

Abstract
Myeloid leukemias in AKXD23 mice contain proviral insertions at Evi1, resulting in transcriptional activation. Although Evi1 is clearly involved in leukemia, gene transfer studies in mice with Evi1 fail to cause leukemia, arguing that cooperating events are necessary. We reanalyzed AKXD-23 tumors for cooperating proviral insertion and found that each tumor had a proviral insertion in Sox4, which encodes an HMG-box transcription factor. RNA analysis revealed these insertions cause increased Sox4 expression. Overexpression of Sox4 in 32Dcl3 cells markedly inhibited cytokine-induced granulocyte maturation, as documented by morphologic and mRNA analysis. Sox4-expressing cells had higher levels of transcripts associated with proliferation, including Evi1. Conversely, in leukemic cells that express Sox4 and bear provirally activated Evi1, suppression of Sox4 with short hairpin RNAs resulted in down-regulation of both Sox4 and Evi1. By cotransfection studies, Sox4 is able to transactivate the AKV long terminal repeat, which likely explains how Sox4 transcriptionally up-regulates provirally activated Evi1; however, Sox4 does not appear to regulate the native Evi1 promoter. We propose that Sox4 proviral activation is selected for in the setting of prior proviral activation of Evi1, because it transactivates the relatively weak LTR of AKV leading to higher Evi1 expression and consequent block to differentiation. (Blood. 2006;107:733-741)
Introduction
Acute myeloid leukemia (AML) is a disease characterized by the uncontrolled proliferation of immature hematopoietic precursor cells in the bone marrow. Leukemia, like other cancers, is believed to be a multistep process involving activation of oncogenes and inactivation of tumor suppressor genes.1 Dysregulation of these genes can occur through many mechanisms, including, at least in mice, retroviral mutagenesis. Proviral integration can result in increased production, truncation, or disruption of mRNA transcripts, and using the viral sequences as tags, it is possible to clone the integration sites. The prevalence of the same insertion site in multiple tumors suggests that this integration event plays a causative role in the pathogenesis of the disease.
The Evi1 locus (ecotropic viral integration site 1) was identified as a common site of viral integration in the myeloid tumors of AKXD-23 mice2,3 and has also been found as a common site of viral insertion in a variety of myeloid tumor cell lines derived from the neoplastic spleens of mice infected with other murine leukemia viruses, including the Cas-Br-E and Moloney murine leukemia viruses.3,4 The human EVI1 gene at 3q26 is subject to structural alteration and/or activation by chromosomal translocation in human myeloid leukemias and myelodysplasia.5-7 The Evi1 locus encodes a series of protein isoforms that variably contain up to 10 C2H2 zinc fingers that bind DNA in a sequence-specific manner,8-11 as well as a repression domain that binds CtBP,12 consistent with its demonstrated role in transcriptional repression.13 Two recent reports document the identity of EVI1 target genes, which include Gata2.14,15 The mechanisms by which Evi1 induces leukemia are thought to include block to myeloid differentiation16,17 and stimulation of cell-cycle progression.18-20
Although a link between Evi1 overexpression and AML has been established, it remains unclear exactly how Evi1 functions in the development of the disease. The latency of AML in the AKXD-23 mouse strain (> 400 days) suggests that the pathogenesis of AML is a multistep process involving the cooperation of several genes. It has been demonstrated that engineered overexpression of Evi1in the hematopoietic cells of transgenic mice21 or via bone marrow transplantation22,23 does not result in leukemia, which also suggests the need for multiple oncogenic hits. To date, little is known about the additional factors required for acting in concert with Evi1 in the genesis of AML, and what other molecular genetic mutations have occurred in tumors where Evi1 is overexpressed. Therefore, we have focused our studies on searching for additional sites of proviral integration that occur in retrovirally induced tumors, specifically the AKXD-23 tumors and several myeloid tumors arising in NFS mice. We have identified a second gene locus, Sox4, which is targeted by viral integration in all of the AKXD-23 tumors and at least one of the Cas-Br-MuLV-induced myeloid tumors from NFS mice and present evidence that SOX4 may augment provirally activated Evi1 expression through transactivation of the proviral LTR.
Materials and methods
Identification of retroviral insertion sites
Inverse polymerase chain reaction (PCR) and cloning were performed essentially as reported.24 Sequence analysis of inverse PCR clones was performed by the Keck Foundation Biotechnology Resource Laboratory at Yale University (http://info.med.yale.edu/wmkeck). Proviral insertion sequences trimmed of vector and viral sequences were compared with the Ensembl DNA database using the BLAST function.25 Genes within 50 kilobases (kb) were considered a direct hit and are reported in the upper portion of Table 1.
Table 1.
Sites of proviral insertions in AKXD-23 leukemias
| No. insertions | Chromosome | Location | |
|---|---|---|---|
| Characterized genes | |||
| Sry box containing gene 4* (Sox4) | 11 | 13 | 26,176,056 |
| ATP-binding cassette, sub-family D (ALD), member 2 (Abcd2) | 1 | 15 | 89,886,136 |
| Tumor necrosis factor receptor superfamily, member 13C (Tnfrsf13C) | 1 | 15 | 81,345,397 |
| Formin binding protein 4* (Fnpb4) | 1 | 2 | 89,130,141 |
| Transducer of ERBB2-2 (Tob2) | 1 | 15 | 80,969,800 |
| Neural-salient serine/arginine-rich (Nssr) | 1 | 4 | 131,050,573 |
| Growth factor independent 1* (Gft1) | 1 | 5 | 101,829,265 |
| Ring finger protein 10 (Rnf10) | 1 | 5 | 109,340,985 |
| Putative genes | |||
| Inositol 1,4,5-triphosphate receptor 1 | 1 | 6 | 107,018,039 |
| Cytosolic purine 5′-nucleotidase | 1 | 19 | 44,656,320 |
| Sialic acid binding immunoglobulin-like lectin | 1 | 7 | 35,821,355 |
| Poly (A) polymerase | 1 | 11 | 22,967,435 |
| Novel genes | |||
| Uncharacterized, mCG129661 | 1 | 5 | 116,293,267 |
| Uncharacterized, mCG6830 | 1 | 11 | 125,392,932 |
| No genes hit | |||
| 1 | 11 | 126,342,856 | |
| 1 | 7 | 10,176,626 | |
| 1 | 13 | 62,107,419 | |
| 1 | 6 | 30,234,758 |
Gene has been previously identified as a common site of retroviral insertion.
Cell lines
The NFS-60, NFS-58, NFS-78,26 DA-1,27 32Dcl3,28 32Dwt18,29 and erythroid-myeloid-lymphoid (EML) cells30 were cultured as described in the associated citations. For differentiation of 32Dwt18 cells and 32Dwt18 stable cell clones, cells were washed in PBS and reseeded in IMDM culture media29 lacking WEHI-3B, conditioned media and containing 1 U/mL Epo (Epoetin alfa, Procrit; Amgen, Thousand Oaks, CA).
