

Abstract
The mechanisms underlying aneuploidy in multiple myeloma (MM) are unclear. Centrosome amplification has been implicated as the cause of chromosomal instability in a variety of tumors and is a potential mechanism causing aneuploidy in MM. Using immunofluorescent (IF) staining, centrosome amplification was detected in 67% of monoclonal gammopathies, including monoclonal gammopathy of undetermined significance (MGUS). We also investigated the gene expression of centrosome proteins. Overall, gene expression data correlated well with IF-detected centrosome amplification, allowing us to derive a gene expression-based centrosome index (CI) as a surrogate for centrosome amplification. Clinically, MM patients with high CI (> 4) are associated with poor prognostic genetic and clinical subtypes (chromosome 13 deletion, t(4; 14), t(14;16), and PCLI > 1%, P < .05) and are shown here to have short survival (11.1 months versus 39.1 months, P < .001). On multivariate regression, a high CI is an independent prognostic factor. Given that centrosome amplification is already observed in MGUS and probably integral to early chromosomal instability and myeloma genesis, and patients with more extensive centrosome amplification have shorter survival, the mechanisms leading to centrosome amplification should be investigated because these may offer new avenues for therapeutic intervention.
Introduction
Genomic instability in cancer is broadly categorized into mutational instability (eg, microsatellite, MIN) and chromosome instability (CIN). CIN, characterized by unstable aneuploidy, is the most common form of genetic instability in human cancer. Several pathways are operative in the generation of CIN. One pathway involves telomeric dysfunction with chromosome breakage and the presence of dicentric chromosomes. Another pathway involves gains and losses of intact whole chromosomes and is thought to be due to continuous chromosome mis-segregation during mitosis.1,2 This latter phenotype may result from genetic alterations in genes that delay cell entry into anaphase if chromosomes are misaligned on the bipolar mitotic spindle such as BUB1, MAD2, and BUBR1 (so-called mitotic checkpoint genes) or centrosome amplification.3,4 Few examples of mutations in the mitotic checkpoint genes have been documented in human cancers, despite elegant mechanistic studies showing their putative pathogenetic role.5
Centrosomes are cellular microtubule-organizing centers whose normal function is crucial for chromosome segregation and cytokinesis during mitosis.6 The centrosome consists of 2 centrioles that are surrounded by amorphous pericentriolar material (PCM). The centrosome is duplicated only once during the cell cycle to give rise to 2 centrosomes that function as spindle poles of the dividing cell. Extra copies (aberrant) of the centrosome frequently result in the formation of multipolar spindles and inevitably chromosome mis-segregation and aneuploidy.7-10
Centrosome amplification is characterized by combinations of increase in number, size, abnormal structure, and function. Centrosome amplification has been detected in a broad range of solid tumors2,11-16 and more recently in leukemia17,18 and lymphoma.19-21 In solid tumors, the centrosome abnormalities are associated with advancing stages of the disease, aneuploidy, and an aggressive clinical course.2,11,13-15 The existence of centrosome abnormalities in preinvasive carcinomas suggests they are early events in cellular transformation.22,23
Multiple myeloma (MM) is characterized almost universally by aneuploidy,24 although it is more evident in the 40% to 50% of hyperdiploid cases. Multiple trisomies and monosomies can be observed in the clonal cells. Aneuploidy evolution is still evident at all stages of the disease because there is heterogeneity in the number of cells harboring any specific aneuploid chromosomes. The mechanisms leading to this aneuploidy are at present unclear. The presence of defined stages of progression from monoclonal gammopathy of undetermined significance (MGUS) to symptomatic MM provides an ideal model for the study of disease pathogenesis and evolution. In this study, we investigated the role of centrosome amplification in the pathogenesis of aneuploidy in MM. To do so, we used a combined immunofluorescent (IF) staining and gene expression approach. We showed that centrosomes are not only present in the majority of clonal plasma cells (PCs), but that they are absent in most normal PCs. In addition, centrosome amplification is present in MGUS, suggesting that centrosome amplification occurs early in myeloma genesis. Clinically, patients with high levels of centrosome amplification have short survival.
Patients and methods
Patients
Patients with MGUS, smoldering myeloma (SMM), and MM were included in the study. The diagnoses of the different monoclonal gammopathies were based on the criteria of the International Myeloma Working Group.25 In addition, bone marrow (BM) PCs from healthy donors who underwent hip replacement were obtained for comparison. Research BM samples were obtained following informed consent before the time of routine procurement for clinical samples. These studies were conducted under the Mayo Clinic Institutional Review Board (IRB) approval and followed the Helsinki guidelines for research with human subjects.
Two different cohorts were studied. IF staining for centrin, a centrosome protein, was performed on samples from 10 healthy control subjects, 3 patients with MGUS, 7 patients with SMM, and 14 patients with MM (group 1), whereas gene expression profiling was performed on a separate cohort of 15 healthy control subjects, 23 patients with MGUS, 25 patients with SMM, and 97 patients with MM (71 newly diagnosed and 26 relapsed) (group 2).
