrection of T-cell suppression as a legitimate area of inquiry and the growing appreciation of the role it plays in maintaining tolerance to self antigens, it has become clear that regulatory function can be attributed to cells with diverse phenotypes, origins, and modes of action. Indeed, the in vivo system examined here provides an opportunity to identify factors that influence Treg induction as well as how discrete phases of an effector response may be differentially regulated. However, regardless of the mechanisms contributing to the induction and function of such cells, a major insight from these studies is that measurements made of tumor-specific immune responses reflect the integrated sum of effector cells and Tregs, both of which may be amplified by therapeutic vaccination. Of obvious clinical relevance, the demonstration that tumor-induced Tregs can be expanded by immunization underscores the potential that therapeutic cancer vaccines given in isolation could deepen tumor-specific T-cell tolerance. This concern is amplified by results from a number of tumor models in which vaccination during early stages of tumor growth actually accelerates tumor progression (C.G.D. and G.Z., unpublished data, May 2005). Such observations strongly support the development of treatment paradigms that seek to not only increase the frequency of tumor-specific T cells, but to do so in conjunction with strategies that block the induction or promote the inactivation or removal of Treg populations, tipping the balance in favor of unopposed effector function.
Acknowledgments
We thank R. L. Blosser and A. Tam for assistance in cell sorting and R. Abdulai and D. Monie for technical support.
Prepublished online as Blood First Edition Paper, September 22, 2005; DOI 10.1182/blood-2005-07-2737.
Supported by National Institutes of Health grants 2P01CA15396-23 and 1P50CA96888-01.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21: 137-148. [DOI] [PubMed] [Google Scholar]
- 2.Sotomayor EM, Borrello I, Levitsky HI. Tolerance and cancer: a critical issue in tumor immunology. Crit Rev Oncog. 1996;7: 433-456. [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22: 531-562. [DOI] [PubMed] [Google Scholar]
- 4.Shevach EM. Regulatory T cells in autoimmmunity*. Annu Rev Immunol. 2000;18: 423-449. [DOI] [PubMed] [Google Scholar]
- 5.Maloy KJ, Powrie F. Regulatory T cells in the control of immune pathology. Nat Immunol. 2001;2: 816-822. [DOI] [PubMed] [Google Scholar]
- 6.Fehervari Z, Sakaguchi S. Development and function of CD25+CD4+ regulatory T cells. Curr Opin Immunol. 2004;16: 203-208. [DOI] [PubMed] [Google Scholar]
- 7.Jordan MS, Boesteanu A, Reed AJ, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2: 301-306. [DOI] [PubMed] [Google Scholar]
- 8.Groux H, O'Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389: 737-742. [DOI] [PubMed] [Google Scholar]
- 9.Roncarolo MG, Bacchetta R, Bordignon C, Narula S, Levings MK. Type 1 T regulatory cells. Immunol Rev. 2001;182: 68-79. [DOI] [PubMed] [Google Scholar]
- 10.Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev. 2001;182: 207-214. [DOI] [PubMed] [Google Scholar]
- 11.Antony PA, Restifo NP. Do CD4+ CD25+ immunoregulatory T cells hinder tumor immunotherapy? J Immunother. 2002;25: 202-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones E, Dahm-Vicker M, Golgher D, Gallimore A. CD25+ regulatory T cells and tumor immunity. Immunol Lett. 2003;85: 141-143. [DOI] [PubMed] [Google Scholar]
- 13.Golgher D, Jones E, Powrie F, Elliott T, Gallimore A. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol. 2002;32: 3267-3275. [DOI] [PubMed] [Google Scholar]
- 14.Jones E, Dahm-Vicker M, Simon AK, et al. Depletion of CD25+ regulatory cells results in suppression of melanoma growth and induction of autoreactivity in mice. Cancer Immun. 2002;2: 1. [PubMed] [Google Scholar]
- 15.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163: 5211-5218. [PubMed] [Google Scholar]
- 16.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200: 771-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194: 823-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo EY, Chu CS, Goletz TJ, et al. Regulatory CD4(+) CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61: 4766-4772. [PubMed] [Google Scholar]
