

Abstract
Hepcidin is an antimicrobial peptide secreted by the liver during inflammation that plays a central role in mammalian iron homeostasis. Here we demonstrate the endogenous expression of hepcidin by macrophages and neutrophils in vitro and in vivo. These myeloid cell types produced hepcidin in response to bacterial pathogens in a toll-like receptor 4 (TLR4)-dependent fashion. Conversely, bacterial stimulation of macrophages triggered a TLR4-dependent reduction in the iron exporter ferroportin. In vivo, intraperitoneal challenge with Pseudomonas aeruginosa induced TLR4-dependent hepcidin expression and iron deposition in splenic macrophages, findings mirrored in subcutaneous infection with group A Streptococcus where hepcidin induction was further observed in neutrophils migrating to the tissue site of infection. Hepcidin expression in cultured hepatocytes or in the livers of mice infected with bacteria was independent of TLR4, suggesting the TLR4-hepcidin pathway is restricted to myeloid cell types. Our findings identify endogenous myeloid cell hepcidin production as a previously unrecognized component of the host response to bacterial pathogens.
Introduction
Hepcidin is a key regulator of iron homeostasis and anemia of inflammation.1 A cationic peptide produced in the liver and detectable in blood and urine,2,3 hepcidin is processed from an propeptide precursor into mature forms of 20 to 25 residues. Knockout mice lacking hepcidin developed iron overload in liver and pancreas, but iron deficit in the macrophage-rich spleen.4 Conversely, transgenic animals with constitutive hepcidin gene expression die at birth of severe iron deficiency.5 These studies suggested that hepcidin both inhibits iron absorption in the small intestine and the release of recycled iron from macrophages. Two isoforms of hepcidin (1 and 2) are present in the mouse, but only hepcidin-1 produces changes in iron metabolism when overexpressed.6 The underlying mechanism has been elucidated as hepcidin binds ferroportin, the sole known iron exporter, inducing its internalization and degradation, thus trapping iron in enterocytes, hepatocytes, and macrophages.7
In humans, hereditary hemochromatosis can result from hepcidin deficiency attributed to specific mutations in at least 4 distinct genes, including the hepcidin gene (HAMP) itself.8-10 Emerging data suggest that mutations in 3 genes (HFE, TFR2, and HJV) lead to aberrant regulation of hepcidin expression. Mutations in HFE, a gene encoding a protein of the major histocompatibility complex class I, represent the most prevalent form, while rarer defects are found in TRF2, which mediates cellular uptake of transferrin-bound iron.11 Knockout mice lacking either Hfe12,13 or Tfr214 develop hepatic iron overload as do human patients with mutations in the corresponding genes. Even in the face of iron overload, Hfe- or Tfr2-deficient mice exhibit hepcidin levels that are lower than control mice.15,16 A more severe disease, juvenile hemochromatosis, can result either from mutations in HAMP itself or from mutations in hemojuvelin (HJV). Mouse models of juvenile hemochromatosis have been developed by 2 groups through targeted disruptions of the Hjv gene.17,18
Hepcidin is strongly induced during infections and inflammation, causing intracellular iron sequestration and decreased plasma iron levels, consequently triggering the “anemia of chronic disease.”19 In both humans and mice, inflammatory stimulation of liver hepcidin synthesis is indirect and mediated by macrophage production of cytokines interleukin-6 (IL-6) and IL-1.20,21 Curiously, a recent microarray analysis of murine gene expression within lesions of a cutaneous infection model listed hepcidin among the upregulated genes.22 These results let us hypothesize that hepcidin could be expressed directly by inflammatory cells recruited to the site of infection (eg, neutrophils and macrophages). Here we investigate the intrinsic ability of myeloid cells to produce hepcidin in vitro and in vivo and the involvement of pattern recognition by toll-like receptor 4 (TLR4) in this process.
Materials and methods
Mice and bacterial strains
Inbred mouse strains CH3/HeOuJ and CH3/HeJ were obtained from Jackson Laboratories (Bar Harbor, ME), then bred and handled in our facilities under the approved protocols of the University of California at San Diego Animal Care Committee. Group A Streptococcus strain 5448 (GAS),23 Pseudomonas aeruginosa ATCC (American Type Culture Collection, Manassas, VA) 27853 (PA) and Salmonella typhimuriumATCC 13311 (ST) were propagated using Todd-Hewitt broth (THB) or agar (THA). Bacteria were grown to logarithmic growth phase, pelleted by centrifugation, then washed and resuspended in phosphate-buffered saline (PBS) at known concentration for use in the described in vitro and in vivo challenge experiments.
