

Abstract
Anaplastic large-cell lymphomas (ALCLs) carry chromosome translocations in which the anaplastic lymphoma kinase (ALK) gene is fused to several partners, most frequently, the NPM1 gene. We have demonstrated that the constitutive activation of ALK fusion proteins results in cellular transformation and lymphoid neoplasia. Herein, we specifically down-regulated ALK protein expression by using small hairpin RNA (shRNA) targeting a sequence coding for the catalytic domain of ALK. The ablation of ALK leads to the down-modulation of known ALK downstream effectors, cell growth arrest, and reversion of the transformed phenotype of ALK+ mouse embryonic fibroblasts in vitro and in vivo. In human ALCL cells lentiviral-mediated ALK knock-down leads to G1 cell-cycle arrest and apoptosis in vitro and tumor growth inhibition and regression in vivo. Using a specific approach we have demonstrated that the survival and growth of ALK+ ALCLs are strictly dependent on ALK activation and signaling. Therefore, ALK is a viable target for therapeutic intervention and its inactivation might represent a pivotal approach for the treatment of ALK lymphomas and other ALK-dependent human tumors.
Introduction
Anaplastic large-cell lymphomas (ALCLs) represent a subset of neoplasms with distinctive genetic defects that are immunophenotypically characterized by the sustained expression of CD30.1 In the late 1980s several groups described the unique association of the t(2;5)(p23;q35) chromosome translocation with ALCL. However, the corresponding genes were discovered several years later by Morris and colleagues.2 In the t(2;5)(p23;q35) translocation the ALK (anaplastic lymphoma kinase) gene on chromosome 2 is fused to the NPM1 (nucleophosmin) gene on chromosome 5. Recently, several groups have successfully cloned many additional ALK translocations.3 Translocations of ALK have also been discovered in inflammatory myofibroblastic tumors (IMTs),4 and deregulated expression of ALK has been documented in a subgroup of diffuse large-cell lymphomas (DLCL).5-7 Notably, the constitutive expression of the wild-type ALK receptor (ALK-R) has been shown in several cancer cell lines8 and in primary neuroblastoma and rhabdomyosarcoma.9,10 The ALK gene encodes a tyrosine kinase receptor whose physiologic expression in mammals is largely limited to neuronal cells.11,12 At present, the physiologic role of ALK is still largely unclear. Nevertheless, ALK and its ligand play an important role in the formation of visceral muscle in Drosophila,13,14 and in the regulation of neurologic synapses in Caenorhabditis elegans.15 All ALK fusion proteins maintain the intracytoplasmic moiety of the ALK-R at their C terminus. This region, which contains the catalytic domain, is required for cellular transformation,16 whereas the N-terminal fusion partners act as dimerizing domains.17,18 Activated ALK chimeras bind multiple adaptor proteins such as Grb2, Shc, IRS-1, and p130Cas, which link them to diverse pathways regulating cell proliferation, survival, and cell transformation. Among them, PLC-γ, PI3K, and Jak3 lead to the activation of numerous downstream molecules including cyclin D, Erk1/2, STAT3, and AKT.19,20 Using cell lineage–specific conditional knock-out models, we have recently demonstrated that the genetic ablation of STAT3 in ALK+ cells leads to T- and B-cell death, and prevents the generation of B-cell neoplasms.21 Previously, PLC-γ and AKT have been shown to play an essential role in ALK-mediated transformation, in vitro.16,22-24 Thus, it is conceivable that the inhibition of ALK could induce biologic changes capable of inhibiting cell growth and/or promoting cell death. This hypothesis is supported by a study in which a nonselective, but active drug against ALK was shown to successfully lead to tumor cell death.25 Nonetheless, the specific down-modulation of NPM-ALK mRNA via siRNA does not induce apoptosis, despite a significant reduction in NPM-ALK protein.26 Thus, the formal demonstration that the specific loss of ALK kinase activity has relevant biologic effects is still missing.
Several kinase inhibitors were recently developed and successfully used in the treatment of human tumors. Systematic analyses in vitro and in vivo have shown that some of these inhibitors can efficiently block not only the kinase that was designed to be targeted, but neutralize other enzymes as well. In this context, imatinib, originally developed for Bcr-Abl,27 also efficiently hinders the activity of c-kit and PDGFR molecules. No specific inhibitor(s) against ALK are available, and thus alternative strategies designed to inhibit and/or neutralize ALK may have great relevance. Several approaches to block ALK functional activity including small peptides28 and antisense molecules29 might be envisioned. More recently, silencing the expression of a specific gene has become possible using RNA interference (RNAi).30,31 Targeting of Bcr-Abl via RNAi, for example, has proven feasible in inhibiting growth of leukemic tumor cells27,32 acting similarly to imatinib, the “gold-standard” drug for the treatment of Bcr-Abl+ leukemias.27 Taking advantage of this new approach, novel avenues for the treatment of human diseases might be opened by pivotal trials under investigation.33
To further dissect the role of ALK in the pathogenesis of ALK+ ALCL, we have developed stable small hairpin RNAs (shRNAs) targeting sequences coding for the intracytoplamic region of ALK. This approach efficiently and specifically down-regulates ALK protein expression, resulting in the reversion of ALK-mediated tumor phenotype in vitro and in the impairment of cell growth of ALK-transformed mouse embryonal fibroblasts (MEFs) in vivo. More importantly, ALK shRNA specifically induced cell death of human ALK+ ALCL cell lines, and inhibited their growth in vivo. These data demonstrate that ALK is required for the growth and survival of ALK+ ALCLs. Thus, the shRNA approach is not only a powerful tool to characterize signals that mediate tumorigenesis of ALK+ ALCLs, but also represents a potential strategy for the treatment of ALK+ neoplasms.
Materials and methods
shRNA sequences and plasmid constructs
We designed 14 short hairpin oligonucleotides (Invitrogen, Carlsbad, CA) directed against the cytoplasmic domain of the ALK-R. shALK sequences were identified following the recommendations of Brummelkamp et al34 and/or a dedicated program (Ambion, Austin, TX). The sense strand of the ALK-A5 shRNA sequence is: gatccccGGGCGAGCTACTATAGAAAttcaagagaTTTCTATAGTAGCTCGCCC tttttggaaa. The 19–nucleotide (nt) ALK target sequences are indicated in uppercase letters, whereas the hairpin and the sequences necessary for the directional cloning are depicted in lowercase letters. Restriction endonuclease sequences (5′-BlgII and 3′-HindIII) for directional cloning into the pSuper vector (Oligoengine, Seattle, WA) were included. Single-strand ALK oligonucleotides were first annealed and then cloned into the BglII-HindIII sites of expression vectors as described.34 The fidelity of cloned double-strand DNA was confirmed by DNA sequencing of both DNA strands using specific oligoprimers.