IF staining and slide analysis
Bone marrow samples were subjected to red cell lysis using ammonium chloride following which the sample was centrifuged and washed, and a cell count was performed. Cytospin slides were prepared using 75 000 cells per slide and air dried for a minimum of 24 hours. Extra slides were archived at -20°C for future use. Slides were then fixed in -20°C methanol for 5 minutes and placed in APK (phosphate-buffered detergent) for 5 minutes. Blocking buffer (100 μL; PBS with 5% normal goat serum and 0.1% BSA) was then added to each slide and incubated for 15 minutes at room temperature. After incubation, the blocking buffer was poured off, and the slide was blotted dry before 100 μL diluted (1:500) hCen2.4 ascites antibody (generous gift from Dr J.L. Salisbury, Mayo Clinic Rochester, MN) was applied. The slides were then incubated for 30 minutes at room temperature before being washed with APK 3 times (3 minutes/wash) using gentle agitation. The slides were wiped dry, and 100 μL(2 μg/mL) diluted (1:1000) goat anti-mouse IgG F(ab′)2-Alexa 488 (Molecular Probes, Eugene, OR) was added. Further incubation and washing was done before adding 100 μL (20 μg/mL) of either goat anti-human κ or λ light chain conjugated with 7-amino-4-methylcoumarin-3-acetic acid (AMCA; Vector Labs, Burlingame, CA). The slides were then incubated at room temperature for 30 minutes and washed before applying a coverslip using antifade without PI.
One hundred isotype-restricted PCs were scored per slide. The centrin signals were enumerated and classified as follows: (1) no signal, (2) normal (1-4 signals), or (3) abnormal (> 4 signals). One to 4 centrin signals was considered as consistent with normal mitotic cells because centrin is a component of centriole,26 and in a normal cycling cell, up to 4 centrioles can be present depending on the stage of the cell cycle. We used a cut-off value of 10% of cells with more than 4 signals (based on mean + 3 SD of percentage of normal BM PCs with > 4 centrin signals) as an indicator of centrosome amplification.
Gene expression profiling (GEP)
Gene expression analysis was performed on CD138+ selected PC RNA using the Affymetrix U133A chip (Affymetrix, Santa Clara, CA). RNA isolation, purification, and microarray hybridization has been previously reported.27 Gene expression intensity values were log transformed, normalized to the median, and analyzed using GeneSpring 7 (Agilent Technologies, Palo Alto, CA).
Patients were then classified according to the expression of 7 genes (FGFR3, MMSET, ITGB7, MAF, CCND3, CCND1, and CCND2) as previously described in the translocation and cyclin D (TC) classification.28 This classification identifies all the major cytogenetic categories in MM: t(4;14)(p16;q32) (based on FGFR3/MMSET expression); t(14;16)(q32;q23) and t(14;20) (based on expression of ITGB7 and MAF); t(6;14) assessed by high level of CCND3 expression; t(11;14)(q13;q32) based on high level of CCND1 expression; hyperdiploid MM based on aberrant low level of CCND1 expression (D1 group), or low CCD1 with high CCD2 expression (D1 + D2 group). Two other TC groups, high CCND2 but no CCND1 expression (D2 group) or no CCND1 or CCND2 expression (none group), are biologically poorly defined.
cIg-FISH
Cytoplasmic immunoglobulin-fluorescence in situ hybridization (cIg-FISH) was performed on patients in group 2 as previously described.29 We used probes previously described by us for the detection of 13q deletion (Δ13) and translocations involving the immunoglobulin heavy chain (IgH) locus using the “break apart” strategy.30 In addition, for patients in group 2, commercially available direct-labeled chromosome enumeration probe (CEP) for chromosomes 3, 9, 11, and 15 (Vysis, Downers Grove, IL) and a direct-labeled BAC clone for the p-arm of chromosome 19 (RPCI 11 88I12; BACPAC Resources, Children's Hospital Oakland Research Institute, Oakland, CA) were used to detect chromosomal aneusomy and to assign ploidy status based on an index highly specific for hyperdiploidy.31 Aneusomy is present if greater than 10% of PCs have 3 or more signals or only 1 signal for each CEP probe.
DNA index by flow cytometry
Ploidy for patients in group 1 was assessed by flow cytometry. The total DNA content was analyzed by dual channel flow cytometry using propidium iodide to measure the DNA content and κ and λ light chain antisera to identify the clonal cells as previously described.32 The ploidy subtypes are defined as follows: DNA index 1.06 to 1.74 hyperdiploid; DNA index less than 1.06 or more than 1.74 nonhyperdiploid.
Plasma cell labeling index (PCLI)
The bone marrow PCLI, which reflects proliferative activity of PCs, was determined using a slide-based IF method as previously described.33
Statistical analysis
The Mann-Whitney U test or the Student t test was used to compare continuous variables. For comparison of multiple groups, the Kruskal-Wallis test was used with Dunn multiple testing correction for pair-wise comparisons. Correlation was measured using the Spearman correlation coefficient. Categorical variables were compared using the Fisher exact test. The distribution for overall survival (OS) was estimated using the method of Kaplan and Meier. The log-rank test was used to test for differences in survival between groups. OS was calculated from the date of diagnosis to death. Cox proportional hazards models were used to assess the association of several prognostic factors with OS. Variables were selected using stepwise selection from a list of candidate variables. A P value below .05 was considered significant.
Results
Centrosome amplification is common in all stages of PC neoplasm since MGUS
The pattern of centrin staining was different between PCs from healthy donors and patients with monoclonal gammopathies (Figure 1A-H). The majority of PCs derived from healthy control subjects (mean ± SD, 74.9% ± 18.8%) did not have any centrin signals detectable by IF (Figure 1A). In 8 of the 10 healthy donors, no PCs with more than 4 centrin signals were detected. The other 2 cases have 5% and 8% of PCs with more than 4 centrin signals. This was in sharp contrast to PCs from patients with PC neoplasm in which the majority express 1 to 4 or more than 4 centrin signals (51.5% ± 19.2% and 15.7% ± 14.5%, respectively) (Figure 2A). In addition, structurally abnormal centrosomes, where the shape and configuration of the centrosome were abnormal (Figure 1D-H), were predominantly seen in MM.
Figure 1.