Plasmids
The MIGR1 and MIGR1 p185 (Bcr-Abl) plasmids were gifts of Warren Pear.31 The pTATAluc-SX3x and pTATAluc plasmids were gifts of Michael Wegner.32 The pBabePuro plasmid and pBP-Evi1 plasmids have been described33 (T. Husain, M. Tran, P. Clark, B. Yatsula, V. Tsakraklides, S. Williams, D. Krause, and A.S.P., unpublished data). The pAU-CAT plasmid was obtained from Jack Lenz. The MIGR1-Sox4 plasmid was constructed by annealing these complementary oligos (5′-TCGAGGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGCGTTGAG-3′ and 3′-CCCATTCGGATAGGGATTGGGAGAGGAGCCAGAGCTAAGATGCGCAACTCCTAG-5′) then kinasing and ligating them into XhoI and BamHI-digested pIRES2-EGFP vector (Clontech, Mountain View, CA). The Sox4 cDNA was cloned from mouse BAC DNA (RP23-262C6) by high fidelity PCR (Boehringer Mannheim, Mannheim, Germany) using primers 5′-CTACAAGCTAGCCAAGCCGGGGCCATGGTACAACAG-3′ and 5′-CTGGACCTCGAGGTAGGTGAAGACCAGGTTAGAGATGC-3′, and the product was digested with XhoI and NheI and ligated into XhoI-NheI-digested pV5IRES2-EGFP, and the resultant clones were verified by sequence analysis. The MIGR1-Sox4 construct was created by isolating Sox4-V5 from pSox4V5IRES2-EGFP using NheI-BamHI digestion and ligating this as a blunt-end fragment into a HpaI-linearized MIGR1 vector.
Southern and Northern blot analysis
DNA (20 μg) was digested with 40 U XbaI in a total of 80 μL volume at 37°C for 16 hours, precipitated with EtOH, resuspended in 20 μL TE, heated at 65°C for 10 minutes, separated on a 0.8% agarose 1 × TPE gel, and transferred to Hybond-N membrane (Amersham, Piscataway, NJ) using standard upward capillary transfer.34 Total RNA was isolated from cells using TRIzol Reagent (Invitrogen, Carlsbad, CA) or, for WT18 stable cell clones, RNeasy mini columns (Qiagen, Valencia, CA). Total cellular RNA (10 μg) was separated by formaldehyde-agarose gel electrophoresis and transferred to Hybond-N membrane using standard Northern blotting techniques.34 The Sox4 probe or murine lactoferrin (shown in Figure 1) was labeled with [α-32P] dCTP using random priming (Roche, Indianapolis, IN) and hybridized at 65°C as described.35 Hybridized blots were exposed to Kodak X-Omat film (Kodak, Rochester, NY).
Figure 1.

Identification of retroviral insertions clustered in the 3′ untranslated region of Sox4. Shown is a schematic of the Sox4 locus, containing the retroviral insertions identified in the AKXD-23 tumor DNA samples. The black line represents a 19.3-kb XbaI fragment containing the Sox4 gene with additional enzyme sites shown below. The single exon encoding the SOX4 protein is depicted by the large gray rectangle. The size of the 5′ and 3′ untranslated regions, represented by thick gray lines, was estimated based on the partial Sox4 mRNA sequence (GenBank) and the reported size of Sox4 mRNA.53 The MuLV insertions identified by SacII digestion and inverse PCR are shown as thin vertical lines. The numbers and arrows above each insertion indicate the tumor number from which it was identified and the orientation of the provirus relative to Sox4 transcription. The probe used to detect Sox4 in both Southern and Northern blots analyses is depicted by the dashed line.
Transfections
For transient transfection assays, NIH 3T3 cells were seeded at 105 cells/60-mm dish (in triplicate for each condition) 1 day prior to transfection, and DNA (1.5 μg expression plasmid and 1.0 μg reporter plasmid) was introduced into cells using 7.5 μL FuGene 6 transfection reagent (Roche) according to manufacturer's instructions. Cells were harvested 48 hours following transfection using a Luciferase Reporter Gene Assay, High Sensitivity kit (Roche). Cells were lysed in 250 μL, and 50 μL lysate was assayed using a Berthold LB9501-0 luminometer (Perkin-Elmer, Boston, MA). For stable transfections of 32Dwt18 cells, plasmid DNAs (MIGR1 expression plasmid with pBABEpuro selection plasmid) were electroporated as described.29 GFP+/puromycin cells were cloned by limiting dilution in medium containing 2 μg/mL puromycin. For the chloramphenicol acetyl transferase (CAT) reporter assays, NIH 3T3 cells were plated at 0.5 × 106/60-mm plate, transfected with DNA in a calcium phosphate precipitate,36 and shocked with glycerol. After 48 hours, the cells were harvested and assayed as described.37
Western blotting
Nuclear extract29 (40 μg) was resolved by SDS-polyacrylamide (10%) gel electrophoresis, electroblotted, blocked, and reacted with anti-V5 mouse monoclonal antibody (1:5000 dilution; Invitrogen). Bands were identified with ImmunoPure goat anti-mouse IgG + IgM peroxidase-conjugated secondary antibody (1:4000 dilution; Pierce Rockford, IL) using an electrochemiluminescence (ECL) Western Blotting Detection Reagent (Amersham Pharmacia Biotech, Piscataway, NJ). For Bcr-Abl detection, blots were incubated with anti-abl mouse monoclonal antibody (1:1000 dilution; BD Pharmingen, San Diego, CA).
Microarray studies
Total RNA was isolated from cells by guanidinium/phenol isolation38 using TRIzol reagent (Invitrogen). Glass-based slides bearing 13 400 spots of single-stranded 70mers representing mouse genes were hybridized with labeled cDNAs as described.15 Scanning was done on an Axon scanner at the Yale Microarray Facility (http://info.med.yale.edu/wmkeck/dna_arrays.htm). Quantitation of the hybridization signal was done with GenePix software (Axon Instruments, Foster City, CA). The data were subjected to (1) elimination of low-intensity signals (summed intensities < 100); (2) normalization of data, based on the sum of intensities for each dye; (3) calculation of log2 ratio; (4) adjustment of the log2 ratio so that the median equalled 0.
Quantitative real-time PCR
Quantitation of mRNA levels for Actb, Sox4, and Evi1 transcripts was done using SybrGreen intercalating dye on an iCycler (Bio-Rad, Hercules, CA) thermal cycler as described.15 PCR reactions (20 μL total volume, in triplicate) were set up using a mix from Invitrogen. Cycle threshold values were used to calculate a ratio of β-actin to Evi1 relative transcript levels by the equation 2(Ctβactin - CtEvi1).
RNAi studies
DA-1 cells were infected by spinfection39 with recombinant retrovirus containing a puromycin resistance marker and either short hairpin RNAs specific for Sox4 (sh31 or sh51) or control shRNA against luciferase. The construction of the luciferase virus is described elsewhere (Y.-Y.X., D. Tuck, P. Clark, D. Yue, M. Lopingco, A. Khanna-Gupta, H. Sun, N. Berliner, and A.S.P., manuscript in preparation); construction of sh31 and sh51 involved annealing complementary oligonucleotides and ligating the resultant duplexes into EcoR1-BamHI-cut plasmid pSIREN-RetroQ (BD Biosciences, San Jose, CA). For sh31, the oligonucleotides used were 5′-GATCCGGGCGGCTGCATCGGTCTCTATCAAGAGAAGAGAACGATGCAGCCGCCTTTTTTACGCGTG-3′ and 5′-AATTCACGCGTAAAAAAGGCGGCTGCATCGTTCTCTTCTCTTGATAGAGAACGATGCAGCCGCCCG-3′. For sh51, the oligonucleotides were 5′-GATCCGCCCGGCTCAGGCTCCCACTATCAAGAGAAGTGGGAGCCTGAGCCGGGTTTTTTACGCGTG-3′ and 5′-AATTCACGCGTAAAAAACCCGGCTCAGGCTCCCACTTCTCTTGATAGTGGGAGCCTGAGCCGGGCG-3′. Recombinant retrovirus was produced by cotransfecting the pSIREN-RetroQ vector together with pNCA40 (added to boost viral titers) into Phoenix cells41 using calcium phosphate precipitation.36 Virus supernatants were harvested at 48 hours after transfection and frozen. Cells (1 × 106 in 5 mL) were spinfected as described.39 Forty-eight hours after spinfection cells were culture in puromycin (2 μg/mL) selection media. After 2 weeks in puromycin media the cells were harvested for RNA and real-time PCR was performed for Sox4, Evi1, and β-actin genes.