Cassettes containing the H1 promoter and shALK sequences were subcloned into a pSuperRetro vector (Oligoengine) previously modified by the insertion of a cytomegalovirus–enhanced green-fluorescent protein (CMV-EGFP) (XhoI-blunt) reporter to yield the pSuperRetro-GFP (pSRG).
LV-shALK vectors were constructed by subcloning the H1 promoter-shALK cassette into the EcoRV-XhoI sites of the vector pCCL.sin.PPT. hPGK.GFPWpre, kindly provided by Dr Luigi Naldini (San Raffaele Scientific Institute, Milan, Italy).
NPM-ALK, ATIC-ALK, ALK-R, or TPR-MET expression was achieved via the CMV promoter of the episomal retroviral vector Pallino,35 carrying EGFP under the transcriptional activity of the Moloney leukemia long terminal repeat region (LTR).36
The inducible expression of NPM-ALK was obtained using a Tet-Off bidirectional pBI-EGFP vector (Clontech, Palo Alto, CA) in which NPM-ALK (HindIII-XhoI), after blunting, was subcloned into the MluI cloning site. NPM-ALK expression was repressed by culturing NPM-ALK–transfected MEF-3T3 Tet-Off cells (Clontech) in the presence of doxycycline (1 μg/mL).
Cell culture, transfection, and retroviral and lentiviral infection
HEK-293T (ATCC, Manassas, VA), GP-293 (Clontech) packaging, and MEF Tet-Off cells were cultured under standard conditions (37°C in humidified atmosphere, with 5% CO2) in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum (FCS). Human ALK+ (TS [a subclone of SUP-M2], Karpas 299, SU-DHL-1 [kindly provided by Dr A. Lorenzana, Department of Pediatrics, University of Toronto, ON, Canada]),37 murine ALK (CD4-43), and ALK– (Jurkat and CCRF-CEM) cells were grown in RPMI 1640 medium with 10% FCS.
Transfections of HEK-293T, GP-293, and MEF Tet-Off cells were performed with Effectene reagent (Qiagen, Valencia, CA), according to the manufacturer's instructions. The NPM-ALK MEF Tet-Off cell line was generated by transfecting pBI-EGFP-NPM-ALK vector in Tet-Off MEF cells. Clones were selected based on their lowest basal NPM-ALK protein expression in presence of doxycycline and their highest expression in absence of the drug.
Retroviral supernatants were produced by cotransfection of GP-293 packaging cells with pSRG vectors and pVSV-G vector (Clontech). NPM-ALK Tet-Off MEF cells infected with pSRG retroviruses were enriched by selection with puromycin (1 μg/mL, for 7 days).
High titer lentiviral vector stock was produced in HEK-293T cells by calcium phosphate–mediated transfection of the modified transfer vector and the packaging vectors pMDLg/pRRE, pRSV-Rev, and pMD2.VSVG.38 Supernatants were harvested over 36 to 60 hours, filtrated (0.22-μm pore), and used directly or after concentration by ultracentrifugation (50 000g for 2 hours). Virus titers were assessed by transducing HeLa cells with serial dilutions of viral stocks. Aliquots of virus, plus 8 μg/mL of polybrene, were used to infect exponentially growing cells (1 × 105/mL). Fresh medium was supplemented at 24 hours after the infection. The infectivity was determined (after 72 hours) by FACS analysis of EGFP+ cells.
To study the growth patterns of ALCL cells infected with shRNAs, TS cells were first infected with ALK-A5, ALK-A6, or empty retroviral supernatants. Three days after infection cells were extensively washed with phosphate-buffered saline (PBS) and seeded (1 × 104/well) in different concentration of FCS or in presence of decreasing concentration of bleomycin or cyclophosphamide with 10% FCS. Cells were appropriately fed every 48 hours. Percentages of EGFP+ cells within viable cells were calculated by flow cytometry.
Immunofluorescence
MEF Tet-Off and TS cells were grown in complete cell-culture media onto glass cover-slips, rinsed in PBS, and fixed with ice-cold methanol for 20 minutes at –20°C, followed by permeabilization for 10 minutes with 0.25% Triton X-100 in PBS at room temperature. Immunofluorescence stainings were performed with mouse monoclonal anti-ALK (4C5B8) antibody (Zymed, South San Francisco, CA), and detected with antimouse biotinylated antibodies (Sigma Aldrich, St Louis, MO), followed by streptavidin-Cy3 (Sigma Aldrich). Samples were counterstained with Hoechst 33258 (Sigma Aldrich). Coverslips were mounted in antifading solution and viewed using a Leica TCS SP2 laser-scanning confocal microscope driven by Leica confocal software; the images were acquired at room temperature by means of a 63 × PL APO objective (numeric aperture [NA] 1.32) (Leica, Heidelberg, Germany). Bright-field images were acquired on a Leica DM IRE2 microscope using 40 × (NA 0.55) and 20 × (NA 0.40) objectives, and a DC300F camera, and were analyzed with IM 50 software.
Western blotting
For Western Blot analysis, cells were lysed (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Triton X-100, 5 mM EDTA, 1 mM Na3VO4, 1 mM phenylmethyl sulfonyl fluoride, and protease inhibitors). Total protein lysates (20 μg) after sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) were transferred onto nitrocellulose membranes. Filters were first blocked (5% lowfat milk in PBS with 0.1% Tween 20; 1 hour at room temperature [RT]), and then incubated with primary antibodies for 1 hour at RT. After 3 washes, filters were incubated with horseradish peroxidase–conjugated goat antimouse or antirabbit antibodies (1:5000; Amersham, Piscataway, NJ) for 1 hour at RT. The detection of immunocomplexes was performed with an enhanced chemiluminescence system (ECL; Amersham).
The following primary antibodies were used: mouse anti-ALK (4C5B8), anti-SKP2 (2C8D9), and anti-STAT3 (5G7) from Zymed; mouse anti– α-tubulin (B-5-1-1) from Sigma Aldrich; mouse anti-PCNA (PC10) and cyclin D1 (DCS6) from DAKO (Fremont, CA); rabbit anti–phospho STAT3 Tyr705, phospho AKT Ser473, phospho ERK1/2 Thr202/Thy204, AKT, cleaved caspase 3, cleaved caspase 6, cleaved caspase 7, and cleaved caspase 9 from Cell Signaling Technology (Beverly, MA); mouse anti–cyclin E (HE12) and p53 (DO1), rabbit anti–cyclin E (M20), cyclin A (H-432), cyclin B1 (H-433), Jun B (N-17), ERK1 (C-16), ERK2 (C-14), PARP (H-250), BclX (S-18), and eIF2α (FL-315) from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit anti–phospho eIF2α (Ab1) and survivin (Ab1) from Oncogene Research (San Diego, CA); mouse anti-p21/waf1 (70), p27/kip1 (57), and RB (G3-245) from BD Pharmingen Biosciences (San Diego, CA); and mouse anti–cyclin D3 (DCS-22) from Neomarkers (Fremont, CA).