Different patterns of centrin staining. The isotypic PCs were identified by cytoplasmic κ or λ light-chain antibody conjugated with AMCA (cIg, blue), and centrin was stained with anticentrin2 conjugated with Alexa 488 (arrow; green). (A) Most PCs from healthy donors had no signals. (B-C) Cells with 1 to 4 signals were considered to have normal centrosome. (D-E) Centrosome amplification was seen in typical clonal PCs as well as the rare multinucleated PCs in patients. Various centrosome abnormalities were seen as follows: (D-E) increased number of signals in a cluster, (F) increased signals that were scattered, (G) coalescence of centrins into string-like structures, and (H) centrins staining up as ring-like structure. Abnormal centrosome structure as seen in panels F to H were predominantly seen in MM. The cells were visualized with a Leitz Epifluorescence microscope using a 100 ×/1.4 NA oil objective lens (Leitz, Wetzlar, Germany). The images were captured with the CoolSNAP ES CCD camera (Photometrics, Tucson, AZ) and acquired using either the Vysis smart capture software (Vysis, Downer's Grove, IL) or Oncor imaging system software (Oncor, Gaithersburg, MD). The acquired images were subsequently processed with Adobe Photoshop 7.0 (Adobe Systmes, San Jose, CA).
Figure 2.
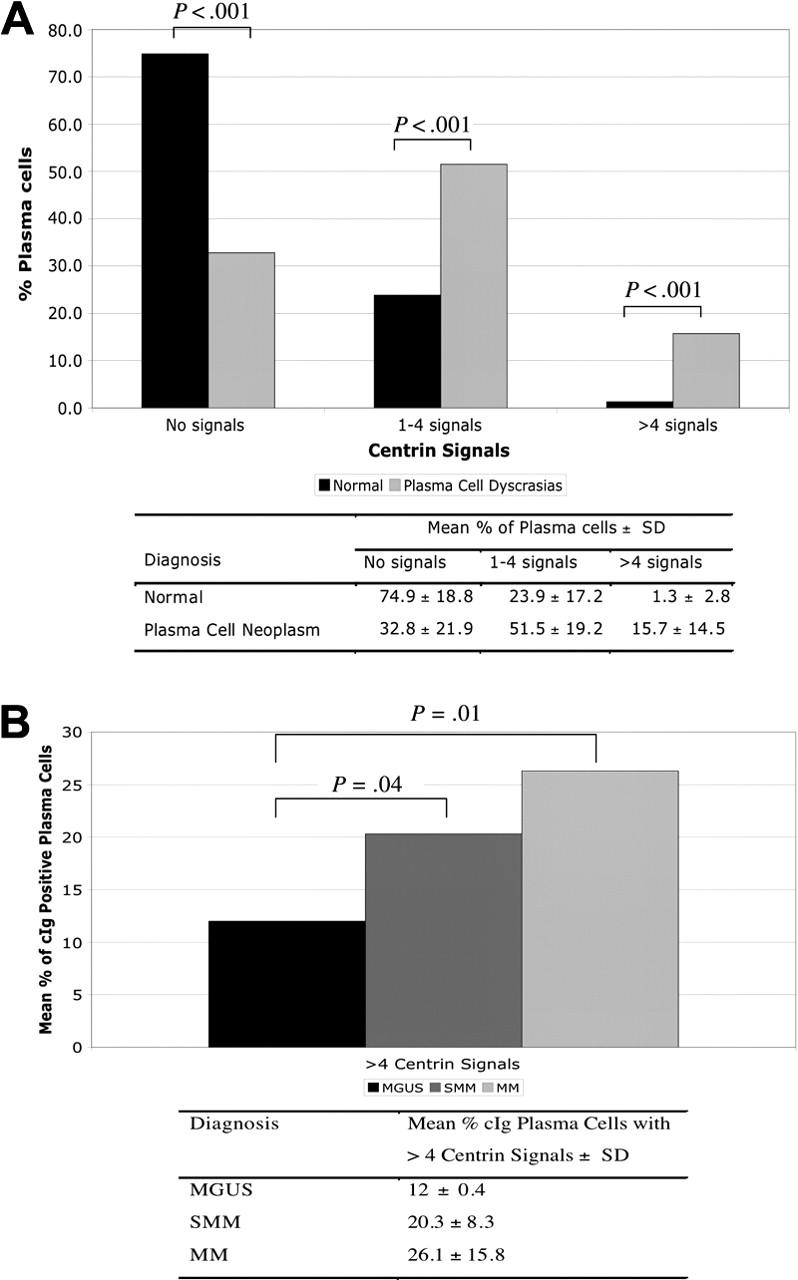
Centrin staining and centrosome amplification in healthy donors and patients with monoclonal gammopathies. (A) Majority of normal PCs had no centrin signals compared with clonal plasma cells (P < .001), whereas centrosome amplification was almost exclusively seen in clonal PCs (P < .001). (B) Percentage of cIg-positive PCs with centrosome amplification increased with more advance stage of PC proliferation.
Centrosome amplification was present in all stages of monoclonal gammopathies. Using predefined criteria, 16 (67%) of 24 patients (2 [67%] of 3 MGUS, 5 [71%] of 7 SMM, and 9 [64%] of 14 MM) had centrosome amplification. In addition, the percentage of PCs with centrosome amplification increased progressively from MGUS to MM (Figure 2B).