Results
Inverse PCR cloning of proviral insertion sites in AKXD-23 myeloid leukemias
With the goal of identifying events that cooperate with Evi1 activation in myeloid leukemogenesis, we analyzed a set of myeloid leukemia DNAs from AKXD-23 mice, all of which were known to have proviral insertions at Evi1.2 We performed inverse PCR using primers specific for ecotropic retroviruses. From 8 tumors, we isolated 28 inverse PCR products for which we determined the DNA sequence. Sequences were analyzed against the mouse genome, using the Ensembl database.25 The results of this analysis are shown in Table 1. Clearly evident in this analysis was that Sox4 was a common site of proviral insertion, occurring in 8 of 8 tumors analyzed. All of the insertions were located in the 3′ UTR, leaving the coding potential of the gene intact (Figure 1). The orientation of the proviral insertions in Sox4 was the same for all insertions, being in the same transcriptional orientation as the gene. For 3 tumors, no. 91, no. 144, and no. 163, we identified 2 different Sox4 insertions (Figure 1). Whether these are at both alleles of Sox4 within all tumor cells or are due to oligoclonality of the tumor mass is not known.
Sox4 is a single exon gene for which the initiation of transcription and polyadenylation sites are not precisely defined. However, the sequence for numerous Sox4 cDNAs have been deposited in the NCBI sequence database, and, on the basis of the comparison of these sequences to the genomic sequence, it is evident that polyadenylation occurs downstream of the most 3′ insertion that we identified (insertion in tumor no. 144; Figure 1).
Analysis of leukemic cell lines reveals Sox4 activation by proviral insertion in NFS-60 cells
We presume that the effect of the proviral insertion is to increase the half-life of the mRNA by causing premature polyadenylation within viral sequences, which likely results in effective removal of an mRNA instability sequence located further 3′ in the UTR. We wanted to test this by analysis of RNA. However, because RNA was not available for the myeloid tumors from the AKXD-23 mice, we screened a number of cell lines that had known proviral insertions at Evi1 to see whether we could identify one with a proviral insertion at Sox4. DNAs from 4 such cell lines were analyzed by Southern blot to detect the presence of a provirus-induced restriction fragment length polymorphism, when hybridized with a probe for Sox4 (Figure 2A). This revealed that the tumor-derived NFS-60 cell line has such a polymorphism, whereas in 2 other tumor cell lines, NFS-58 and DA-1, and in the 32Dcl3 myeloid progenitor cell line, no such polymorphism was detected. It is not possible that the polymorphism seen in NFS-60 is a naturally occurring sequence variation, because all NFS tumors were derived from the same inbred strain (NFS), and this polymorphism is not seen in the other NFS tumors (Figure 2A). From the size of the polymorphism and the restriction enzyme used, it is clear that the proviral insertion is within the 3′ UTR, similar to that seen in the AKXD-23 tumors.
Figure 2.

Survey of hematopoietic cell lines for retroviral insertions in Sox4 and expression of Sox4 mRNA. (A) Southern blot analysis of the NFS-58, DA-1, NFS-60, and EML cell lines. XbaI-digested DNA was separated by agarose gel electrophoresis, transferred to nylon membrane, and probed with the 3′ UTR Sox4 fragment shown in Figure 1. XbaI digest of wild-type alleles of murine Sox4 is predicted to yield a 20-kb band. The arrows indicate provirus-induced polymorphisms that are only seen in the NFS-60 DNA. (B) Northern blot analysis of logarithmically growing NFS-58, NFS-60, DA-1, 32Dcl3, and EML cell lines. Total RNA (10 μg) from each cell line was separated by formaldehyde agarose gel electrophoresis, transferred to nylon membrane, and sequentially probed with the 3′ UTR Sox4 fragment (Figure 1) and a murine β-actin fragment. Size markers are indicated on the right. (C) Assessment of Evi1 mRNA expression in EML cells (top row) by Northern blot analysis using a radiolabeled probe for Evi1. EML cells are transfected with either vector or with Evi1 expression plasmid as indicated. The same blot was hybridized for β-actin (bottom row).
Because the NFS-60 cell line harbored a proviral insertion at Sox4, we analyzed its RNA for Sox4 expression by Northern blot analysis. This clearly revealed an up-regulation of the gene because of the proviral insertion (Figure 2B). Interestingly, 3 transcripts are observed: 1 at the normal size of 4.4 kb, and 2 additional transcripts of 9.5 kb and 3.0 kb. These likely represent mRNAs from the provirally activated locus, with the smaller transcript being derived from polyadenylation in the 5′ LTR, and the longer one from polyadenylation in the 3′ LTR, with removal of the gag and pol region by splicing out of the retroviral intron. Expression of normally sized Sox4 mRNA transcript was present in the DA-1 myeloid leukemia cell line, which has a proviral insertion at Evi1 but no detectable Sox4 insertion within a 20-kb region of the locus (Figure 2A), and in the EML cell line, a nontumorigenic multipotent progenitor cell line that can differentiate into erythroid, myeloid, and lymphoid cells.30 EML cells do not express Evi1 (T. Husain, M. Tran, P. Clark, B. Yatsula, V. Tsakraklides, S. Williams, D. Krause, and A.S.P., manuscript submitted) and have no proviral insertion at Evi1 (data not shown).
Sox4 blocks myeloid differentiation of 32Dwt18 cells
We were interested in determining the mechanism by which Sox4 overexpression contributed to leukemia. Two effects were considered: a loss of cell-cycle control and a block to differentiation. To perform these studies, we first developed a retroviral vector with which to transfer Sox4 with facility into cells of interest. We thus inserted a Sox4 cDNA engineered with a V5 epitope tag at the C terminus into the MIGR1 retroviral vector (Figure 3A). To test whether this construct expressed functional Sox4 protein, we performed cotransfection studies with a reporter construct that has concatemerized SOX4 binding sites upstream of a minimal promoter and a luciferase reporter. Although empty vector showed no ability to activate the reporter, our MIGR1-Sox4 construct had potent transactivating ability, indicating that biologically active protein was being produced.
Figure 3.

Analysis of the Sox4 retroviral vector. (A) Schematic of the MIGR1-based retroviral vectors. The complete cDNA of Sox4 (light gray rectangle) including a C-terminal V5 epitope tag (dark gray square) was inserted into the MIGR1 vector. The MIGR1 and the MIGR1-p185 vectors were gifts of Warren S. Pear. The MIGR1-p185 vector contains the complete Bcr-abl cDNA (dotted rectangle). All vectors contain an IRES (internal ribosome entry site) and the EGFP cDNA. The arrow depicts the start of transcription. (B) Results of transient cotransfection assays. NIH 3T3 cells were transiently cotransfected with either 5 μg MIGR1 or MIGR1-Sox4 expression plasmid (as indicated on the x-axis) and 1 μg of either reporter plasmid, pTATAluc (▪) or pTATAluc Sx3X (□). The pTATAluc Sx3X contains 3 copies of the SOX4 consensus binding site. Cells were harvested 48 hours following FuGene transfection, and lysates from an equal number of cells were analyzed for relative luciferase units (RLUs). The results are presented as the RLU value of cells transfected with both reporter and expression plasmids divided by cells transfected by reporter alone. Results are presented as an average of 2 experiments with the standard error indicated. (C) Western blot of 32Dwt18 clones. Single-cell-derived 32Dwt18 clones expressing GFP were isolated and expanded into clonal cell lines (Sox4 lines A-C, MIGR1 lines A-B, and p185 lines A-B). Nuclear extracts were isolated from an equal number of cells from each cell line. Equal volumes of extract were separated by SDS-PAGE electrophoresis and transferred to nitrocellulose membrane. Membranes were sequentially probed with anti-V5 and anti-abl antibodies. The anti-V5 antibody detects an approximately 47-kDa band in the Sox4 clones (top row), and the anti-abl antibody detects an approximately 185-kDa band in the p185 clones.