Tumor growth in immunocompromised mice
NPM-ALK MEF Tet-Off cells were infected with pSRG or pSRG-A5 supernatants and puromycin-selected in the presence of doxycycline for 7 days; NPM-ALK expression was achieved by the removal of doxycycline and confirmed by Western blotting. Enriched EGFP+ cells (1 × 106; > 90%) were cultured in the absence of doxycycline (48 hours) and then injected subcutaneously into athymic Nu/Nu mice (Charles River Laboratories, Wilmington, MA). Tumor growth was scored weekly over a period of 4 weeks by determining the volumes of tumor masses. Lentivirus-infected TS cells (100% EGFP+) were injected (2 × 106) into FoxChase (C.B-17) severe combined immunodeficient (SCID) mice (Charles River Laboratories) and tumor growth was determined over time. Animals carrying tumor masses of 1 cm3 were injected intratumorally with 50 μL (1 × 108 transduction mits [TU]) of ALK-A5 or control concentrated lentivirus preparations every alternate day. Mice were ultimately killed 1 day following the third viral injection. Animals were housed in the animal facility of the New York University School of Medicine and treated under the NIH animal guidelines. Animal studies were approved by the NYU animal committee.
Cell cycle and BrdU cell growth analyses
For DNA content studies, 5 × 105 cells were fixed for 1 hour in 70% ethanol at 4°C. After washing, cells were treated with RNase (0.25 mg/mL) and stained with propidium iodide (50 μg/mL) or with 7-aminoactinomycin D (7-AAD; BD Pharmingen Biosciences). The G1/G0-phase fraction was calculated using the CellQuest program (BD Pharmingen Biosciences).
For BrdU labeling, 5 × 105 cells were incubated in complete cell-culture media in the presence of BrdU (10 mM) for 45 minutes at 37°C in 5% CO2. BrdU incorporation was determined by flow cytometry, using the APC BrdU flow kit (BD Pharmingen Biosciences) according to the manufacturer's instructions.
Analysis of apoptosis by flow cytometry and TUNEL assay
Apoptosis was measured by flow cytometry after staining with annexin V or with the mitochondrion-permeable voltage-sensitive dye tetrametylrodamine methyl ester (TMRM; Molecular Probes, Eugene, OR).39 Cells (5 × 105) were washed once in PBS, incubated for 15 minutes at 37°C in HEPES buffer solution (10mM HEPES pH 7.4, 140mM NaCl, 2.5mM CaCl) with 2 μL biotin-conjugated annexin V (BD Pharmingen Biosciences), or with 200 nM TMRM. annexin V binding was revealed by additional incubation with streptavidin-PE (BD Pharmingen Biosciences). Cells were analyzed by FACScan using CellQuest Program. TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling) assay was performed following manufacturer's instructions (Roche, Indianapolis, IN).
Results
Identification of ALK shRNA
We designed shRNA spanning the cytoplasmic domain of ALK-R since this region is conserved in all oncogenic fusion proteins.3 We reasoned that a functional shRNA specifically recognizing the cytoplasmic moiety would result in broad-spectrum inhibition of ALK expression, targeting all ALK chimeras and ALK-R. This strategy appears advantageous in view of the fact that the expression of ALK-R is also deregulated in nonlymphoid neoplasms. Fourteen shRNA sequences were generated. ALK shRNA pSuper (pS) and NPM-ALK cassettes were first cotransfected into HEK-293T cells. We identified a single construct (ALK-A5) that potently down modulated (> 90%) the ectopic expression of NPM-ALK (Figure 1A). Partial abrogation was observed with a second shRNA, also recognizing a sequence within the intracytoplasmic domain (A7). Expression of a p53 shRNA pS cassette34 did not result in changes of ALK expression, despite robust down-modulation of the p53 (data not shown). Similar findings were obtained using a pS carrying a shRNA specific for the break point of TPR-MET fusion protein (Figure 1B).38 To validate the efficacy of ALK-A5 against other ALK fusion proteins and ALK-R, we ectopically expressed ATIC-ALK and ALK-R in HEK-293T cells transfected with several ALK shRNAs. In this set of experiments, ALK-A5 efficiently abrogated the expression of both ATIC-ALK fusion and of the full-length ALK-R proteins (Figure 1C-D).
Figure 1.

Selection of ALK shRNA. (A) ALK-A5 efficiently inhibits NPM-ALK protein expression. HEK-293T cells were cotransfected with Pallino NPM-ALK (0.2 μg) and 1 of 6 different pSUPER vectors carrying shRNA specific for ALK sequences (pS-A1-6; 0.8 μg). Untransfected cells were also included (–). Lysates (72 hours) were immunoblotted using a specific anti-ALK antibody recognizing the cytoplasmic region of the ALK-R. Protein loading was normalized using anti–α-tubulin antibody. (B) shRNA ALK-A5 does not modulate the expression of TPR-MET. HEK-293T cells transfected with TRP-MET and ALK-A5 pSUPER vectors express unmodified levels of TPR-MET. However, TPR-MET expression is down-modulated after cotransfection with a specific TPR-MET (TM2) shRNA construct (top panel).37 HEK-293T cells transfected NPM-ALK and the TPR-MET shRNA cassette expressed unmodified levels of NPM-ALK (bottom panel). (C) ALK5 inhibits ATIC-ALK protein expression. HEK-293T cells were cotransfected with Pallino ATIC-ALK and ALK-pSUPER shRNA interfering sequences. Expression of ATIC-ALK protein was determined by Western blot analysis as described in “Materials and methods.” (D) ALK-A5 inhibits ALK-R protein expression. HEK-293T cells were cotransfected with Pallino ALK-R and pSUPER-ALK shRNA interfering sequences. Protein loading was normalized using a rabbit polyclonal antibody to CDK2.
ALK shRNA inhibits NPM-ALK–mediated transformation.
We next determined whether ALK-A5 shRNA could abrogate or revert the transforming properties of NPM-ALK, ectopically expressed in MEF cells. We employed an inducible expression system in which ALK-mediated transformation is tightly regulated after doxycycline withdrawal. MEFs carrying the NPM-ALK Tet-Off expression system were infected with the ALK-A5 pSRG virus in the presence of doxycycline. The percentages of infected cells before and after puromycin selection were monitored using EGFP as a reporter. Figure 2A shows a significant down-regulation of NPM-ALK protein expression in the absence of doxycycline when the cells were transduced with ALK-A5 shRNA. The degree of protein ablation was proportional to the percentages of EGFP-positive cells (percentages of infected cells). Moreover, the loss of NPM-ALK expression lead to decreased levels of phosphorylation of Stat3, a known ALK substrate,40 and to the down-modulation of JunB, a protein highly expressed in ALCLs,41 whose transcription is positively modulated via NPM-ALK (R. C. and G. I., manuscript in preparation).
Figure 2.