Gene expression analysis of centrosome protein genes
Together with centrin, pericentrin and γ-tubulin are also major centrosome protein and components of the PCM.9,34 We assessed the expression levels of genes encoding these proteins in patients in group 2. The expression of these genes were significantly correlated and increased progressively from MGUS to MM (Figure 3; Table 1), similar to results from other studies that showed concordant results when these different proteins are stained.13,17 We then calculated a gene expression-based centrosome index (CI) by adding the normalized expression value of each of these 3 genes. The CI increased significantly from MGUS to MM (Figure 4A), consistent with our IF results. CI more than 4 (mean + 2 SD CI of normal PCs) was highly correlated with centrosome amplification by IF. In 20 patients with MM in group 2 (10 CI > 4 and 10 CI < 4) 100% of patients with CI more than 4 have centrosome amplification, whereas only 20% with CI less than 4 have centrosome amplification (P = .001). Furthermore, the CI is strongly correlated with the percentage of clonal cells with centrosome amplification in each patient (Figure 4B). This suggests that the CI can be used as a surrogate for expression of centrosome proteins. Overall, the data suggest that centrosome amplification occurs early in myeloma genesis and increases with disease progression.
Figure 3.

Expression levels of centrosome proteins are closely correlated. Expression levels of centrin were correlated with (A) pericentrin (r = 0.16; P = .046) and (B) γ-tubulin (r = 0.47; P < .001). (C) Pericentrin was also correlated with γ-tubulin (r = 0.35; P < .001).
Table 1.
Comparison of centrin, pericentrin, and γ-tubulin gene expression between MGUS/SMM and MM
| MGUS/SMM | MM | P | |
|---|---|---|---|
| No. | 48 | 97 | |
| Centrin expression | 0.93 (0.43-1.69) | 1.06 (0.25-2.37) | .004 |
| Pericentrin expression | 0.87 (0.40-1.77) | 1.06 (0.37-2.32) | .003 |
| γ-tubulin expression | 0.78 (0.14-1.78) | 1.16 (0.07-3.16) | < .001 |
Figure 4.
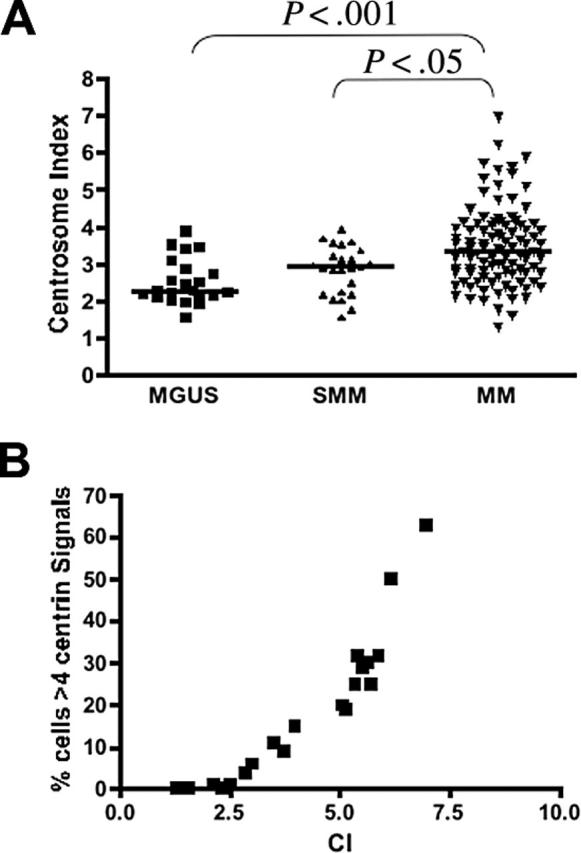
Gene expression based centrosome index (CI) in PC neoplasm. (A) CI increased progressively from MGUS (n = 23; median CI, 2.27; range, 1.56-3.90) to SMM (n = 25; median CI, 2.91; range, 1.58-3.91) to MM (n = 97; median CI, 3.36; range, 1.29-6.97). The pairs represented by the brackets were significantly different. (B) In the 20 patients with MM in group 2 in which IF was also performed, the CI was highly correlated with the percentage of clonal plasma with abnormal (> 4) centrin signals (Spearman correlation coefficient r = 0.97; P < .001).
Centrosome amplification and ploidy category (hyperdiploid versus not)
No relation between ploidy categories of PC neoplasm and centrosome amplification was found. In patients in group 1, neither the frequency of cases with centrosome amplification (67% versus 75%) nor the percentage of cells with centrosome amplification is different between the 2 ploidy subtypes of MM (32.8% ± 22.1% versus 19.9% ± 7.9%, P = .11). Similarly, in patients in group 2, the CI is not different between hyperdiploid and nonhyperdiploid patients identified by a FISH-based index (data not shown).
Centrosome amplification and aneuploidy
To see whether centrosome abnormalities increase with aneuploidy, the number of aneusomic chromosomes was correlated with the CI. In 97 patients with MM in group 2, no correlation between the number of aneuploid chromosomes as detected by FISH and the centrosome index was demonstrated (data not shown).
Patients with MM with high centrosome index have poor prognostic features and survival
Next, we investigated the clinical significance of centrosome amplification in the cohort of 97 patients with MM in group 2. High CI (CI > 4) is associated with established poor prognostic features such as Δ13, high PCLI (> 1%). Limited information also allowed us to find more extreme centrosome amplification (CI > 4) among patients with 4p16 + MAF compared with other TC groups. However, there was no difference in the distribution of CI across the International Staging System (ISS) stages (Table 2). When treated as a continuous variable, the PCLI was also significantly higher in patients with high CI (3.6% [range, 0%-9.4%] versus 0.35% [range, 0%-4.8%], P < .001). Consistent with this, patients with high CI had significantly poorer survival. When we limited the analysis to newly diagnosed cases studied by GEP with survival data and a median follow-up of 25.5 months (n = 67), the median survival was 11.1 months for those with high CI compared with 39.1 months for the others (log-rank P = .001; Figure 5). The important clinical characteristics of these patients are shown in Table 3. Of note, the median age (58.5 years [range, 49-81 years] versus 62 years [range, 38-82 years], P = .97) and percentage of patients treated with stem cell transplantation (50% versus 59%, P = .58) were similar between patients with high CI and others.