We then sought to address what effect Sox4 overexpression has on myeloid cell growth and differentiation. As a test cell line, we used the 32Dwt18 cell line, a derivative of 32Dcl329 that expresses the ectodomain of the erythropoietin receptor fused to the cytoplasmic domain of the G-CSF receptor. These cells differentiate to granulocytes on treatment with erythropoietin, while maintaining a high viability (Figure 4). We transduced the 32Dwt18 cell line with MIGR1-Sox4 and isolated several single-cell-derived clones that expressed the GFP marker and, by Western blot, also expressed high levels of SOX4 protein, as detected by using the V5 epitope tag (Figure 3C). As a negative control in this experiment, we also developed clonally derived 32Dwt18 cells transduced with empty MIGR1 vector, and these showed no immunodetectable V5 protein on Western blot (Figure 3C). To contrast the effect of Sox4 against an established myeloid oncogene, we included in this experiment a MIGR1-Bcr-Abl construct. Two 32Dwt18-derived cell lines were established that showed p185 expression on Western blot using an anti-Bcr-Abl antibody (Figure 3C).
Figure 4.
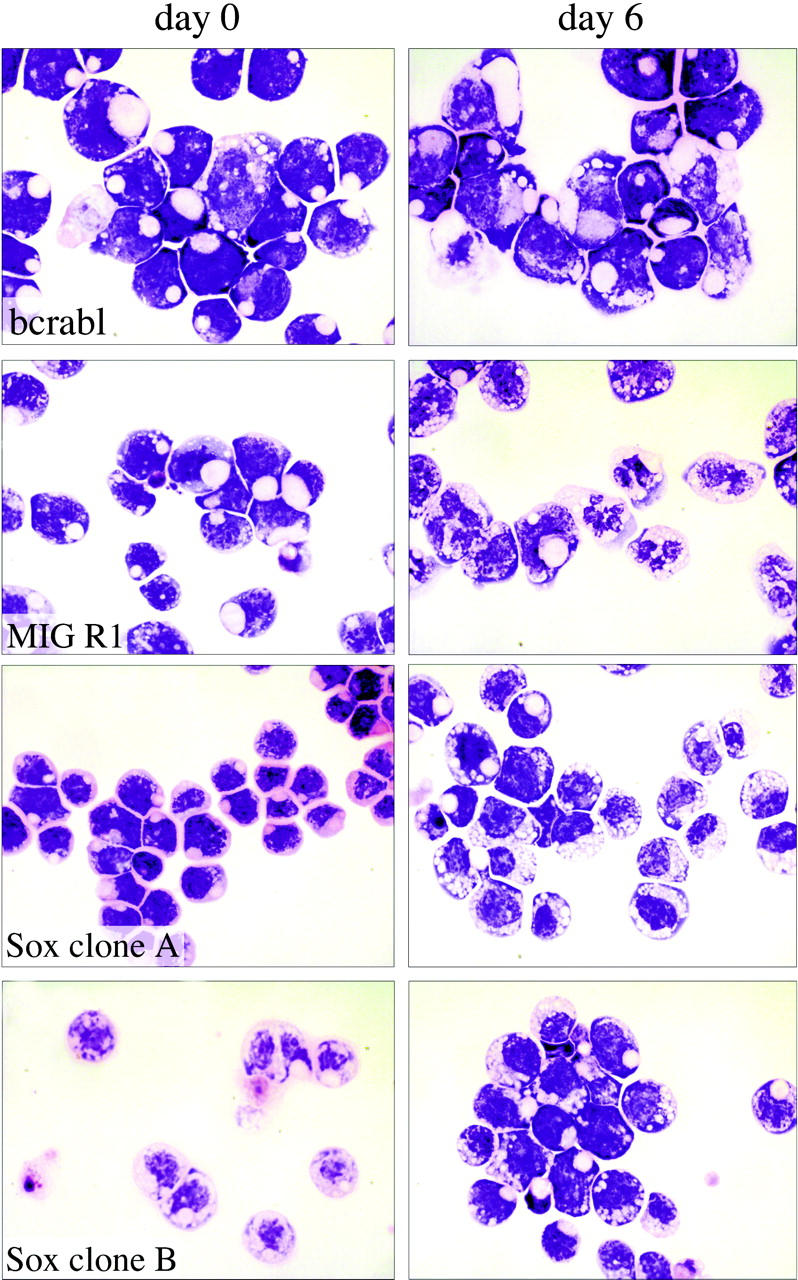
Analysis of the effects of Sox4 on myeloid cell differentiation. Single-cell-derived 32Dwt18 clones (MIGR1-Bcr-abl, MIGR1, MIGR1-Sox4 Clone A, MIGR1-Sox4 Clone C) were treated to differentiate in the presence of Epo (6 U/mL). Prior to treatment with Epo, a fraction of the culture was harvested and washed with PBS, and cytospin preparations were made. The rest of the culture was washed with PBS and replaced in media containing Epo and lacking IL-3. Each culture was refed every 48 hours. At day 6, samples of the cells were washed with PBS, and cytospin preparations were made. Slides were stained with Wright-Giemsa stain and analyzed by light microscopy to visualize morphologic changes. Images were acquired digitally on an Olympus Vanox microscopes (Olympus, Tokyo, Japan) equipped with a SPIano 100× oil immersion lens (NA = 1.40) and a ProgRes camera (Kontron America, Poway, CA) using AutoCyte image-capture software (AutoCyte, Burlington, NC). Images were cropped and assembled using Adobe Photoshop (Adobe, San Jose, CA).
Having developed cell lines expressing SOX4, BCR-ABL with 2 empty vector-transduced lines as controls, we then tested whether Sox4 had any effect on cell proliferation or differentiation. Accordingly, we tested cell proliferation by recording cell number in cultures of exponentially growing cells. Although BCR-ABL had a demonstrable effect in increasing the proliferative potential of the cells, Sox4 expression had no measurable effect, being essentially the same as the vector control cells (data not shown).
We next tested whether Sox4 affected the ability of the 32Dwt18 cells to differentiate into mature granulocytes when exposed to Epo, because this is an effect that is seen with a number of myeloid leukemia genes.42 To assess this, we first examined morphologic differentiation of the cells. Cultures of Sox4-expressing and Bcr-Abl-expressing clones, as well as vector control cells, were washed in PBS and placed in media containing 6 U/mL Epo. At various days thereafter, Giemsa-stained cytospin smears were examined for morphologic evidence of differentiation. During the 12-day induction, a large proportion of the vector-transduced cells acquired the features of differentiated myeloid cells, including a lobated nucleus and the presence of cytoplasmic granules, which are evident at day 6 in some of the cells as depicted in Figure 4. However, the Sox4- and Bcr-Abl-expressing clones exhibited a complete absence of morphologic maturation and remained as a uniform population of immature cells with a large nuclear-to-cytoplasmic ratio, and no cytoplasmic evidence of differentiation (Figure 4). This phenomenon was seen in all 3 of the Sox4 clones tested, as well as in all of the BcrAbl clones, clearly indicating that Sox4, as with BcrAbl, causes a block to myeloid maturation, as assessed in this cell culture model.