Mouse model of PA and GAS infection
In systemic challenge experiments, 6- to 8-week-old mice were given intraperitoneal injections of mid-log phase (approximately 107 colony-forming units [cfu]) PA in 200 μL PBS or PBS alone as a control. After 4 or 24 hours, spleen, liver, and blood (retroorbital bleed) were collected for histopathologic analysis or iron studies. In certain experiments, mice were fed an iron-poor feed (4 parts per million iron; Harlan Teklad, Indianapolis, IN) prior to PA infection. For GAS challenges, we used a well-established model of necrotizing soft-tissue infection.24,25 Briefly, log-phase GAS strains were resuspended in PBS, and mixed 1:1 with Cytodex beads (50 mg/100 mL; Sigma, St Louis, MO). An inoculum of 107 cfu GAS in 200 μL was then injected into the right flank of 6- to 8-week-old mice. At 96 hours, all animals were killed, and biopsies of skin, spleen, bone marrow, and liver were performed for histopathologic assessment.
Macrophages and neutrophil harvest, Western blot
Neutrophils were isolated from the peritoneal cavity 3 hours after injection of thioglycollate as previously described.26 To isolate bone marrow (BM)-derived macrophages, the marrow of femurs and tibias of wild-type (WT) and Tlr4Lps-d mice were collected. Cells were plated in Dulbecco-modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 30% conditioned medium (a 7-day supernatant of fibroblasts from cell line L-929 stably transfected with an macrophage colony-stimulating factor [M-CSF] expression vector). Mature adherent BM cells were harvested by gentle scraping after 7 days in culture. Neutrophils or macrophages exposed to bacteria for 4 hours were harvested, washed (PBS), and proteins extracted with RIPA buffer. Nuclear or cytoplasmic extracts (20 μg) were loaded on a 12% Tris-tricine gel in a MES buffer (Invitrogen, Carlsbad, CA) for hepcidin and ferroportin Western blot using standard methodologies. The primary antibodies used were anti-mouse hepcidin (Hepc-25) and rabbit anti-mouse ferroportin (Alpha Diagnostics, San Antonio, TX) at a concentration of 1/1000.
Immunohistochemistry, iron staining, and immunofluorescence
Tissues were fixed in 10% formalin sectioned in paraffin, then subjected to microwave antigen retrieval and immunohistochemistry (AP-blue kit; Vector Laboratories, Burlingame, CA) with primary antibodies against hepcidin (1:200) and neutrophils (1:100; Accurate Chemical, Westbury, NY). Bone was decalcified in Cal-Ex (Fisher Scientific, Hampton, NH) for 2 days prior to fixation in formalin. Iron detection was performed with Perl Prussian blue and nuclear red counterstain. For immunofluorescence, macrophages were plated on chamber slides (Nalge Nunc International, Naperville, IL) and incubated for 4 hours with PA or GAS. Cells were fixed with 4% paraformaldehyde for 25 minutes at 4°C and permeabilized with 0.2% Triton X-100 for 5 minutes. Rabbit anti-mouse hepcidin antibodies (1:200) were added overnight at 4°C and detected with Alexa Fluor 488 goat antirabbit antibodies (Molecular Probes, Eugene, OR).
Antibody control experiments with blocking peptides
Anti-mouse hepcidin antibodies (5 μL) were added to 100 μL PBS with or without 50 μg mouse HEPC control/blocking peptide (Alpha Diagnostic) and gently mixed. Both tubes were incubated at 37°C for 2 hours, followed by 2 hours at 4°C. The tubes were centrifuged for 15 minutes at 4°C (13 400g) to pellet immune complexes and the supernatant was carefully removed. After centrifugation, the volume of supernatant was replenished to 5 mL with buffer (Tris-buffered saline [TBS] + 0.2% Tween + 5% nonfat dried milk) for Western blot or to 1 mL with PBS for immunohisto-chemistry. Hepcidin antibody with or without control/blocking peptide was used for Western blot and immunohistochemical detection.