Table 2.
Associations of patients with different centrosome index (CI)
| CI above 4, no./no. total (%) | CI below 4, no./no. total (%) | P | |
|---|---|---|---|
| Relapse cases | 7/26 (27) | 21/71 (27) | .80 |
| Hyperdiploid cases | 12/25 (48) | 43/71 (61) | .25 |
| Δ13 | 17/26 (65) | 27/70 (38) | .02 |
| TC classes | |||
| 4p16 + MAF | 6/26 (23) | 4/71 (6) | .02 |
| 11q13 + 6p21 | 6/26 (23) | 10/71 (14) | .36 |
| D1 + (D1 + D2) | 8/26 (31) | 40/71 (56) | .04 |
| D2 + None | 6/26 (23) | 17/71 (24) | > .999 |
| PCLI greater than 1 | 18/26 (69) | 19/71 (27) | < .001 |
| ISS stage | |||
| I | 7/19 (37) | 23/59 (39) | > .999 |
| II | 6/19 (32) | 23/59 (39) | .60 |
| III | 6/19 (32) | 13/59 (22) | .54 |
Figure 5.

Kaplan-Meier curve for OS of newly diagnosed MM in patients (n = 72) stratified by CI. Patients with CI more than 4 (n = 18) had significantly shorter survival compared with patients with CI less than 4 (n = 49) (median OS, 11.1 months versus 39.1 months; log-rank P < .001).
Table 3.
Clinical characteristics of newly diagnosed group 2 MM patients included in survival analysis (n = 67)
| Mayo ID | Sex | Age, y | TC class | Heavy chain | Light chain | 13 del | PCLI | ISS stage | CI | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|
| M025 | F | 49 | D2 | L | Kappa | 0 | 0 | III | 6.19 | BMT |
| M452 | F | 49 | 4p16 | G | Kappa | 1 | 4.2 | I | 5.89 | BMT |
| M413 | M | 54 | 11q13 | L | Lambda | 1 | 4.2 | III | 5.52 | BMT |
| M227 | M | 50 | D1 | L | Kappa | 1 | 0 | I | 5.08 | BMT |
| M263 | F | 53 | 11q13 | L | Kappa | 1 | 6.2 | III | 4.74 | BMT |
| M427 | M | 53 | 11q13 | N | N | 1 | 0 | I | 4.48 | BMT |
| M310 | M | 56 | 4p16 | A | Kappa | 1 | 1.8 | I | 4.15 | BMT |
| M822 | M | 67 | D1 + D2 | A | Lambda | 1 | 2.2 | I | 4.12 | BMT |
| M163 | M | 60 | D1 | A | Kappa | 0 | 0.4 | II | 4.06 | BMT |
| M288 | M | 57 | None | G | Lambda | 0 | 9.4 | II | 5.73 | Dex |
| M196 | F | 68 | MAF | L | Kappa | 1 | 4.4 | — | 5.43 | M + P |
| M814 | M | 67 | MAF | A | Kappa | 1 | 5.2 | III | 4.99 | M + P |
| M037 | M | 81 | D2 | G | Kappa | 1 | 0.2 | III | 4.23 | M + P |
| M024 | M | 81 | 4p16 | G | Kappa | 1 | 3.6 | II | 4.04 | M + P |
| M146 | M | 76 | D2 | L | Kappa | 0 | 1.6 | II | 4.29 | Multiple |
| M318 | M | 51 | D1 | L | Lambda | 0 | 4 | I | 4.52 | Dex |
| M806 | M | 76 | D1 | G | Kappa | 0 | 0 | — | 4.04 | Dex |
| M249 | F | 73 | D1 | A | Kappa | 1 | 5.1 | II | 4.08 | Thal |
| M350 | F | 59 | 6p21 | L | Kappa | — | 0.4 | I | 3.91 | BMT |
| M804 | F | 38 | 11q13 | L | Lambda | 1 | 0 | II | 3.83 | BMT |
| M419 | F | 65 | D2 | A | Lambda | 1 | 0.2 | III | 3.68 | BMT |
| M157 | F | 52 | D2 | L | Lambda | 1 | 0 | I | 3.61 | BMT |
| M200 | M | 65 | D2 | A | Kappa | 0 | 3.8 | II | 3.58 | BMT |
| M380 | F | 51 | D1 | G | Kappa | 0 | 2 | I | 3.57 | BMT |
| M797 | F | 49 | D2 | A | Lambda | 0 | 1.4 | I | 3.47 | BMT |
| M300 | M | 59 | None | G | Kappa | 1 | 1.2 | I | 3.33 | BMT |
| M307 | F | 68 | D1 + D2 | A | lambda | 0 | 0 | III | 3.27 | BMT |
| M058 | M | 70 | D1 | G | lambda | 1 | 0 | III | 3.25 | BMT |
| M198 | M | 53 | D1 | A | Kappa | 0 | 0.4 | II | 3.17 | BMT |
| M387 | F | 62 | D2 | L | Kappa | 0 | 0.2 | II | 3.16 | BMT |
| M407 | M | 58 | D1 | A | Kappa | 0 | 0.6 | I | 3.00 | BMT |
| M700 | M | 59 | D1 | G | Kappa | 1 | 0.8 | I | 2.92 | BMT |
| M687 | M | 62 | D2 | L | lambda | 1 | 0 | I | 2.79 | BMT |
| M059 | M | 61 | D1 | L | Kappa | 0 | 0 | I | 2.79 | BMT |
| M233 | F | 71 | D2 | G | Lambda | 0 | 0.3 | II | 2.74 | BMT |
| M167 | M | 53 | D2 | G | Kappa | 0 | 0.7 | I | 2.64 | BMT |
| M245 | F | 62 | D1 | G | Kappa | 0 | 0 | I | 2.53 | BMT |
| M304 | M | 60 | D1 + D2 | G | Lambda | 0 | 0 | I | 2.53 | BMT |