ALK siRNA reverts NPM-ALK–mediated transformation of MEF cells in vitro and in vivo. (A) Suppression of NPM-ALK expression leads to down-regulation of known downstream targets of ALK. Lysates from NPM-ALK Tet-Off MEFs infected with pSRG-A5 virus (semiconfluent cells in absence of doxycycline) before (40% EGFP+) or after (> 90% EGFP+) puromycin selection were immunoblotted with the indicated antibodies. (B) NPM-ALK MEF cells lose their transformed phenotype in absence of NPM-ALK expression. Cell cultures in absence of doxycyline are rescued from the ALK-mediated transformed phenotype by the expression of ALK-A5 shRNA (contrast phase microscopy, left panels). Control and pSRG-A5–infected NPM-ALK Tet-Off MEF cells were stained with anti-ALK antibodies, followed by biotin-conjugated horse antimouse antibody and streptavidin-Cy3. Cells were counterstained with Hoechst 33258 and visualized with a Leica fluorescence microscope using a 63× objective (right panels). (C) pSRG-A5 expression prevents NPM-ALK MEF cell growth in immunocompromised mice. NPM-ALK Tet-Off MEF cells transduced with pSRG-A5 or control pSRG were first selected with puromycin (> 90% EGFP+), inoculated (106 cells/mouse) subcutaneously into athymic Nu/Nu mice recipients (3 mice for each construct). • indicates pSRG; ▪, pSRG-A5. Tumor growth was monitored over time. These findings are representative of 2 experiments. Error bars indicate SD.
Since NPM-ALK expression in MEF cells also leads to reproducible morphologic changes, including cell elongation and bipolarization, and loose adhesion to the tissue culture plate (Figure 2B, top panels), we next studied the phenotypic changes associated with ablation of NPM-ALK. NPM-ALK+ MEFs infected with ALK-A5 shRNA (pSRG-ALK-A5 NPM-ALK MEFs) expressed undetectable levels of NPM-ALK by immunofluorescence and became large and oval, with rounded nuclei, and strongly adhered to the plate in the absence of doxycycline (Figure 2B, bottom right panel). These features were virtually identical to those seen in NPM-ALK MEF cells grown in the presence of doxycycline and lacking ALK expression. ALK-A5 pSRG NPM-ALK MEFs not only lack the transformed phenotype, but also the ability to grow in soft agar (data not shown). Finally, these cells (> 95% EGFP+) when injected into athymic Nu/Nu mice (1 × 106 cells/mouse), generated significantly smaller tumors than those derived form cells infected with the control pSRG vector (Figure 2C).
ALK-shRNA inhibits cell growth and induces cell death of human ALCL cells in vitro
ALK shRNA was subsequently tested in multiple human ALK-positive ALCL cell lines. To determine if the down-regulation of NPM-ALK expression would lead to inhibition of cell growth, we transduced human ALCL cells with pSRG, pSRG-A5, pSRG-A6, or pSRG-TM2 retroviral supernatants. Jurkat and CCRF-CEM cell lines were used as ALK– controls. Percentages of the EGFP+ cells ranged from 15% to 70% in the different cell lines, with TS (ALCL, ALK+) being the most efficiently transduced cell line (Figure 3A). After infection, the overall percentages of EGFP ALK-A5+ cells decreased overtime in all ALK+ cell lines. Similar findings were obtained with several murine NPM-ALK–positive cell lines derived from primary T lymphoblastic lymphomas of NPM-ALK Tg mice (Figure 3E).42 On the other hand, EGFP+ CCRF-CEM and Jurkat cells (ALK–, Figure 3B-F; and data not shown) remained unchanged following transduction with controls or pSRG-A5 viruses. Likewise, the percentages of EGFP+ ALCL cells infected with TM2, ALK-A6, or empty control vectors remained unvaried over time (data not shown). BrdU incorporation assays showed that pSRG-A5 ALCL cells displayed a lower percentage of proliferating cells compared with uninfected cells within the same cultures or with control infected cells (Figure 3G-H).
Figure 3.

ALK shRNA inhibits the growth of human ALCL cells. (A) Percentages of human (TS, 80%) and murine (CD4-43, 8%) ALK+ cells transduced with pSRG-A5 vector as measured by EGFP expression by FACS analysis. (B-F) Human and murine ALCL ALK-A5+ cells decrease over time. Percentages of EGFP+ cells in pSRG-A5 or pSRG retrovirally transduced human ALCL cells SU-DHL-1, TS, and Karpas 299, human lymphoblastoid cells CCRF-CEM (ALK–), and murine NPM-ALK (CD4-43) cells were determined over time. These findings are representative of at least 3 different experiments. In B-F, ♦ indicates pSRG-A6; ▪, pSRG-A5. (G) Percentages of BrdU/ALK-A5+ ALCL cells decrease over time. ALK-A5–infected TS cells were cultured over time and the percentages of BrdU+ cells within EGFP– and + cells were calculated at different intervals. These findings are representative of at least 3 different experiments. Numbers represent the percentages of positive cells for each quadrant. (H) Percentages of BrdU+ TS cells over time after transduction with pSRG-A5 and pSRG-A6 viruses. TS cells were transduced with retroviral ALK-A5 or ALK-A6 pSRG vectors. BrdU incorporation of EGFP+ and EGFP– cells was established as described in “Materials and methods.” ▪ indicates pSRG-A5; □, pSRG-A6. Percentages of total are indicated. Normalized ratio of BrdU+ cells among EGFP+ or EGFP– cells was calculated over time. (A-F, H) Error bars indicate SD.
To study whether ALK-A5 shRNA could sensitize infected cells to stress conditions, we first cultured recently transduced (3 days) ALCL cells in suboptimal concentrations of FCS. As shown in Figure 4A, the percentages EGFP+ ALK-A5–transduced TS cells cultured with low concentrations of FCS decreased rapidly as a percentage of total. In contrast, the loss of EGFP was less evident and required longer periods of time when the same cells were cultured in 10% FCS. Moreover, under the same conditions, EGFP– cells displayed a higher percentage of BrdU incorporation (data not shown). Similar findings were observed when ALK-A5+ cells, but not control cells, were grown in the presence of low doses of chemotherapeutic agents such as bleocin or cytoxan (Figure 4B-C).
Figure 4.

Growth of ALK-A5 retrovirally infected cells is inhibited in presence of low FCS concentration or cytotoxic agents. (A) ALK-A5 or ALK-A6 pSRG– or pSRG-infected cells were cultured in the presence of different concentrations of FCS. The percentages of EGFP+ cells were calculated over time. Data are reported as ratios of ALK-A5 versus ALK-A6 pSRG EGFP+ cells. (B) Retrovirally infected cells (3 days after transduction) were cultured with different concentration of bleomycin (B) or cyclophosphamide (C). Percentages of EGFP+ cells were obtained by flow cytometry and ratios between ALK-A5 versus ALK-A6 pSRG EGFP+ cells were calculated. In A-C, ▪ represents pSRG-A5; □, pSRG-A6.