To confirm this impression that Sox4 abrogates Epo-induced myeloid differentiation, we used Northern blot analysis to assess the expression of mlf, which encodes lactoferrin, a component of the secondary granules, and a key marker of myeloid maturation. This study revealed that indeed, with Epo treatment of the Sox4-transduced 32Dwt18 cells, there was little if any induction of mlf expression, confirming at the biochemical level what the morphology had indicated (Figure 5). A failure to activate mlf was also seen in Epo-treated cultures of 32Dwt18 cells expressing BcrAbl, whereas in vector control cells, there was clearly evident activation of mlf after 3 to 6 days of treatment, with some variability from clone to clone (Figure 5).
Figure 5.

Northern blot analysis of MIGR1-Sox4 clones during myeloid differentiation. Single-cell-derived 32Dwt18 clones (Sox4 A-C, MIGR1 A-C, and Bcr-abl A-B) and wild-type 32Dwt18 cells were treated to differentiate in the presence of Epo (6 U/mL). Cultures were harvested from prior to differentiation treatment (day 0) or at days 3 and 6 following placement in media containing Epo and lacking IL-3. RNA was extracted from cells using RNAeasy (Qiagen). Total RNA (10 μg) was separated on a formaldehyde agarose gel, transferred to nylon membrane, and sequentially probed with murine lactoferrin (mlf) and β-actin.
Microarray analysis of Sox4 clones reveals significant differences in transcriptional response of 32Dwt18 cells to Epo
We were interested in obtaining a broader view of the effect of Sox4 expression on Epo-induced myeloid differentiation, and so we performed analysis of the spectrum of genes expressed in these cells at 4 days following Epo treatment, comparing these to vector-transduced cells. Two hybridizations were performed, with RNA from 2 different clones, and the data were remarkably consistent. Figure 6 shows an x-y scatter plot comparing the log2 ratio (Sox4 versus vector control) of expression level for each gene obtained in experiment 1 (slide 74; y-axis) versus experiment 2 (slide 198; x-axis). This revealed that for the majority of genes, there was excellent correlation between the 2 experiments. We focused our attention on genes that had a log2 ratio of at least 1 or -1 and less in both experiments; Table 2 lists these genes.
Figure 6.

XY scatter plot of cDNA microarrays. MIGR1 and Sox4 cell lines were compared on a 13.4K mouse oligonucleotide cDNA microarray. Genes of interest indicating up-regulation or down-regulation because of induction were calculated statistically using log base 2. Those with a ratio greater than 1 were considered of interest. An XY scatter plot of 2 independent microarrays (slide 74 and slide 198) showed a linear trend. Those data points at either end indicate those genes of greatest interest in 2 independent experiments. The values on the x- and y-axes indicate the ratio calculated.
Table 2.
Differentially expressed genes identified by microarray
| Ratio (log2) Sox4/vector | |
|---|---|
| Genes higher in vector control | |
| Granulocyte differentiation markers | |
| Pira1 | −3.1 |
| Pira3 | −3.1 |
| Prg2 (eosinophil major basic protein) | −2.0 |
| Ltf (Lactoferrin) | −4.7 |
| Ngp (Neutrophilic granule protein) | −3.2 |
| Lzp-s (P lysozyme structural) | −3.0 |
| Cmkbr5 (Chemokine receptor 5) | −2.4 |
| Lrg (Leucine-rich α-2 glycoprotein) | −2.4 |
| Other genes | |
| Chi311 (Chitinase 3-line 1) | −4.1 |
| Idb2 (Inhibitor of DNA binding 2) | −3.1 |
| Nif311 (Ngg1 interacting factor 3-like 1) | −3.1 |
| Genes higher in Sox4 | |
| Cytokines | |
| II4 (Interleukin-4) | 2.3 |
| II6 (Interleukin-6) | 1.4 |
| Transcription factors | |
| Tcfec (Transcription factor TFE-C) | 1.5 |
| Zfpm1 (FOG-1) | 1.8 |
| Evi1 | 1.4 |
| Signaling molecules | |
| Lat (Linker for activation of T cells) | 1.1 |
| Mapk12 (Mitogen-activated protein kinase 12) | 2.0 |
In vector control cells, there was higher expression of a number of genes that are known to be up-regulated in granulocytic differentiation. These include Prg2 which encodes the major basic protein of eosinophils; Ltf, encoding lactoferrin; Ngp, encoding neutrophil granule protein; Lrg, encoding leucine-rich α2 glycoprotein43; and Lzp-s, encoding P lysozyme. Interestingly, 7 members of the Pira family of genes are higher in the vector-transduced cells after Epo treatment relative to the Sox4-expressing cells. These genes encode activating receptor partners for a variety of cell-surface receptors, including the Fc receptor. Thus, one would expect these genes to be activated in granulopoiesis. These data are completely consistent with those presented in Figure 5, and reflect the block to differentiation imposed by Sox4.
In contrast to the genes expressed at higher levels in vector control cells, the genes expressed 2-fold or more in Sox4-expressing 32Dwt18 cells at 4 days after Epo administration are much more varied in function and in typical tissue expression. This group includes 2 cytokine genes (Il4 and Il6), a receptor (F2r, encoding the thrombin receptor), several signaling molecules (Lat, involved in T-cell receptor signaling; and Mapk12, encoding a MAP kinase family member), a gene encoding an actin sequestering protein (Tmsb10, encoding thymosin beta 10), a serine protease inhibitor (serpin) family member (Spi2-1), and 3 transcription factors: Zfpm1, encoding the GATA1-interacting protein FOG-1; Tcfec, encoding the helix-loop-helix protein TCF-C; and, interestingly, Evi1.
Sox4 causes up-regulation of Evi1 mRNA levels in 32Dwt18 cells
We were interested in pursuing the possibility that the increase in Evi1 expression in Sox4-transduced cells provided the mechanism by which Sox4 may cooperate with Evi1 in leukemogenesis. It is known that 32Dwt18 cells have a proviral insertion at Evi1 and overexpress it at the mRNA and protein level,44 and considered it possible that Sox4 may cause up-regulation of Evi1 expression. In 32Dwt18 cells, Evi1 is activated via proviral insertion but that this level of Evi1 is not enough to block cytokine-induced maturation.45 Further overexpression of Evi1 in 32Dcl3 cells is able to block differentiation.16,44 Thus, the capacity of the cells to differentiate on cytokine induction appears to depend on the amount of EVI1 present in the cell; in normal 32Dcl3 or 32Dwt18 cells, the level of EVI1 is below the threshold needed to block differentiation, but further overexpression causes that block.
To confirm our microarray results that Sox4 overexpression causes up-regulation of Evi1, we performed quantitative PCR analysis of Evi1 mRNA levels in Sox4-transduced and control cells. This revealed that in 2 different Sox4-transduced clones, Evi1 is up-regulated: in clone 1, it is 4.1-fold higher than vector-transduced cells, whereas in clone 2 it is 2.6-fold higher (Figure 7A).
Figure 7.

SOX4 transactivates EVI1 via AKV LTR. (A) Quantitative PCR analysis of Evi1 expression in 32Dwt18 cells. Two different Sox4-transduced clones are compared with 1 vector-transduced line and parental cells. mRNA levels are expressed as ratio to β-actin. (B) Effect of shRNA for SOX4 on mRNAs for Sox4 (top) and Evi1 (bottom) is assessed by quantitative PCR. (C) Cotransfection studies in NIH 3T3 cells using a AKV-CAT reporter transfected with increasing amounts of Sox4 expression plasmid.