Reverse transcription and real-time quantitative PCR
Macrophages were exposed to bacteria (multiplicity of infection [MOI] = 10 bacteria/cell) for 1 hour. Gentamicin (100 μg/mL) and penicillin (5 μg/mL) were added and the cells incubated for an additional 3 hours. As a further control, 100 ng/mL lipopolysaccharide (LPS; Sigma) was added for 6 hours before RNA harvest (Tri-Reagent; Molecular Research Center, Cincinnati, OH). In iron stimulation studies, diferric transferrin (Sigma) was added to macrophage monolayers for 24 hours at 30 μM. Hepatocytes were exposed to bacteria (MOI = 10) for 4 hours or 100 ng/mL LPS for 6 hours. First-strand synthesis and real-time polymerase chain reaction (PCR) were performed as described24 in a TaqMan-Universal Mastermix SYBR Green (Applied Biosytems, Foster City, CA) using specific primers with the following sequences (rates normalized to the expression level of ribosomal RNA): mouse FN1-Forward 5′-CTACCATTAGAAGGATTGACCAGCTA-3′, mouse FN1-Reverse 5′-ACTGGAGAACCAAATGTCATAATCTG-3′; and mouse Hepcidin-Forward 5′-TGTCTCCTGCTTCTCCTCCT-3′, mouse Hepcidin-Reverse 5′-CTCTGTAGTCTGTCTCATCTGTTG-3′.
The hepcidin primers recognize both isoforms of murine hepcidin (HEPC1 and HEPC2). Specific primer sequences distinguishing mouse hepcidin-1 from hepcidin-2 were identical to those published by Ilyin et al.27
Image analysis
Stained sections were examined with a Leitz MRB microscope (Leica, Bensheim, Germany) equipped with an RT Color Spot camera (Diagnostic Instruments, Sterling Heights, MI). Image processing was completed with Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA). The objective lenses used were Leica HC PL Fluotar 5 ×/0.12 NA, 20 ×/0.5 NA, 50 ×/0.7 NA or Leica HCX PL APO 60 ×/1.4-0.6 NA oil objectives.
Serum iron measurement
Serum iron was quantitated colorimetrically in an assay based on the chromophore Ferene (Iron-SL; Diagnostic Chemicals, Oxford, CT) using a 595-nm endpoint and calculated against a standard curve.
Statistical analysis
All values in the figures are expressed as mean plus or minus standard deviation. Student t test (unpaired, 2-tailed) was used for comparison between experimental groups.
Results
Hepcidin production is induced in neutrophils and macrophages by bacterial pathogens
To explore potential new linkages between the immune response and iron regulation, we examined whether exposure to bacteria might trigger hepcidin production in myeloid cells. By immunofluorescence, macrophages exposed to either the Gram-positive bacterium GAS or the Gram-negative bacterium PA were found to produce hepcidin (Figure 1A). Bacterial induction of hepcidin in both macrophages and neutrophils was confirmed by Western blot (Figure 1B). Specificity of the antigen detection was supported by antigen blocking, as the observed approximately 7-kDa and approximately 16-kDa bands were no longer detected when the antibodies were neutralized by addition of excess hepcidin peptide (Figure 1B). It is important to note that the sequence of the mature form of hepcidin (hepcidin-25) would predict a mass of approximately 3 kDa. While a specific antibody against human hepcidin can detect a mature form of approximately 3 kDa in human urine specimens (T. Ganz, oral communication, December 2005), available murine antibody reagents detect only precursor forms (S. Vaulont, oral communication, December 2005). The antibody we used was generated to a linear peptide motif within the mature peptide sequence, but final folding may eliminate recognition. Detection of a band migrating with an apparent mass of approximately 7 to 10 kDa is consistent with pro-hepcidin as reported in other published Western blot studies.28,29
Figure 1.

Hepcidin production is induced in neutrophils and macrophages by bacterial pathogens. (A) Immunofluorescence using anti-mouse hepcidin antibody on normal murine macrophages at baseline and upon exposure to Group A Streptococcus (GAS) or P aeruginosa (PA); magnification × 630. (B) Western blot analysis of hepcidin in murine macrophages and neutrophils at baseline and upon PA exposure. Addition of 10 μg blocking hepcidin peptide per milliliter of antibody was used to check for the specificity of the anti-mouse hepcidin antibody.
TLR4-dependency of bacterial-induced hepcidin mRNA expression
TLRs function in innate immune recognition of invading microorganisms and activation of signaling pathways leading to transcriptional activation of inflammatory response genes. To determine whether the myeloid cell hepcidin response was transcriptionally regulated, we performed real-time reverse transcriptase (RT)-PCR analysis of hepcidin transcript in WT murine macrophages and those possessing a spontaneous mutation in TLR4, the lipopolysaccharide response locus (Tlr4Lps-d).30 Results were normalized first to 18S ribosomal RNA and then to mRNA levels of the housekeeping protein β-actin. Upon exposure to the Gram-negative pathogens PA or ST, macrophages isolated from WT (C3H/HeOuJ) mice showed strong induction (20- to 40-fold) of hepcidin mRNA, while this response was absent in Tlr4Lps-d (C3H/HeJ) mice (Figure 2A). LPS, the TLR4 ligand from Gram-negative bacteria, also induced hepcidin in the macrophages (Figure 2A). A variety of Gram-positive cytolytic toxins are now recognized to also activate macrophages via TLR4.31,32 We found WT GAS produced a 9-fold TLR4-specific activation of hepcidin mRNA expression, a response absent in macrophages challenged with an isogenic GAS mutant25 lacking the cytolysin streptolysin S (Figure 2A). Control RT-PCR experiments using primers to discriminate hepcidin isoforms show the TLR4-specific induction in macrophages is almost exclusively hepcidin-1 (Figure 2B).