The amount of time required to achieve detectable biologic effects after retroviral infection of ALK-A5 might be due to the long half-life of the NPM-ALK protein, and/or to low-level shRNA expression. In fact, the protein levels of NPM-ALK in these cells were only slightly decreased (data not shown). Since concentrated VSV-G lentiviruses can efficiently infect the majority of the target cells, and allow a more potent expression of transgenes, we generated a lentivirus cassette carrying the ALK-A5 shRNA under the H1 promoter.43 An empty EGFP+ lentivector or a lentivector carrying shRNA with mutated ALK-A5 (ALK-A5M) or specific for TPR-MET (TM2) sequences were used as controls. Lentivirus infection led to more than 99% transduction (based on EGFP expression) of all human ALK+ cells and the relative intensity of EGFP+ cells increased proportionally with higher viral titers (data not shown). TS cells transduced with ALK-A5 lentivirus showed an increased expression of EGFP over time, and this was associated with a down-modulation of ALK protein expression, detectable as early as 72 hours after infection (Figure 5A-B). Known targets of NPM-ALK signaling, such as the phosphorylation of STAT3 and AKT, and the expression of JunB, were concomitantly down-modulated in ALK-A5+ cells (Figure 5C).
Figure 5.

Lentiviral ALK shRNA abrogates NPM-ALK expression and signaling. (A) Lentivirus infection with ALK-A5 leads to an efficient down-regulation of NPM-ALK protein expression. TS cells were transduced with ALK sh-RNA lentivirus (300 μL) and then harvested to determine the NPM-ALK protein expression over time. Immunoblotting with anti-GFP antibodies was used to check the expression of the report gene within the lentiviral cassette. (B). Loss of NPM-ALK is specifically observed in cells infected with ALK-A5. TS cells were transduced with ALK-A5 or TM2 viruses, cultured for 96 hours, and then stained with anti-ALK antibody. Immune complexes were visualized using biotin-conjugated rabbit antimouse antibody followed by streptavidin-Cy3 (right panels). Nuclei were identified using Hoechst 33258 (left panels). EGFP expression is shown in middle panels. Objective magnification, 40×. (C) TS cells transduced with ALK-A5 show phospho-Stat3, phospho-AKT, and Jun B down-modulation. EV indicates empty vector.
Moreover, transduction with ALK-A5 lentivirus induced G0/G1 arrest by 96 hours after infection (Figure 6A). Cell-cycle arrest was sustained by the concomitant down-modulation of cyclin A, cyclin B, and Skp2, by the up-regulation of p27 and p21, which resulted in hypophosphorylation of RB (Figure 6B). In parallel with the growth arrest, NPM-ALK silencing was associated with an increased number of apoptotic cells at 4 and 5 days after infection (Figure 6C). Accordingly, activation of effector caspases known to precede overt apoptosis, together with the down-regulation of the antiapoptotic protein survivin, were distinctively detected in ALK+ ALCL cells 4 days after transduction with ALK-A5 (Figure 6D).
Figure 6.
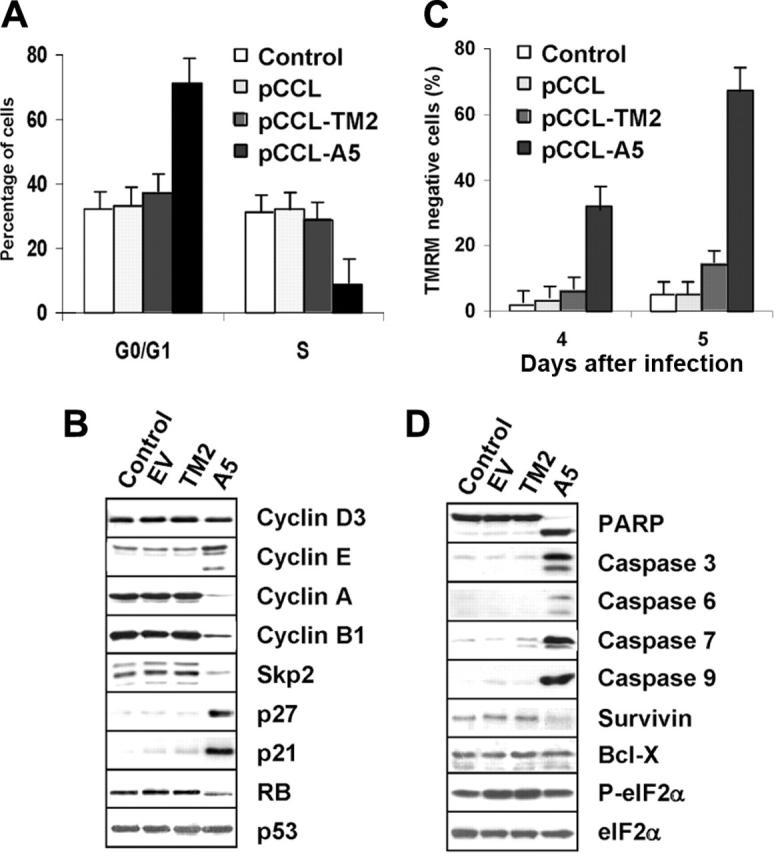
Lentiviral ALK shRNA leads to cell-cycle arrest and apoptosis of ALCL cells. (A-B) Anti–ALK-A5 leads to G1 cell-cycle arrest. TS cells transduced with the indicated constructs were harvested 96 hours after infection and stained with propidium iodide (PI) to determine the corresponding fractions in G0/G1, S, and G2-M phases. Lysates were analyzed by Western blotting with the indicated antibodies. (C) Anti–ALK-A5 leads to cell death. Percentages of TMRM– cells were calculated in TS cells 4 and 5 days after infection. (A, C) Error bars indicate SD. (D) Loss of NPM-ALK leads to caspase activation. TS cells transduced with empty vector, TM2, or ALK were harvested 96 hours after transduction and immunoblotted with the indicated antibodies. These findings are representative of 4 different experiments.
Finally, to verify that “off-target” silencing effects were not responsible for the observed decrease in cell growth and viability, we performed a functional rescue experiment. We designed an NPM-ALK expression construct carrying 4 conservative nucleotide mutations, without any changes in the corresponding translated amino acids, within the ALK-A5 target sequence. TS cells ectopically expressing the mutated NPM-ALK construct were resistant to the ALK-A5 silencing, remained viable, and continued to grow normally (data not shown). The specificity of the shRNA ALK-A5 findings was further validated, demonstrating that the protein expression of known genes, whose regulation is modulated in response to interferon-mediated responses, was not significantly changed (Figure 6D, P-eIF2α and eIF2α).