Next, we assessed this in DA-1, a myeloid leukemia cell line that is known to express both Evi1 and Sox4. In this cell line, Evi1 is activated via proviral insertion2 and expresses high levels of mRNA.15,45 Our studies here showed that DA-1 cells express high levels of Sox4 (Figure 2), but do not show a polymorphism within the 20-kb XbaI fragment. However, our analysis of proviral insertions in primary tumors showed that insertions can occur at least up to 50 kb away from Sox4 (Table 1), and so our Southern blot analysis may have failed to reveal a proviral insertion at the locus. We postulated that the high expression of Sox4 resulted in up-regulation of Evi1 in DA-1 cells. If this were the case, then suppression of Sox4 expression by short hairpin RNAs (shRNAs) would cause down-regulation of Evi1 mRNA levels. We developed 2 different shRNAs against Sox4 (sequences 31 and 51) and inserted them into retroviral vectors downstream of a polIII promoter; retroviral supernatants containing sh31 or sh51 were used to infect DA-1 cells. These shRNAs caused significant suppression of Sox4 expression, to 42% and 43% of normal levels, in sh31- and sh51-transduced cells, respectively (Figure 7B). We then examined Evi1 levels by quantitative real-time PCR and found them to be suppressed to roughly half the values in cells transduced with a luciferase-specific control shRNA (Figure 7). These data support the idea that Sox4 overexpression results in up-regulation of Evi1.
We considered 2 possible mechanisms by which SOX4 could cause increased expression of Evi1: either SOX4 is activating the endogenous Evi1 promoter, or it is acting on the proviral insertion to cause up-regulation of Evi1. To evaluate the former possibility, we analyzed Evi1 expression levels in EML cells that express high levels of Sox4 (Figure 2). By both Northern blot (Figure 2) and Western blot (Dong xian Yue and A.S.P., December 4, 1997, unpublished observation), we find no evidence of Evi1 expression in these cells, indicating that SOX4 does not activate Evi1 expression via its native promoter, making it likely that it activates the proviral LTR.
To assess whether SOX4 could transactivate the retroviral LTR, we performed cotransfection studies in NIH 3T3 cells with a SOX4 expression plasmid and a AKV-CAT reporter construct that contains the LTR from the AKV virus (the endogenous virus present in the AKXD-23 mouse line) linked to a chloramphenicol acetyl transferase gene (Figure 7C). Cells were transfected with 1 μg AKV-CAT reporter and increasing amounts of pSG5-SOX4-V5 expression plasmid, and assays for CAT performed at 48 hours after transfection revealed a greater than 3-fold increase in CAT activity (Figure 7C), which is comparable to the increase in Evi1 expression seen in 32Dwt18 cells on transfection of SOX4 (Figure 7A). These data provide a mechanistic link between overexpression of Sox4 and cooperativity with proviral activation of Evi1: SOX4-mediated transactivation of the proviral LTR inserted at Evi1, leading to increased expression of Evi1, leading to a greater leukemic effect of EVI1.
Discussion
In this article we document the coexistence of proviral insertions at Evi1 and Sox4 in 8 of 8 myeloid leukemias derived from the AKXD-23 recombinant inbred strain of mouse. In addition, we identified a proviral insertion at Sox4 and Sox4 overexpression in the myeloid leukemia cell line, NFS-60, which is known to have a proviral insertion at Evi1.45 These findings strongly suggest cooperativity between Evi1 and Sox4 in myeloid leukemogenesis and may help to explain why assays testing the leukemogenic potential of Evi1 have thus far been negative.17,21 From our analysis of the NFS-60 cell line it is clear the effect of proviral insertion at Sox4 is to increase Sox4 transcription, most likely through altering mRNA structure leading to increased mRNA half-life and thereby increased protein level. Because of the lack of reliable anti-SOX4 antisera, we were not able to assess whether there is in fact increased SOX4 protein in NFS-60 cells.
SOX4 protein is in Group C within the family of HMG-box proteins that have been shown to bind in a sequence-specific manner in the minor groove of the DNA. Within the HMG-box family are important factors involved in sex determination (Sry) and in β-catenin signaling (Tcf/Lef), whereas Group C consists of SOX4, SOX11, SOX22, and SOX24 (reviewed in Wegner46). Although the exact regulatory role of SOX4 has not been defined, it has been shown that IL-5 signaling may involve the activation of SOX4, through the intermediary signaling protein syntenin.47 Sox4 homozygous null animals show no detectable abnormalities in myelomonocytic cells, but they do display markedly defective B-cell development, having decreased numbers of mature cells, decreased IL-7-responsive colony-forming units in the fetal liver, and diminished responsiveness to lipopolysaccharide.48 On the basis of these findings, it is thought that Sox4 plays a critical role in pro-B-cell expansion.48 Sox4 has been implicated in the genesis of carcinoma of the breast,49 colon,50 and liver,51 as well as medulloblastoma.52 It has also been found as a site of proviral insertion in B-cell tumors.24,53 The mechanism by which it contributes to malignant transformation in these settings is unknown.
The main motivation for embarking on these studies was to identify genetic events that cooperate with Evi1 activation in myeloid leukemogenesis. Although we have obtained genetic evidence for cooperativity between Sox4 and Evi1, what is lacking is a reconstruction experiment to demonstrate that this is indeed the case. We have attempted to show that the 2 genes cooperate in the formation of anchorage-independent colonies of Rat1 cells in soft agar, but we saw no increased number in Sox4 + Evi1-transduced cells over what was obtained with Evi1 alone. However, a negative result here may be meaningless, given that this may be an inappropriate cell-culture model for the study of myeloid leukemia. We have also attempted cooperativity studies in retrovirally transduced murine primary bone marrow cells that were transplanted into irradiated recipients. These experiments revealed that, although transplantation of Sox4-transduced cells resulted in leukemia in the recipient mice, Evi1-transduced cells remained healthy. Furthermore, Evi1 failed to accelerate Sox4-induced leukemia in this model system (S. Spence, A.S.P., N.A.J., and N.G.C., unpublished results, June 2003). When Sox4-induced leukemias were examined for where the Sox4-containing provirus had integrated, it was found that, although 2 of 13 had insertions at Mef2c and 3 of 13 had insertions at Sfpi1, Evi1 was not identified as a site of insertion.54 This is consistent with our proposed mechanism, in that only certain LTRs are transcriptionally activated by SOX4 and that, in tumors with insertions at both genes, insertions at Evi1 occur first, and Sox4 insertions happen subsequently, selected for by their ability to up-regulate provirally-activated Evi1 expression.
In this study, we provide evidence that SOX4 can transactivate the AKV LTR and thereby increase the expression of provirally activated Evi1. This conclusion is based on 3 findings: (1) When Sox4 is overexpressed in 32Dwt18 cells (which have a provirally activated Evi1), there is an accompanying increase in Evi1 expression; (2) when Sox4 expression is knocked down with shRNA in DA-1 cells (which harbor a proviral insertion at Evi1 and express high levels of Sox4), there is a concomitant decrease in Evi1 expression; and (3) Sox4 can transactivate the AKV LTR in cotransfection studies. This, together with the observation that EML cells express abundant Sox4 but no Evi1 lead us to conclude that Sox4 increases Evi1 expression via transactivation of the proviral LTR rather than the endogenous Evi1 promoter. Indeed, it has been shown that several Sry-like proteins are able to activate expression of the human retrovirus-like element LINE.55 In cotransfection studies, Sry-box family members were able to activate a LINE-luciferase construct greater than 10-fold, and binding sites for SOX proteins are present within the LINE promoter region. Sequence analysis of the LTR regions of murine ecotropic retroviruses reveals 1 to 4 putative Sry binding sites, revealed by TransFac analysis. The most impressive of these is the AKV LTR, which has 4 sites, having scores between 90 and 95 (data not shown). Interestingly, the AKV virus is the endogenous retrovirus in the AKXD strain, in which most of the leukemias analyzed in this study arose. The cell lines analyzed were derived from mice infected with either Cas-Br-E (NFS-60) or Moloney (DA-1) retroviruses. However, the exact nature of the proviral insertions has not been determined; the virus inserted into Evi1 in 32Dwt18 cells has not been characterized. It could be that the failure of Evi1 and Sox4 to cooperate in murine bone marrow transplantation studies was due to the inability of SOX4 to transactivate the LTR of MSCV, the retroviral vector used in those studies.