| M134 | M | 52 | D2 | N | N | 0 | 0 | I | 2.43 | BMT |
| M073 | M | 45 | 11q13 | G | Lambda | 1 | 0 | II | 2.42 | BMT |
| M412 | M | 71 | D1 | G | Kappa | 0 | 0.2 | II | 2.38 | BMT |
| M271 | M | 66 | D2 | A | Kappa | 0 | 0 | II | 2.30 | BMT |
| M410 | F | 64 | None | A | Kappa | 0 | 0 | I | 2.20 | BMT |
| M795 | M | 55 | D1 | G | Lambda | 0 | 0 | II | 2.13 | BMT |
| M373 | M | 53 | D1 | G | lambda | 0 | 0 | I | 2.11 | BMT |
| M338 | F | 45 | D1 | G | Kappa | 0 | 2.8 | II | 1.78 | BMT |
| M678 | M | 70 | 11q13 | A | Kappa | 1 | 0 | II | 1.29 | BMT |
| M467 | M | 44 | D1 | A | Lambda | 0 | 0 | I | 3.82 | Dex |
| M813 | M | 53 | D2 | G | Kappa | 0 | 0.2 | I | 3.60 | Dex |
| M061 | M | 71 | D1 | L | Kappa | 0 | 0 | II | 3.46 | Dex |
| M272 | F | 68 | MAF | L | Lambda | 1 | 1 | III | 3.36 | Dex |
| M356 | M | 74 | MAF | G | Kappa | 1 | 0.6 | I | 2.65 | Dex |
| M264 | F | 56 | D1 | G | Kappa | 1 | 0.6 | III | 2.53 | Dex |
| M032 | M | 78 | D2 | A | Lambda | 0 | 0.2 | II | 3.97 | M + P |
| M218 | F | 69 | D1 | A | Lambda | 0 | 0 | II | 3.94 | M + P |
| M279 | M | 77 | D1 | A | Kappa | 0 | 1.2 | III | 3.57 | M + P |
| M321 | F | 82 | 11q13 | D | Lambda | 1 | 0 | — | 3.35 | M + P |
| M142 | M | 81 | D1 | A | Kappa | 0 | 0.2 | II | 3.20 | M + P |
| M820 | M | 74 | 11q13 | G | Lambda | 1 | 0 | III | 2.84 | M + P |
| M672 | F | 41 | D1 | G | Kappa | 0 | 0 | — | 2.76 | M + P |
| M337 | M | 75 | D1 | A | Kappa | 1 | 0 | II | 2.51 | M + P |
| M135 | M | 78 | D1 | G | Kappa | 0 | 0 | II | 2.33 | M + P |
| M359 | M | 49 | D1 | G | Kappa | 0 | 0.2 | II | 2.06 | M + P |
| M383 | M | 71 | D1 | G | Kappa | 0 | 0.2 | II | 3.74 | Multiple |
| M390 | F | 64 | 11q13 | L | Kappa | 1 | 0.6 | I | 1.59 | Multiple |
| M230 | M | 58 | D1 | A | Kappa | 1 | 1 | III | 2.98 | Multiple |
| M250 | M | 52 | 11q13 | G | Kappa | 1 | 0.8 | II | 2.85 | Thal |
L indicates light chain only; 0, absent; BMT, single autologous stem cell transplantation; G, IgG; I, present; N, nonsecretory MM; A, IgA; Dex, dexamethasone-based treatment; M + P, melphalan and prednisolone; Multiple, multiple nontransplantation-based treatments including novel agents; Thal, thalidomide-based treatment; D, IgD; and —, unknown, due to inadequate information.
To further characterize the prognostic significance of high CI, we performed multivariate regression analysis using the following dichotomized parameters that were significant prognostic factors on univariate analysis (data not shown): PCLI greater than 1%, 4p16 or MAF TC class, CI more than 4, Δ13, and ISS stage III. Including only samples with all of the above-mentioned information available (n = 63), CI more than 4 emerged as an independent prognostic factor for poor survival together with ISS stage III and 4p16 or MAF TC class (Table 4).
Table 4.
Cox proportional hazard regression model for OS (n = 63)
| Variable | Estimate | Hazard ratio | P |
|---|---|---|---|
| 4p16 or MAF TC class | 1.09877 | 3.00 | .038 |
| CI greater than 4 | 0.86838 | 2.38 | .037 |
| ISS stage III | 1.30521 | 3.69 | .002 |
Discussion
In this comprehensive study of centrosome abnormalities in PC neoplasm using IF for protein expression and GEP, we have shown that centrosome amplification is present because of MGUS, suggesting it plays an early role in genomic instability and myeloma pathogenesis. Our results obtained with gene expression analysis suggest good concordance between protein and gene expression data. This allows us to derive a gene expression-based CI that is highly correlated with the presence of centrosome amplification. Clinically, a high CI is associated with poor prognostic features and a very short survival.