ALK-A5 shRNA impairs the growth of human ALK positive cells in vivo
To study whether the loss of NPM-ALK could modulate the growth of human ALCL cells in vivo, we used 2 different strategies. In the first approach, ALCL cells were infected with ALK-A5 or control virus and then, after 48 hours of culture (> 95% EGFP- and < 10% annexin V+ cells), were injected into FoxChase (C.B-17) SCID mice. Tumor growth was monitored over time. TS cells in which NPM-ALK expression was silenced grew far less efficiently than controls (Figure 7A). In a second series of experiments, we treated large (1-cm) xenograft ALCL tumor masses with multiple injections (3 injections on alternate days) of concentrated ALK-A5 or control lentiviral preparations (0.5 to 1 × 108 TU) and showed that the tumor growth of neoplastic cells was significantly inhibited by lentiviruses carrying the ALK-A5 shRNA construct compared with the control ALK-A6 vector (Figure 7B) (P < .001). Flow cytometric analysis of EGFP+ tumor cells showed that approximately 50% of transplanted cells were efficiently transduced in vivo. Notably, the very large majority of EGFP ALK-A5+ cells were also annexin V positive, in contrast to a smaller fraction of control (ALK-A6) cells (Figure 7C, left quadrants). Finally the histologic sections of the tumors (obtained 1 day after the last injection) demonstrated that tumor masses treated with ALK-A5 viral preparations were largely necrotic, and in more viable areas, many apoptotic cells were interspersed among viable cells. At this time, no significant tissue remodeling or fibrosis could be observed. On the contrary, control samples were largely viable (Figure 7C, middle panels). TUNEL analyses further confirmed these findings, demonstrating that the very large majority of the ALK-A5+ cells display DNA genomic breaks (Figure 7C, right panels).
Figure 7.

NPM-ALK silencing impairs the growth of human ALK+ cells in vivo. (A) TS cells transduced with ALK-A5 do not generate xenograph tumors in immunocompromised animals. TS cells transduced with ALK-A5 or with the control ALK-A6 lentivirus were injected into FoxChase (C.B-17) SCID mice 48 hours after infection. Tumor growth was observed over time. A total of 12 mice (6 for each group) was studied. (B) ALCL tumor mass growth is hindered by the intratumoral injection of lentiviruses carrying the ALK-A5 shRNA. FoxChase (C.B-17) SCID mice injected with TS cells (2 × 106) were treated intratumorally when the tumor masses were about 1 cm3 with 50μL (0.5-1 × 108 TU) of concentrated lentivirus preparations carrying ALK-A5 or control ALK-A6 shRNA, every alternative day, 3 times. Tumor growth was determined over time. (A-B) Error bars indicate SD. (C) ALK-A5 shRNA leads to cell death in vivo. After treatment, tumor cells were isolated and stained with PE–annexin V and evaluated by flow cytometry. Percentages of EGFP+ cells are indicated (left panels). Representative histologic sections (hematoxylin and eosin [H&E]) of tumor masses derived from animals treated with LV-A6 or LV-A5 preparations are shown as indicated (second panels from left; objective magnification, 20×). The same paraffin-embedded tissue sections were also stained for DNA breaks with a TUNEL method. Hoechst counterstain identifies the nuclei (third panels from left; objective magnification, 40×). DNA break points are demonstrated by the positive FITC stains (fourth panels from left; objective magnification, 40×). Variable percentages of positive cells were observed in different areas within the tumor cells treated with ALK-A5 lentiviral preparations. These findings are representative of 3 different experiments with a total of 12 mice for each group.
Discussion
Small interfering RNAs have proven to be a useful tool for gene silencing and represent a promising approach for studying the tumorigenic role of many genes. To validate ALK as a target for therapeutic intervention, we designed a specific ALK shRNA construct which effectively down-modulated ALK protein expression. NPM-ALK ablation led to cell growth inhibition, loss of the tumor phenotype of transformed MEFs, and cell death of human ALK+ ALCL lymphocytes in vitro and in vivo. These findings clearly demonstrate that ALK fusion proteins are suitable targets for therapeutic intervention in ALK+ ALCLs and possibly for other human tumors in which ALK-R could play a pathogenetic role. Accordingly, silencing ALK tyrosine kinase activity via RNAi technologies or alternative approaches, such as antisenseoligonucleotides, tyrosine kinase inhibitors, or peptidomimetics should be actively pursued for the treatment of ALK+ tumors.
Several studies have demonstrated that RNAi targeting oncogenic chimeric RNA sequences can be successfully used to down-modulate the expression of oncoproteins.27,32,44 Since ALK-R expression is a feature of some nonlymphoid tumors including neuroblastomas, rhabdomyosarcomas, and other mesenchymal tumors, we designed a series of shRNA constructs directed against sequences encoding the cytoplasmic domain of the ALK oncogene. Based on the physiologic expression pattern of ALK-R,11,12 and the absence of a striking phenotype in knockout mice,45 the inhibition of ALK-R in normal cells is expected to produce only minor to nil effects. In this perspective, we should consider that ALK-R mRNA transcripts have been recently detected in many human tumors.9,10 If these findings are confirmed and a pathogenetic role of ALK in these tumors is demonstrated, the availability of ALK inhibitors and or RNAi will have considerable clinical implications.
RNAi approaches have also been proven to be specific in vivo46,47 and recently a novel clinical trial using chemically modified siRNA was initiated.48 In our study, we confirmed that shRNA leads to the specific loss of ALK. No changes in ALK protein expression were seen when unrelated RNA interfering sequences were transduced in ALK+ cells and no detectable biochemical and/or biologic effects were observed when ALK-A5 shRNA was expressed in several ALK– cells. More importantly, the absence of significant interferon-mediated responses and the functional rescue experiments excluded the possibility that unintentional silencing of nontarget genes was responsible for the observed reduction in cell viability and cell growth.
This study demonstrates that the sustained ablation of NPM-ALK protein expression promotes human lymphoid cells carrying the ALK fusion protein to undergo apoptosis. These findings strongly suggest that ALK is absolutely necessary for the survival of ALK+ lymphoma cells and essential to maintain their overall growth. These findings are in concordance with the data obtained using a pharmacologic approach,25 which, however, has a relatively large range of action.