- 19.Woo EY, Yeh H, Chu CS, et al. Cutting edge: regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168: 4272-4276. [DOI] [PubMed] [Google Scholar]
- 20.Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169: 2756-2761. [DOI] [PubMed] [Google Scholar]
- 21.Wang HY, Lee DA, Peng G, et al. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 2004;20: 107-118. [DOI] [PubMed] [Google Scholar]
- 22.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10: 942-949. [DOI] [PubMed] [Google Scholar]
- 23.Levitsky HI, Montgomery J, Ahmadzadeh M, et al. Immunization with granulocyte-macrophage colony-stimulating factor-transduced, but not B7–1-transduced, lymphoma cells primes idiotype-specific T cells and generates potent systemic antitumor immunity. J Immunol. 1996;156: 3858-3865. [PubMed] [Google Scholar]
- 24.Zhou G, Lu Z, McCadden JD, Levitsky HI, Marson AL. Reciprocal changes in tumor antigenicity and antigen-specific T cell function during tumor progression. J Exp Med. 2004;200: 1581-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299: 1057-1061. [DOI] [PubMed] [Google Scholar]
- 26.Staveley-O'Carroll K, Sotomayor E, Montgomery J, et al. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95: 1178-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sotomayor EM, Borrello I, Tubb E, Allison JP, Levitsky HI. In vivo blockade of CTLA-4 enhances the priming of responsive T cells but fails to prevent the induction of tumor antigen-specific tolerance. Proc Natl Acad Sci U S A. 1999;96: 11476-11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sotomayor EM, Borrello I, Rattis FM, et al. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood. 2001;98: 1070-1077. [DOI] [PubMed] [Google Scholar]
- 29.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4: 337-342. [DOI] [PubMed] [Google Scholar]
- 30.Berendt MJ, North RJ. T-cell-mediated suppression of anti-tumor immunity: an explanation for progressive growth of an immunogenic tumor. J Exp Med. 1980;151: 69-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuenca A, Cheng F, Wang H, et al. Extra-lymphatic solid tumor growth is not immunologically ignored and results in early induction of antigen-specific T-cell anergy: dominant role of cross-tolerance to tumor antigens. Cancer Res. 2003;63: 9007-9015. [PubMed] [Google Scholar]
- 32.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299: 1033-1036. [DOI] [PubMed] [Google Scholar]
- 33.Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol. 2004;5: 508-515. [DOI] [PubMed] [Google Scholar]
- 34.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol. 2002;3: 756-763. [DOI] [PubMed] [Google Scholar]
- 35.Sotomayor EM, Borrello I, Tubb E, et al. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med. 1999;5: 780-787. [DOI] [PubMed] [Google Scholar]
- 36.Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med. 2003;198: 249-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein L, Khazaie K, von Boehmer H. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc Natl Acad Sci U S A. 2003;100: 8886-8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298: 850-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174: 2591-2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alpan O, Bachelder E, Isil E, Arnheiter H, Matzinger P. “Educated” dendritic cells act as messengers from memory to naive T helper cells. Nat Immunol. 2004;5: 615-622. [DOI] [PubMed] [Google Scholar]
- 41.Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297: 2060-2063. [DOI] [PubMed] [Google Scholar]
- 42.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421: 852-856. [DOI] [PubMed] [Google Scholar]
- 43.Dittmer U, He H, Messer RJ, et al. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20: 293-303. [DOI] [PubMed] [Google Scholar]
- 44.Lin CY, Graca L, Cobbold SP, Waldmann H. Dominant transplantation tolerance impairs CD8+ T cell function but not expansion. Nat Immunol. 2002;3: 1208-1213. [DOI] [PubMed] [Google Scholar]
- 45.Chen ML, Pittet MJ, Gorelik L, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A. 2005;102: 419-424. [DOI] [PMC free article] [PubMed] [Google Scholar]