Centrosome expression is different between normal and malignant PCs
Using IF detection of centrin, an integral centrosome protein associated with and required for the duplication of centrioles,26 we showed for the first time that most normal PCs do not have centrosome. This is consistent with the notion that normal PCs are generally recognized as terminally differentiated nondividing cells.31 In contrast, most malignant PCs have centrosomes. The finding indicates that centrosomes are either retained in the initial steps of malignant transformation of PCs or that centrosome reactivation occurs as an integral part of the oncogenesis in MM. As reactivation of centrosome as a consequence of malignant transformation has never been reported, we believe it is most likely that malignant transformation of PCs occurs prior to loss of centrosomes. An indirect conclusion would be that in most cases of MM, the majority of the cells that form the MM clone have replicative potential (ie, those with centrosomes). Although this study cannot demonstrate the long-term viability of such cells and thus disprove the theory of a MM stem cell, it certainly argues strongly in favor of the aforementioned replicative potential of malignant PCs. This is consistent with the data generated by the xenograft models reported by Yaccoby and Epstein.35
Centrosome amplification is observed in MGUS
Centrosome amplification detected by IF is prevalent in all stages of PC neoplasm, confirming in a larger number of patients a previous report showing frequent centrosome abnormalities in MGUS and MM.36 Our finding that centrosome amplification is frequently present, even in the premalignant MGUS stage, suggests that centrosome abnormalities are present early during disease evolution and may contribute toward increasing genomic instability and disease progression. This adds to the accumulating evidence that centrosome amplification may be important in tumorigenesis rather than reflecting a malignant phenotype.16,17,22 This is further substantiated by the fact that the percentage of plasma cells with centrosome amplification increases through stages of clonal PC expansion. Although a possible explanation for these differences could be the dilution of clonal cells with polyclonal cells in samples from patients with MGUS and SMM, our use of concurrent cIg staining to identify clonotypic plasma cells should minimize this. These results are in agreement with studies in solid tumors that showed higher level of centrosome abnormalities in more advance disease.2,12-17,19
Centrosome amplification and aneuploidy
Chromosomal abnormalities and aneuploidy are pervasive in MM. Because the 2 main ploidy subtypes of MM differ with regard to the predominant karyotypic abnormalities: whole chromosome gains and losses for hyperdiploid MM and structural abnormalities, including translocations for nonhyperdiploid MM,37 we hypothesized that the prevalence of centrosome abnormalities may differ between the 2 subtypes (being more common in hyperdiploid MM). However, no difference in centrosome abnormalities was detected. This suggests that, although the spectrum of defining genetic abnormalities is different between the 2 ploidy categories of MM, centrosome amplification is involved in mediating ongoing genomic instability in both instances. In particular it should be noted that aneuploidy is also evident in the nonhyperdiploid MM, predominantly based on the presence of monosomies, although trisomies may also be present.
In addition, no correlation between the CI and number of aneusomies from a panel of 5 chromosomes as detected by FISH was found. This may be due to the extensive and complex cytogenetic abnormalities in MM. Most karyotypes contain more than 5 abnormalities, and the aneusomies may involve any of the chromosomes. Therefore, the panel used in our study may be too limited. Karyotypic analysis would be best, but it is well known that informative karyotypes are obtained in only about one sixth of patients tested, and even then they do not fully represent the chromosomal complexity of the disease.38 In our cohort, too few patients have complete karyotypes for meaningful correlation.
High CI is associated with poor prognostic features and short survival
Clinically, patients with a high CI are associated with poor prognostic features such as t(4;14), t(14;16), Δ13, and high PCLI and very short survival. Of note, a high CI is associated with poor outcome independent of these other factors. Centrosome amplification and cell-cycle deregulation are closely linked. Therefore, tumors with high CI may be more genomically unstable, have greater proliferative potential, and are associated with more aggressive subtypes of MM.
Conclusion
In conclusion, we have shown that centrosome amplification is common in all stages of PC neoplasm including MGUS and is therefore an early event in myeloma genesis and is probably integral to disease pathogenesis and genomic instability in MM. In patients with MM, a high CI is associated with poor prognostic genetic subtypes and portends a short survival. Further work to establish mechanism underlying centrosome amplification and its connection to cell-cycle deregulation may identify potential therapeutic targets.
Acknowledgments
R.F. is a clinical investigator of the Damon Runyon Cancer Research Fund.
Prepublished online as Blood First Edition Paper, December 22, 2005; DOI 10.1182/blood-2005-09-3810.