Figure 2.
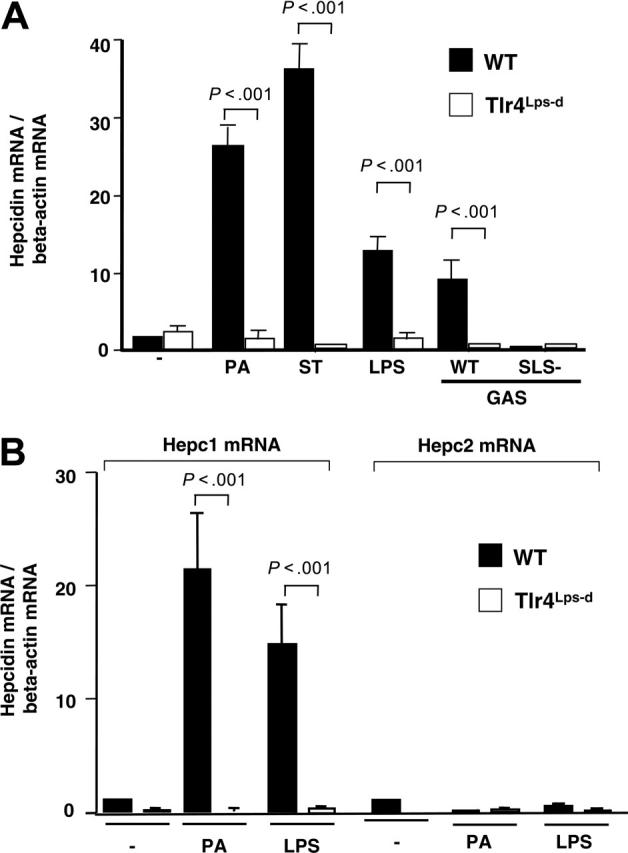
TLR4 dependency of bacterial-induced hepcidin mRNA expression by neutrophils and macrophages. (A) Real-time PCR for hepcidin mRNA on WT (CH3/HeouJ) and Tlr4Lps-d (CH3/HeJ) murine macrophages stimulated with P aeruginosa (PA), S typhimurium (ST), LPS, or Group A Streptococcus (GAS) producing or lacking the cytotoxin streptolysin S (SLS). Hepcidin mRNA was normalized to the expression level of β-actin. Quantitative assays performed in triplicate and representative of 3 repeated experiments. (B) Real-time PCR for hepcidin-1 and hepcidin-2 mRNA in WT (CH3/HeouJ) and Tlr4Lps-d (CH3/HeJ) macrophages stimulated with PA or LPS. Data are shown as mean of values ± standard deviation (SD).
TLR4-dependent suppression of ferroportin in neutrophils and macrophages exposed to bacterial pathogens
Binding and degradation of the iron exporter ferroportin by hepcidin increases intracellular iron stores, triggering hypoferremia.7 We found that isolated macrophages exposed to PA exhibited decreased ferroportin levels (Figure 3A) consistent with induction of endogenous hepcidin (Figure 1A-B). More strikingly, RT-PCR found a strong TLR4-dependent suppression of ferroportin mRNA production in response to PA (Figure 3B). These findings reveal a coordinated macrophage innate immune response to reduce ferroportin iron export at both the transcriptional and posttranslational levels.
Figure 3.

TLR4-dependent ferroportin suppression by neutrophils and macrophages exposed to bacterial pathogens. (A) Western blot of ferroportin production in murine bone marrow derived macrophages stimulated with PA. (B) Real-time PCR for ferroportin mRNA using WT and Tlr4Lps-d bone marrow derived macrophages infected with PA. Quantitative assays performed in triplicate and representative of 3 repeated experiments. Data are shown as mean of values ± SD.