The precise mechanisms leading to NPM-ALK transformation and the requirements of ALK for tumor growth are still unclear. ALK is known to trigger multiple signaling pathways, which lead to the activation of known antiapoptotic molecules, such as AKT,22,23 Bcl-xL,40,49 and survivin.40,50 Thus, the loss of positive antiapoptotic signals alone may be sufficient to drive cells into apoptosis. It is also conceivable that more complex scenarios are at work. For example, we have recently demonstrated that ALK efficiently activates Ras and its downstream effectors, and through Stat3, up-regulates cyclin D1 and c-myc. Moreover, loss of Stat3 via antisense oligonucleotides or shRNA leads invariably to cell death and growth inhibition in ALK+ tumors.21
Overall, our findings justify the search for specific inhibitors of ALK and/or the development of novel strategies, such as oligonucleotide-based antisense targeting, for the clinical treatment of patients carrying ALK+ neoplasms. Interestingly, a moderate down-regulation of ALK protein expression appears to be sufficient to produce measurable biologic effects, even when it is not sufficient to directly induce cell death. While ALK-A5 shRNA sequences delivered using retroviruses resulted only in a moderate reduction of NPM-ALK protein levels, the infected cells displayed a lower rate of proliferation and impaired biologic features. Cell growth disadvantage was further apparent when cells were challenged with suboptimal doses of chemotherapeutic drugs or cultured in stress conditions. Both conditions specifically promote the cell death of ALCL cells infected with ALK-A5 shRNA, but at appropriate concentrations, had very little effect on control cells. These findings suggest that an effective treatment of ALCL can be achieved using a combination approach: avoiding unnecessary toxicity accompanied by moderate- to high-level dosing with the same reagents. The association of low doses of chemotherapeutic drugs in combination with ALK RNAi might also result in a lower incidence of tumoral drug resistance.51,52 This is an important consideration particularly in view of the fact that RNAi resistance can occur.53 Notably, cells infected with a low copy number of ALK-A5 were not resistant to ALK shRNA, since upon reinfection with a high titer of ALK-A5 lentivirus, they underwent apoptosis (data not shown). Even though our in vitro model might not fully reproduce an in vivo scenario, our findings are encouraging and support further investigation. Finally, the data for intratumoral delivery of a high viral titer of shRNA lentivirus demonstrate the possible therapeutic feasibility of RNAi anti-ALK in the treatment of ALCL. In these experiments we have shown that we could induce massive cell death even though we did not observe, immediately after the treatment, the complete disappearance of the tumor masses. The significance of this approach should be further evaluated by studying whether this method alone could improve the overall survival of the animals or, alternatively, if combined treatment with conventional drugs might be required. Nonetheless, many challenges still lay ahead prior to the clinical use of the RNAi technology in cancer treatment. Despite the fact that intratumoral delivery of shRNA cassettes may be feasible in some instances, the targeting of shRNAs to the proper cells remains a challenge. Various research fronts are working together to overcome the obstacles efficiently, and safely target viral shuttle vectors to limited cellular targets.54
In conclusion, we have shown that stable shRNA targeting ALK is a new a powerful approach to investigate and dissect the pathogenetic role of ALK in ALCL lymphomagenesis. Based on these findings, the ablation of ALK should be actively pursued as a new and efficient strategy for the clinical treatment of patients with ALK+ tumors.
Note added in proof. While this paper was in press, a new study55 confirmed that ALK is a molecular target for ALCLs by using specific kinase inhibitors.
Acknowledgments
We thank Dr L. Naldini (San Raffaele Scientific Institute, Milan, Italy) for reagents. Sequences of the shRNA oligonucleotides are available on request.
Prepublished online as Blood First Edition Paper, September 27, 2005; DOI 10.1182/blood-2005-05-2125.
Supported by National Institutes of Health (NIH) grant no. R01-CA64033, Sixth Research Framework Program of the European Union, Project RNA Interference Technology as Human Therapeutic Tool (RIGHT) (LSHB-CT-2004-005276), Ministero dell'Università e Ricerca Scientifica (MIUR), Regione Piemonte, Compagnia di San Paolo, Torino (Progetto Oncologia), and Associazione Italiana per la Ricerca sul Cancro (AIRC). R.P. is supported by the MIUR program “Incentivazione alla mobilità di studiosi residenti all'estero” and by a contribution from Comitato Regionale Piemontese Gigi Ghirotti. W.S. is funded through an NIH Postdoctoral Fellowship in Molecular Immunology and Oncology.
R.P., R.C., and A.D.M. have equally contributed to the paper.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Stein H, Foss HD, Durkop H, et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96: 3681-3695. [PubMed] [Google Scholar]
- 2.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263: 1281-1284. [DOI] [PubMed] [Google Scholar]
- 3.Duyster J, Bai RY, Morris SW. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene. 2001;20: 5623-5637. [DOI] [PubMed] [Google Scholar]
- 4.Cessna MH, Zhou H, Sanger WG, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002;15: 931-938. [DOI] [PubMed] [Google Scholar]
- 5.Onciu M, Behm FG, Downing JR, et al. ALK-positive plasmablastic B-cell lymphoma with expression of the NPM-ALK fusion transcript: report of 2 cases. Blood. 2003;102: 2642-2644. [DOI] [PubMed] [Google Scholar]
- 6.Gascoyne RD, Lamant L, Martin-Subero JI, et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood. 2003;102: 2568-2573. [DOI] [PubMed] [Google Scholar]
- 7.De Paepe P, Baens M, van Krieken H, et al. ALK activation by the CLTC-ALK fusion is a recurrent event in large B-cell lymphoma. Blood. 2003;102: 2638-2641. [DOI] [PubMed] [Google Scholar]
- 8.Dirks WG, Fahnrich S, Lis Y, Becker E, MacLeod RA, Drexler HG. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer. 2002;100: 49-56. [DOI] [PubMed] [Google Scholar]
- 9.Lamant L, Pulford K, Bischof D, et al. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol. 2000;156: 1711-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pulford K, Morris SW, Turturro F. Anaplastic lymphoma kinase proteins in growth control and cancer. J Cell Physiol. 2004;199: 330-358. [DOI] [PubMed] [Google Scholar]
- 11.Iwahara T, Fujimoto J, Wen D, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14: 439-449. [DOI] [PubMed] [Google Scholar]
- 12.Morris SW, Naeve C, Mathew P, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene. 1997;14: 2175-2188. [DOI] [PubMed] [Google Scholar]
- 13.Englund C, Loren CE, Grabbe C, et al. Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature. 2003;425: 512-516. [DOI] [PubMed] [Google Scholar]
- 14.Lee HH, Norris A, Weiss JB, Frasch M. Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature. 2003;425: 507-512. [DOI] [PubMed] [Google Scholar]
- 15.Liao EH, Hung W, Abrams B, Zhen M. An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature. 2004;430: 345-350. [DOI] [PubMed] [Google Scholar]