Evi1 proviral activation has been associated with both malignant and premalignant or nonmalignant expansion of the myeloid compartment in retrovirally infected mice56 and monkeys.57 In tissue culture, retroviral infection of primary bone marrow cells can result in immortalization, and characterization of proviral insertion sites reveals that 25% of immortalized lines have proviral insertions at Evi1.58 Interestingly, these lines are not tumorigenic. These findings strongly suggest that activation of Evi1 has a potent effect on cell growth and senescence but is not by itself sufficient for leukemogenesis. In the setting of Evi1-driven nonmalignant myeloid expansion, it is likely that the chance of transforming proviral insertion (such as at Sox4) is increased. Thus, the expectation that a reconstruction experiment such as cotransduction of Evi1 and Sox4 retroviruses may be unrealistic, because there is no cooperativity in terms of oncogenic pathways, but only in that Evi1 activation provides the requisite expansion of the target cell population.
Acknowledgments
We thank Sharon Lin for logistical support; Olga Mironenko for technical assistance with the microarrays, Ephat Harel for database searches with DNA sequence, John Wheeler for thoughtful comments and advice, Michael Wegner for the gift of the reporter plasmids pTATAluc and pTATAluc Sx3X, Warren Pear for the bcr-abl retrovirus, and Jack Lenz for the AU-CAT reporter. We thank Daniel Link for the 32Dwt18 cells.
Prepublished online as Blood First Edition Paper, October 4, 2005; DOI 10.1182/blood-2003-05-1626.
Supported by the National Institutes of Health (R01 CA81216 [A.S.P.] and 1F32 CA88467-01 [K.E.B.]).
K.E.B. initiated the project, designed and executed the experiments shown in Figures 1, 2, 3, 4, and 5 and Table 1, contributed to the writing of the manuscript; Y.-Y.X. conceived of and executed the experiments described in Figure 7; K.F. performed the CAT assays and Western blot in Figure 7; A.P. executed experiments described in Figure 6 and Table 2; N.G.C. and N.J. designed the mouse experiment, provided mouse tumor DNA, and contributed significantly to the conceptualization of experiments and the discussion; A.S.P. was involved in the design of the experiments, provided laboratory space and reagents, and wrote most of the manuscript.
K.E.B. and Y.-Y.X. contributed equally to this study.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Adams J, Cory S. Oncogene cooperation in leukaemogenesis. Cancer Surv. 1992;15: 119-141. [PubMed] [Google Scholar]
- 2.Mucenski ML, Taylor BA, Ihle JN, Hartley JW, Morse HC III, Jenkins NA, Copeland NG. Identification of a common ecotropic viral integration site, Evi-1, in the DNA of AKXD murine myeloid tumors. Mol Cell Biol. 1988;8: 301-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mucenski ML, Taylor BA, Jenkins NA, Copeland NG. AKXD recombinant inbred strains: models for studying the molecular genetic basis of murine lymphomas. Mol Cell Biol. 1986;6: 4236-4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergeron D, Poliquin L, Kozak C, Rassart E. Identification of a common viral integration region in Cas-Br-E murine leukemia virus-induced non-T-, non-B-cell lymphomas. J Virol. 1991;65: 7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy ER, Parganas E, Morishita K, et al. DNA rearrangements proximal to the EVI1 locus associated with the 3q21q26 syndrome. Blood. 1994;83: 1348-1354. [PubMed] [Google Scholar]
- 6.Morishita K, Parganas E, Willman CL, et al. Activation of EVI1 gene expression in human acute myelogenous leukemias by translocations spanning 300-400 kilobases on chromosome band 3q26. Proc Natl Acad Sci U S A. 1992;89: 3937-3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzukawa K, Parganas E, Gajjar A, et al. Identification of a breakpoint cluster region 3′ of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EVI1 gene in acute myelogenous leukemias with inv(3)(q21q26). Blood. 1994;84: 2681-2688. [PubMed] [Google Scholar]
- 8.Perkins AS, Fishel R, Jenkins NA, Copeland NG. Evi-1, a murine zinc finger proto-oncogene, encodes a sequence-specific DNA-binding protein. Mol Cell Biol. 1991;11: 2665-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delwel R, Funabiki T, Kreider B, Morishita K, Ihle J. Four of the seven zinc fingers of the Evi-1 myeloid transforming gene are required for sequence-specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol Cell Biol. 1993;13: 4291-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsugi T, Morishita K, Ihle JN. Identification, nuclear localization, and DNA-binding activity of the zinc finger protein encoded by the Evi-1 myeloid transforming gene. Mol Cell Biol. 1990;10: 1259-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perkins A, Kim J. Zinc fingers 1-7 of EVI1 fail to bind to the GATA motif by itself but require the core site GACAAGATA for binding. J. Biol. Chem. 1996;271: 1104-1110. [DOI] [PubMed] [Google Scholar]
- 12.Turner J, Crossley M. Cloning and characterization of mCtBP2, a co-repressor that associates with basic Kruppel-like factor and other mammalian transcriptional regulators. EMBO J. 1998;17: 5129-5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartholomew C, Kilbey A, Clark A, Walker M. The Evi-1 proto-oncogene encodes a transcriptional repressor activity associated with transformation. Oncogene. 1997;14: 569-577. [DOI] [PubMed] [Google Scholar]
- 14.Yuasa H, Oike Y, Iwama A, et al. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005;24: 1976-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yatsula B, Lin S, Read A, et al. Identification of binding sites of EVI1 in mammalian cells. J. Biol Chem. 2005;280: 30712-30722. [DOI] [PubMed] [Google Scholar]
- 16.Morishita K, Parganas E, Matsugi T, Ihle JN. Expression of the Evi-1 zinc finger gene in 32Dcl3 myeloid cells blocks granulocytic differentiation in response to granulocyte colony-stimulating factor. Mol Cell Biol. 1992;12: 183-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kreider B, Orkin S, Ihle J. Loss of erythropoietin responsiveness in erythroid progenitors due to expression of the Evi-1 myeloid transforming gene. Proc Natl Acad Sci U S A. 1993;90: 6454-6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurokawa M, Ogawa S, Tanaka T, et al. The AML1/Evi-1 fusion protein in the t(3;21) translocation exhibits transforming activity on Rat1 fibroblasts with dependence on the Evi-1 sequence. Oncogene. 1995;11: 833-840. [PubMed] [Google Scholar]
- 19.Kilbey A, Bartholomew C. Evi-1 ZF1 DNA binding activity and a second distinct transcriptional repressor region are both required for optimal transformation of Rat1 fibroblasts. Oncogene. 1998;16: 2287-2291. [DOI] [PubMed] [Google Scholar]
- 20.Palmer S, Brouillet J, Kilbey A, et al. Evi-1 transforming and repressor activities are mediated by CtBP co-repressor proteins. J. Biol. Chem. 2001;276: 25834-25840. [DOI] [PubMed] [Google Scholar]
- 21.Louz D, van den Broek M, Verbakel S, et al. Erythroid defects and increased retrovirally-induced tumor formation in Evi1 transgenic mice. Leukemia. 2000;14: 1876-1884. [DOI] [PubMed] [Google Scholar]
- 22.Buonamici S, Li D, Chi Y, et al. EVI1 induces myelodysplastic syndrome in mice. J Clin Invest. 2004;114: 713-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cuenco G, Ren R. Both AML1 and EVI1 oncogenic components are required for the cooperation of AML1/MDS1/EVI1 with BCR/ABL in the induction of acute myelogenous leukemia in mice. Oncogene. 2004;23: 569-579. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Shen H, Himmel KL, et al. Leukaemia disease genes: large-scale cloning and pathway predictions. Nat Genet. 1999;23: 348-353. [DOI] [PubMed] [Google Scholar]
- 25.European Bioinformatics Institute (EBI) and Wellcome Trust Sanger Institute (WTSI). Ensembl DNA database. http://www.ensembl.org/Mus_musculus/.