Myeloid cell hepcidin expression in a systemic model of infection
We next asked whether bacterial infection could stimulate myeloid cell expression of hepcidin in vivo. In systemic infection 4 hours following intraperitoneal injection of PA, strong hepcidin expression was seen in the splenic reticuloendothelial system of normal mice, whereas little or no hepcidin was induced in Tlr4Lps-d mice (Figure 4A). Thus, TLR4 seems essential to the hepcidin production by myeloid cells in response to bacterial infection both in vitro and in vivo. In contrast, liver hepatocytes in Tlr4Lps-d mice are not deficient in their ability to produce hepcidin in vivo (Figure 4B), a finding consistent with other studies showing no or a very low expression of TLR4 on the hepatocyte cell surface.33 Real-time RT-PCR analysis performed on liver and spleen samples from these mice confirmed that while absolute levels of induced hepcidin-1 gene expression are significantly greater in liver than spleen, TLR4-specific induction was only appreciated in the macrophage-rich splenic tissue (Figure 4C). These studies were performed in animals fed a low-iron diet because the unusually high iron content of standard rat chows can block the normal hepcidin response to inflammatory stimuli.21
Figure 4.
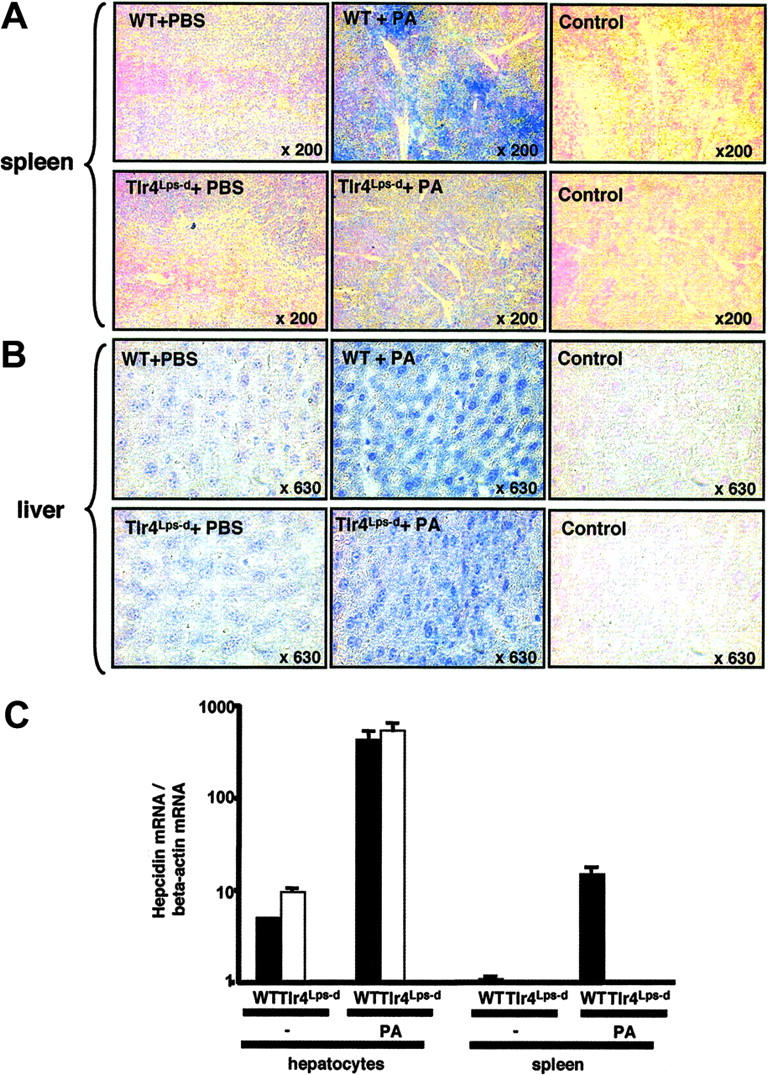
Hepcidin expression in spleen and liver of mice upon systemic infection. Immunohistochemistry using anti-mouse hepcidin antibodies on spleens (A) and livers (B) of mice 4 hours after intraperitoneal challenge with Pseudomonas aeruginosa or PBS control. (C) Real-time PCR for hepcidin-1 mRNA in liver and spleen of WT (CH3/HeouJ) and Tlr4Lps-d (CH3/HeJ) mice challenged for 4 hours with PA. Data are shown as mean ± SD
The TLR4-specific induction of hepcidin by PA correlated with functional changes in iron distribution and with increased iron content in splenic macrophages (Figure 5A). Following infection with PA in mice fed a low-iron diet, a significant decrease in serum iron was detected at 4 hours in WT mice compared with their Tlr4Lps-d counterparts (Figure 5B). This effect is even more striking since the liver of the Tlr4Lps-d mice was not affected in its general ability to produce hepcidin. The absence of infection-associated hypoferremia in the Tlr4-deficient mice may thus be directly correlated to the inability of their myeloid cells to produce hepcidin. Previous studies have indicated that whereas iron overload induces hepcidin expression in vivo,34 iron fails to up-regulate hepcidin transcription in isolated hepatocytes.34,35 We found that addition of iron in the form of diferric transferrin suppressed hepcidin production in both WT and Tlr4Lps-d macrophages (Figure 5C), pointing to differential effects of infection and iron overload on the macrophage contribution to the host hepcidin response.