Supported by the International Waldenström Macroglobulinemia Foundation, the National Cancer Institute (grants R01 CA83724-01, SPORE P50 CA100707-01, and P01 CA62242), the Fund to Cure Myeloma, and by an international fellowship from the Agency for Science, Technology and Research (A*STAR) (W.J.C.).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396: 643-649. [DOI] [PubMed] [Google Scholar]
- 2.Pihan GA, Purohit A, Wallace J, et al. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998;58: 3974-3985. [PubMed] [Google Scholar]
- 3.Balmain A, Gray J, Ponder B. The genetics and genomics of cancer. Nat Genet. 2003;33(suppl): 238-244. [DOI] [PubMed] [Google Scholar]
- 4.Pihan G, Doxsey SJ. Mutations and aneuploidy: co-conspirators in cancer? Cancer Cell. 2003;4: 89-94. [DOI] [PubMed] [Google Scholar]
- 5.Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392: 300-303. [DOI] [PubMed] [Google Scholar]
- 6.Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol. 2002;14: 25-34. [DOI] [PubMed] [Google Scholar]
- 7.D'Assoro AB, Lingle WL, Salisbury JL. Centrosome amplification and the development of cancer. Oncogene. 2002;21: 6146-6153. [DOI] [PubMed] [Google Scholar]
- 8.Kramer A, Neben K, Ho AD. Centrosome replication, genomic instability and cancer. Leukemia. 2002;16: 767-775. [DOI] [PubMed] [Google Scholar]
- 9.Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer. 2002;2: 815-825. [DOI] [PubMed] [Google Scholar]
- 10.Sluder G, Nordberg JJ. The good, the bad and the ugly: the practical consequences of centrosome amplification. Curr Opin Cell Biol. 2004;16: 49-54. [DOI] [PubMed] [Google Scholar]
- 11.Gustafson LM, Gleich LL, Fukasawa K, et al. Centrosome hyperamplification in head and neck squamous cell carcinoma: a potential phenotypic marker of tumor aggressiveness. Laryngoscope. 2000;110: 1798-1801. [DOI] [PubMed] [Google Scholar]
- 12.Kuo KK, Sato N, Mizumoto K, et al. Centrosome abnormalities in human carcinomas of the gall-bladder and intrahepatic and extrahepatic bile ducts. Hepatology. 2000;31: 59-64. [DOI] [PubMed] [Google Scholar]
- 13.Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci U S A. 1998; 95: 2950-2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lingle WL, Salisbury JL. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am J Pathol. 1999;155: 1941-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res. 2001;61: 2212-2219. [PubMed] [Google Scholar]
- 16.Sato N, Mizumoto K, Nakamura M, et al. Centrosome abnormalities in pancreatic ductal carcinoma. Clin Cancer Res. 1999;5: 963-970. [PubMed] [Google Scholar]
- 17.Giehl M, Fabarius A, Frank O, et al. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia. 2005;19: 1192-1197. [DOI] [PubMed] [Google Scholar]
- 18.Neben K, Tews B, Wrobel G, et al. Gene expression patterns in acute myeloid leukemia correlate with centrosome aberrations and numerical chromosome changes. Oncogene. 2004;23: 2379-2384. [DOI] [PubMed] [Google Scholar]
- 19.Duensing S, Lee BH, Dal Cin P, Munger K. Excessive centrosome abnormalities without ongoing numerical chromosome instability in a Burkitt's lymphoma. Mol Cancer. 2003;2: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kramer A, Schweizer S, Neben K, et al. Centrosome aberrations as a possible mechanism for chromosomal instability in non-Hodgkin's lymphoma. Leukemia. 2003;17: 2207-2213. [DOI] [PubMed] [Google Scholar]
- 21.Ventura RA, Martin-Subero JI, Knippschild U, et al. Centrosome abnormalities in ALK-positive anaplastic large-cell lymphoma. Leukemia. 2004; 18: 1910-1911. [DOI] [PubMed] [Google Scholar]
- 22.Lingle WL, Barrett SL, Negron VC, et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A. 2002;99: 1978-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pihan GA, Wallace J, Zhou Y, Doxsey SJ. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003;63: 1398-1404. [PubMed] [Google Scholar]
- 24.Zandecki M, Lai JL, Facon T. Multiple myeloma: almost all patients are cytogenetically abnormal. Br J Haematol. 1996;94: 217-227. [DOI] [PubMed] [Google Scholar]
- 25.The International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121: 749-757. [PubMed] [Google Scholar]
- 26.Salisbury JL. Centrin, centrosomes, and mitotic spindle poles. Curr Opin Cell Biol. 1995;7: 39-45. [DOI] [PubMed] [Google Scholar]
- 27.Abraham RS, Ballman KV, Dispenzieri A, et al. Functional gene expression analysis of clonal plasma cells identifies a unique molecular profile for light chain amyloidosis. Blood. 2005;105: 794-803. [DOI] [PubMed] [Google Scholar]
- 28.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J Jr. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106: 296-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmann GJ, Jalal SM, Juneau AL, et al. A novel three-color, clone-specific fluorescence in situ hybridization procedure for monoclonal gammopathies. Cancer Genet Cytogenet. 1998;101: 7-11. [DOI] [PubMed] [Google Scholar]
- 30.Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101: 4569-4575. [DOI] [PubMed] [Google Scholar]
- 31.Chng WJ, Van Wier SA, Ahmann GJ, et al. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS. Blood. 2005;106: 2156-2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greipp PR, Trendle MC, Leong T, et al. Is flow cytometric DNA content hypodiploidy prognostic in multiple myeloma? Leuk Lymphoma. 1999;35: 83-89. [DOI] [PubMed] [Google Scholar]
- 33.Greipp PR, Lust JA, O'Fallon WM, Katzmann JA, Witzig TE, Kyle RA. Plasma cell labeling index and beta 2-microglobulin predict survival independent of thymidine kinase and C-reactive protein in multiple myeloma. Blood. 1993;81: 3382-3387. [PubMed] [Google Scholar]
- 34.Wang Q, Hirohashi Y, Furuuchi K, et al. The centrosome in normal and transformed cells. DNA Cell Biol. 2004;23: 475-489. [DOI] [PubMed] [Google Scholar]
- 35.Yaccoby S, Epstein J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood. 1999;94: 3576-3582. [PubMed] [Google Scholar]
- 36.Maxwell CA, Keats JJ, Belch AR, Pilarski LM, Reiman T. Receptor for hyaluronan-mediated motility correlates with centrosome abnormalities in multiple myeloma and maintains mitotic integrity. Cancer Res. 2005;65: 850-860. [PubMed] [Google Scholar]
- 37.Smadja NV, Fruchart C, Isnard F, et al. Chromosomal analysis in multiple myeloma: cytogenetic evidence of two different diseases. Leukemia. 1998;12: 960-969. [DOI] [PubMed] [Google Scholar]
- 38.Fonseca R, Barlogie B, Bataille R, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64: 1546-1558. [DOI] [PubMed] [Google Scholar]