- 16.Bai RY, Dieter P, Peschel C, Morris SW, Duyster J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol Cell Biol. 1998;18: 6951-6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuefer MU, Look AT, Pulford K, et al. Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood. 1997;90: 2901-2910. [PubMed] [Google Scholar]
- 18.Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non-Hodgkin's lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol. 1997;17: 2312-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kutok JL, Aster JC. Molecular biology of anaplastic lymphoma kinase-positive anaplastic large-cell lymphoma. J Clin Oncol. 2002;20: 3691-3702. [DOI] [PubMed] [Google Scholar]
- 20.Ambrogio C, Voena C, Manazza AD, et al. p130cas mediates the transforming properties of the anaplastic lymphoma kinase. Blood. 2005;106: 3907-3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiarle R, Simmons WJ, Cai H, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11: 623-629. [DOI] [PubMed] [Google Scholar]
- 22.Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J. Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood. 2000;96: 4319-4327. [PubMed] [Google Scholar]
- 23.Rassidakis GZ, Feretzaki M, Atwell C, et al. Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood. 2005;105: 827-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slupianek A, Nieborowska-Skorska M, Hoser G, et al. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001;61: 2194-2199. [PubMed] [Google Scholar]
- 25.Turturro F, Arnold MD, Frist AY, Pulford K. Model of inhibition of the NPM-ALK kinase activity by herbimycin A. Clin Cancer Res. 2002;8: 240-245. [PubMed] [Google Scholar]
- 26.Ritter U, Damm-Welk C, Fuchs U, Bohle RM, Borkhardt A, Woessmann W. Design and evaluation of chemically synthesized siRNA targeting the NPM-ALK fusion site in anaplastic large cell lymphoma (ALCL). Oligonucleotides. 2003;13: 365-373. [DOI] [PubMed] [Google Scholar]
- 27.Wilda M, Fuchs U, Wossmann W, Borkhardt A. Killing of leukemic cells with a BCR/ABL fusion gene by RNA interference (RNAi). Oncogene. 2002;21: 5716-5724. [DOI] [PubMed] [Google Scholar]
- 28.Walensky LD, Kung AL, Escher I, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305: 1466-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rieber M, Strasberg-Rieber M. Induction of tumor cell apoptosis and chemosensitization by antisense strategies. Methods Mol Med. 2004;106: 205-214. [DOI] [PubMed] [Google Scholar]
- 30.McManus MT, Haines BB, Dillon CP, et al. Small interfering RNA-mediated gene silencing in T lymphocytes. J Immunol. 2002;169: 5754-5760. [DOI] [PubMed] [Google Scholar]
- 31.Timmons L. The long and short of siRNAs. Mol Cell. 2002;10: 435-437. [DOI] [PubMed] [Google Scholar]
- 32.Scherr M, Battmer K, Ganser A, Eder M. Modulation of gene expression by lentiviral-mediated delivery of small interfering RNA. Cell Cycle. 2003;2: 251-257. [PubMed] [Google Scholar]
- 33.Forte A, Cipollaro M, Cascino A, Galderisi U. Small interfering RNAs and antisense oligonucleotides for treatment of neurological diseases. Curr Drug Targets. 2005;6: 21-29. [DOI] [PubMed] [Google Scholar]
- 34.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296: 550-553. [DOI] [PubMed] [Google Scholar]
- 35.Piva R, Liu J, Chiarle R, Podda A, Pagano M, Inghirami G. In vivo interference with Skp1 function leads to genetic instability and neoplastic transformation. Mol Cell Biol. 2002;22: 8375-8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grignani F, Kinsella T, Mencarelli A, et al. High-efficiency gene transfer and selection of human hematopoietic progenitor cells with a hybrid EBV/retroviral vector expressing the green fluorescence protein. Cancer Res. 1998;58: 14-19. [PubMed] [Google Scholar]
- 37.Morgan R, Smith SD, Hecht BK, et al. Lack of involvement of the c-fms and N-myc genes by chromosomal translocation t(2;5)(p23;q35) common to malignancies with features of so-called malignant histiocytosis. Blood. 1989;73: 2155-2164. [PubMed] [Google Scholar]
- 38.Taulli R, Accornero P, Follenzi A, et al. RNAi technology and lentiviral delivery as a powerful tool to suppress Tpr-Met-mediated tumorigenesis. Cancer Gene Ther. 2005;12: 456-463. [DOI] [PubMed] [Google Scholar]
- 39.Rasola A, Geuna M. A flow cytometry assay simultaneously detects independent apoptotic parameters. Cytometry. 2001;45: 151-157. [DOI] [PubMed] [Google Scholar]
- 40.Zamo A, Chiarle R, Piva R, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21: 1038-1047. [DOI] [PubMed] [Google Scholar]
- 41.Mathas S, Hinz M, Anagnostopoulos I, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002;21: 4104-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiarle R, Gong JZ, Guasparri I, et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood. 2003;101: 1919-1927. [DOI] [PubMed] [Google Scholar]
- 43.Piva R, Gianferretti P, Ciucci A, Taulli R, Belardo G, Santoro MG. 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis in human malignant B cells: an effect associated with inhibition of NF-kappa B activity and down-regulation of antiapoptotic proteins. Blood. 2005;105: 1750-1758. [DOI] [PubMed] [Google Scholar]
- 44.Damm-Welk C, Fuchs U, Wossmann W, Borkhardt A. Targeting oncogenic fusion genes in leukemias and lymphomas by RNA interference. Semin Cancer Biol. 2003;13: 283-292. [DOI] [PubMed] [Google Scholar]
- 45.Pulford K, Lamant L, Espinos E, et al. The emerging normal and disease-related roles of anaplastic lymphoma kinase. Cell Mol Life Sci. 2004;61: 2939-2953. [DOI] [PubMed] [Google Scholar]
- 46.Heidel JD, Hu S, Liu XF, Triche TJ, Davis ME. Lack of interferon response in animals to naked siRNAs. Nat Biotechnol. 2004;22: 1579-1582. [DOI] [PubMed] [Google Scholar]
- 47.Soutschek J, Akinc A, Bramlage B, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432: 173-178. [DOI] [PubMed] [Google Scholar]
- 48.Sirna Therapeutics. Sirna Therapeutics announces updated phase I clinical trial data for Sirna-027: Results presented at the American Academy of Ophthalmology demonstrate clinical benefit of a siRNA compound in humans. http://phx.corporate-ir.net/phoenix.zhtml?c=141787&p=irol-newsArticle&ID=769878&highlight. Accessed December 8, 2005.
- 49.Coluccia AM, Perego S, Cleris L, et al. Bcl-XL down-regulation suppresses the tumorigenic potential of NPM/ALK in vitro and in vivo. Blood. 2004;103: 2787-2794. [DOI] [PubMed] [Google Scholar]
- 50.Amin HM, Medeiros LJ, Ma Y, et al. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene. 2003;22: 5399-5407. [DOI] [PubMed] [Google Scholar]
- 51.Hochhaus A, La Rosee P. Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistance. Leukemia. 2004;18: 1321-1331. [DOI] [PubMed] [Google Scholar]
- 52.Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev Drug Discov. 2004;3: 1001-1010. [DOI] [PubMed] [Google Scholar]
- 53.Das AT, Brummelkamp TR, Westerhout EM, et al. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol. 2004;78: 2601-2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431: 371-378. [DOI] [PubMed] [Google Scholar]
- 55.Wan W, Albom MS, Lu L, et al. Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large cell lymphoma cells. Blood. Prepublished on October 27, 2005, as DOI 10.1182/blood-2005-08-3254. [DOI] [PubMed]