Figure 5.

Splenic iron and measure of hypoferremia in WT and Tlr4Lps-d mice upon systemic infection. (A) Iron staining of splenic sections by Perl blue method 24 hour after intraperitoneal challenge with P aeruginosa (PA) or PBS control. (B) Serum iron measurement in WT and Tlr4Lps-d Tlr4Lps-d challenged with PA. (C) Hepcidin mRNA levels in WT and Tlr4Lps-d macrophages upon exposure to iron in the form of diferric transferrin.
Myeloid cell hepcidin expression in a subcutaneous model of infection
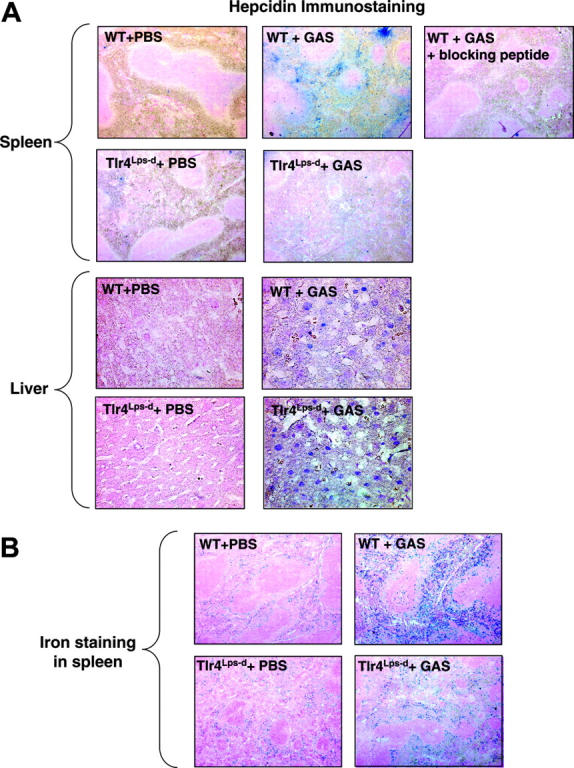
To examine myeloid cell hepcidin expression in localized foci of infection, we injected GAS subcutaneously to produce necrotic ulcer with marked neutrophil influx.24,25 In skin biopsies of the infected lesions, strong levels of hepcidin staining were observed in direct colocalization with recruited neutrophils (Figure 6A). Hepcidin staining was absent in the skin and subcutaneous tissues of the control mice given injections of PBS plus Cytodex beads alone. No hepcidin staining was detected in skin lesions of Tlr4Lps-d mice infected with GAS, once again confirming the TLR-4 dependence of induction of hepcidin in myeloid cells (Figure 6A).ATLR-4 dependent increase in hepcidin expression was also observed in the bone marrows of WT mice infected with GAS compared with controls (Figure 6B). Specificity of staining in all studies was confirmed by antibody neutralization with blocking peptides (Figure 6A-B).
Figure 6.

Myeloid cell hepcidin expression in a subcutaneous model of infection. (A) Immunohistochemistry using anti-mouse hepcidin and anti-polymorphonuclear leukocyte (PMN) antibodies on skin of WT and Tlr4Lps-d mice challenged with group A Streptococcus (GAS); controls include omission of primary antibody and addition of blocking peptide to the hepcidin antibody. (B) Immunohistochemistry using anti-mouse hepcidin antibodies on bone marrow of mice challenged with GAS; control corresponds to the addition of hepcidin blocking peptide to the hepcidin antibody. Magnification, × 400.