- 26.Holmes K, Palaszynski E, Frederickson T, Morse H, Ihle J. Correlation of cell-surface phenotype with the establishment of interleukin 3-dependent cell lines from wild-mouse murine leukemia virus-induced neoplasms. Proc Natl Acad Sci U S A. 1985;82: 6687-6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ihle J, Rein A, Mural R. Immunologic and virologic mechanisms in retrovirus-induced murine leukemogenesis. In: Klein G, ed. Advanced Oncology. Vol 4. New York, NY: Raven; 1984: 95-137. [Google Scholar]
- 28.Greenberger J, Eckner R, Sakakeeny M, et al. Interleukin-3-dependent hematopoietic progenitor cell lines. Fed Proc. 1983;42: 2762. [PubMed] [Google Scholar]
- 29.Khanna-Gupta A, Zibello T, Kolla S, Neufeld EJ, Berliner N. CCAAT displacement protein (CDP/cut) recognizes a silencer element in the lactoferrin gene promoter. Blood. 1997;90: 2784-2795. [PubMed] [Google Scholar]
- 30.Tsai S, Bartelmez S, Sitnicka E, Collins S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 1994;8: 2831-2841. [DOI] [PubMed] [Google Scholar]
- 31.Pear WS, Miller JP, Xu L, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92: 3780-3792. [PubMed] [Google Scholar]
- 32.Kuhlbrodt K, Herbarth B, Sock E, Enderich J, Hermans-Borgmeyer I, Wegner M. Cooperative function of POU proteins and SOX proteins in glial cells. J Biol Chem. 1998;273: 16050-16057. [DOI] [PubMed] [Google Scholar]
- 33.Morgenstern J, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucl Acids Res. 1990;18: 3587-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrook J, Russell D. Molecular Cloning: A Laboratory Manual. 3rd ed. Woodbury, NY: Cold Spring Harbor Laboratory Press; 2000.
- 35.Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci U S A. 1984;81: 1991-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52: 456-467. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989.
- 38.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162: 156-159. [DOI] [PubMed] [Google Scholar]
- 39.Cepko C. Retrovirus infection of cells in vitro and in vivo. In: Ausubel FM, Brent R, Kingston RE, et al, eds. Current Protocols in Molecular Biology. Vol 1. New York, NY: Greene Publishing Associates and John Wiley and Sons; 1993: 9.14.1-9.14.3. [Google Scholar]
- 40.Colicelli J, Goff S. Sequence and spacing requirements of a retrovirus integration site. J Mol Biol 1988;199: 47-59. [DOI] [PubMed] [Google Scholar]
- 41.Pear W, Scott M, Nolan G. Generation of high titre, helper-free retroviruses by transient transfection. In: Robbins P, ed. Methods in Molecular Medicine: Gene Therapy Protocols. Totowa, NJ: Humana Press; 1997: 41-57. [DOI] [PubMed]
- 42.Patel G, Kreider B, Rovera G, Reddy E. v-myb blocks granulocyte colony-stimulating-factor-induced myeloid differentiation but not proliferation. Mol Cell Biol. 1993;13: 2269-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Donnell L, Druhan L, Avalos B. Molecular characterization and expression analysis of leucine-rich alpha2-glycoprotein, a novel marker of granulocytic differentiation. J Leukoc Biol. 2002;72: 478-485. [PubMed] [Google Scholar]
- 44.Khanna-Gupta A, Lopingco M, Savinelli T, Zibello T, Berliner N, Perkins A. Retroviral insertional activation of the Evi1 oncogene does not prevent G-CSF-induced maturation of the murine pluripotent myeloid cell line, 32DCl3. Oncogene. 1996;12: 563-569. [PubMed] [Google Scholar]
- 45.Morishita K, Parker DS, Mucenski ML, Jenkins NA, Copeland NG, Ihle JN. Retroviral activation of a novel gene encoding a zinc finger protein in IL3-dependent myeloid leukemia cell lines. Cell. 1988;54: 831-840. [DOI] [PubMed] [Google Scholar]
- 46.Wegner M. From head to toes: the multiple facets of Sox proteins. Nucl Acids Res. 1999;27: 1409-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geijsen N, Uings IJ, Pals C, et al. Cytokine-specific transcriptional regulation through an IL-5Ra interacting protein. Science. 2001;293: 1136-1138. [DOI] [PubMed] [Google Scholar]
- 48.Schilham M, Oosterwegel M, Moerer P, et al. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature. 1996;380: 711-714. [DOI] [PubMed] [Google Scholar]
- 49.McGowan E, Clarke C. Effect of overexpression of progesterone receptor A on endogenous progestin-sensitive endpoints in breast cancer cells. Mol Endocrinol. 1999;13: 1657-1671. [DOI] [PubMed] [Google Scholar]
- 50.McCracken S, Kim C, Xu Y, Minden M, Miyamoto N. An alternative pathway for expression of p56lck from type I promoter transcripts in colon carcinoma. Oncogene. 1997;15: 2929-2937. [DOI] [PubMed] [Google Scholar]
- 51.Ahn S, Cho G, Jeong S, Rhim H, Choi J, Kim I. Identification of cDNAs for Sox-4, an HMG-Box protein, and a novel human homolog of yeast splicing factor SSF-1 differentially regulated during apoptosis induced by prostaglandin A2/delta 2-PGJ2 in Hep3B cells. Biochem Biophys Res Commun. 1999;260: 216-221. [DOI] [PubMed] [Google Scholar]
- 52.Lee C-J, Appleby V, Orme A, Chan W-I, Scotting P. Differential expression of SOX4 and SOX11 in medulloblastoma. J Neuro-Oncol. 2002;57: 201-214. [DOI] [PubMed] [Google Scholar]
- 53.Hansen GM, Skapura D, Justice MJ. Genetic profile of insertion mutations in mouse leukemias and lymphomas. Genome Res. 2000;10: 237-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du Y, Spence SE, Jenkins NA, Copeland NG. Cooperating cancer-gene identification via oncogenic-retrovirus-induced insertional mutagenesis. Blood. 2005;106: 2495-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tchenio T, Casella J, Heidmann T. Members of the SRY family regulate the human LINE retro-transposons. Nucleic Acids Res. 2000;28: 411-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kustikova O, Fehse B, Modlich U, et al. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science. 2005;308: 1171-1174. [DOI] [PubMed] [Google Scholar]
- 57.Calmels B, Ferguson C, Laukkanen M, et al. Recurrent retroviral vector integration at the Mds1/Evi1 locus in nonhuman primate hematopoietic cells. Blood. 2005;106: 2530-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du Y, Jenkins N, Copeland N. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood. Prepublished on August 18, 2005, as DOI 10.1182/blood-2005-03-1113. [DOI] [PMC free article] [PubMed]