The subcutaneous injection of GAS is associated with bacterial seeding of the bloodstream,24,25 and invoked a mild acute-phase hepcidin response. Hepcidin expression was detected in the splenic reticuloendothelial system of WT mice infected with GAS, whereas little or no hepcidin was induced in Tlr4Lps-d mice (Figure 7A). This result correlates with a TLR4-specific increase of iron content in the splenic reticuloendothelial system of infected mice (Figure 7B). Thus, it appears that myeloid cells can participate directly in the acute hepcidin response in vivo restricting the availability of extracellular iron. A modest increase in liver production of hepcidin was also induced by GAS infection (Figure 7A). However, this response of the hepatocytes was TLR4 independent, since hepatocytes from infected Tlr4Lps-d produced similar levels of hepatic hepcidin as their WT counterparts (Figure 7A).
Figure 7.
Hepcidin expression in liver and spleen of mice infected with GAS and iron staining. (A) Immunohistochemistry using anti-mouse hepcidin antibodies on livers and spleens of mice challenged with group A Streptococcus (GAS), magnification × 50 (spleen) and × 400 (liver). (B) Iron staining of splenic sections by Perl blue method following similar mouse challenge.
Discussion
Hepcidin functions as a key regulator of iron homeostasis and anemia of inflammation. To our knowledge, these studies represent the first demonstration of endogenous hepcidin production by isolated myeloid cells. We have found that myeloid cells, which play a central role in inflammation, produce hepcidin intrinsically, and do so through the action of TLR4, the key pattern recognition receptor for LPS. This finding, however, must be understood in the context of liver hepcidin production, which still greatly exceeds that produced by splenic macrophages. As the primary biosynthetic reservoir, the liver certainly plays a key role in the hepcidin response to systemic infection. However, we found hepatocytes in liver from bacteria-challenged Tlr4Lps-d mice were equivalent to WT controls in their ability to produce hepcidin. Thus, the absence of infection-associated iron deposition in splenic macrophages and hypoferremia in TLR4-deficient mice may reflect the inability of their myeloid cells to contribute additively to the host hepcidin response. TLR4-specific hepcidin production by macrophages is likely to play its largest role in localized foci of infection and inflammation or in reticuloendothelial organs, where these cell types are present in abundance.
Inflammation, including that provoked by LPS administration, is known to decrease intestinal iron absorption and increase macrophage iron retention through the actions of hepcidin. Mice with deficiencies in the hemachromatosis gene product HFE appear to induce hepcidin appropriately and develop hypoferremia in response to LPS challenge,36 although 1 study suggests the opposite.37 Cytokine-induced inflammatory activation of hepcidin appears to occur independent of HJV.18 The evidence would appear to indicate that the pathways for iron regulation versus inflammatory activation of hepcidin could be quite distinct. Our demonstration of TLR4-dependent hepcidin production in myeloid cells may prove useful to investigators working in these genetic model systems to expand the scope of their investigations and perhaps reconcile data into a unified model.
We found a strong TLR4-dependent suppression of ferroportin mRNA production in response to bacteria. Our data support recently published findings showing inflammation-induced down-regulation of mononuclear-phagocyte system (MPS) ferroportin 1 mRNA38 and protein,38,39 the former being independent of hepcidin and the latter being IL-6- and hepcidin-dependent. We show here that the suppression of ferroportin mRNA in response to PA involves TLR4. Mice with defects in nuclear factor (NF)-κB signaling respond to LPS with hypoferremia and FPN1 down-regulation,38 suggesting that TLR4-mediated signaling to suppress ferroportin could be independent of the NF-κB pathway.
The mechanisms underlying of the stimulatory effect of iron overload on hepcidin synthesis remain obscure. Hepatocytes appear to respond to iron with hepcidin induction in vivo, but exposure of cultured hepatocytes to ferric iron or iron-saturated transferrin does not increase hepcidin mRNA, and it may even reduce it.35 Thus, it is tempting to speculate that the role of iron sensing may fall to other cells, which in turn send signals such as IL-6 to stimulate liver hepcidin production. A recent study excluded liver reticuloendothelial macrophages (eg, Kupffer cells) for this function, as adminstration of gadolinium (III) chloride to inhibit Kupffer cells phagocytosis and activation did not affect hepatic hepcidin activation during iron overload.40 The role of Kupffer cells in inflammatory activation of hepcidin remains controversial.40,41 Our finding that iron treatment functioned to suppress rather than stimulate macrophage hepcidin production suggests that myeloid cells do not themselves contribute to the elevated hepcidin levels characteristic of iron overload conditions in vivo.
In conclusion, the alterations in iron metabolism produced by hepcidin are hypothesized to play a role in host defense by limiting the availability of iron to invading microorganisms. Our discovery of endogenous macrophage and neutrophil hepcidin production suggests this process could begin in infected tissue microenvironments to which myeloid cells are recruited or in reticuloendothelial organs where resident macrophages serve a microbial filtering function. Further examination of the myeloid cell contribution to hepcidin biology and iron homeostasis could provide considerable new insight into mammalian iron homeostasis and innate immunity.
Prepublished online as Blood First Edition Paper, January 3, 2006; DOI 10.1182/blood-2005-06-2259.
Supported by grants from the National Institutes of Health AI48694 (V.N.) and CA82515 (R.S.J.), the Swiss National Foundation PBZHB108365 (A.S.Z.), and an Edward J. Mallinckrodt, Jr., Foundation Scholar Award (V.N.).
All authors participated in designing and performing the research, and in controlling and analyzing the data. C.P., R.S.J., and V.N. wrote the paper, and all authors checked the final version of the manuscript.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Ganz T. Hepcidin—a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract Res Clin Haematol. 2005;18: 171-182. [DOI] [PubMed] [Google Scholar]
- 2.Krause A, Neitz S, Magert HJ, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000; 480: 147-150. [DOI] [PubMed] [Google Scholar]
- 3.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276: 7806-7810. [DOI] [PubMed] [Google Scholar]
- 4.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001; 98: 8780-8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicolas G, Bennoun M, Porteu A, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci U S A. 2002;99: 4596-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lou DQ, Nicolas G, Lesbordes JC, et al. Functional differences between hepcidin 1 and 2 in transgenic mice. Blood. 2004;103: 2816-2821. [DOI] [PubMed] [Google Scholar]
- 7.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306: 2090-2093. [DOI] [PubMed] [Google Scholar]
- 8.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996; 13: 399-408. [DOI] [PubMed] [Google Scholar]
- 9.Muckenthaler M, Roy CN, Custodio AO, et al. Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat Genet. 2003;34: 102-107. [DOI] [PubMed] [Google Scholar]
- 10.Papanikolaou G, Tzilianos M, Christakis JI, et al. Hepcidin in iron overload disorders. Blood. 2005; 105: 4103-4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25: 14-15. [DOI] [PubMed] [Google Scholar]
- 12.Bahram S, Gilfillan S, Kuhn LC, et al. Experimental hemochromatosis due to MHC class I HFE deficiency: immune status and iron metabolism. Proc Natl Acad Sci U S A. 1999;96: 13312-13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou XY, Tomatsu S, Fleming RE, et al. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc Natl Acad Sci U S A. 1998;95: 2492-2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleming RE, Ahmann JR, Migas MC, et al. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc Natl Acad Sci U S A. 2002;99: 10653-10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawabata H, Fleming RE, Gui D, et al. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood. 2005;105: 376-381. [DOI] [PubMed] [Google Scholar]
- 16.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105: 1803-1806. [DOI] [PubMed] [Google Scholar]
- 17.Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115: 2187-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest. 2005; 115: 2180-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood. 2002;100: 3776-3781. [DOI] [PubMed] [Google Scholar]
- 20.Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci U S A. 2005;102: 1906-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113: 1271-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Motley ST, Morrow BJ, Liu X, et al. Simultaneous analysis of host and pathogen interactions during an in vivo infection reveals local induction of host acute phase response proteins, a novel bacterial stress response, and evidence of a host-imposed metal ion limited environment. Cell Microbiol. 2004;6: 849-865. [DOI] [PubMed] [Google Scholar]
- 23.Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun. 2000;68: 6362-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peyssonnaux C, Datta V, Cramer T, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115: 1806-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datta V, Myskowski SM, Kwinn LA, et al. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol Microbiol. 2005;56: 681-695. [DOI] [PubMed] [Google Scholar]
- 26.Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112: 645-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ilyin G, Courselaud B, Troadec MB, et al. Comparative analysis of mouse hepcidin 1 and 2 genes: evidence for different patterns of expression and co-inducibility during iron overload. FEBS Lett. 2003;542: 22-26. [DOI] [PubMed] [Google Scholar]
- 28.Kulaksiz H, Gehrke SG, Janetzko A, et al. Pro-hepcidin: expression and cell specific localisation in the liver and its regulation in hereditary haemochromatosis, chronic renal insufficiency, and renal anaemia. Gut. 2004;53: 735-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallace DF, Summerville L, Lusby PE, Subramaniam VN. Prohepcidin localises to the Golgi compartment and secretory pathway in hepatocytes. J Hepatol. 2005;43: 720-728. [DOI] [PubMed] [Google Scholar]
- 30.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282: 2085-2088. [DOI] [PubMed] [Google Scholar]
- 31.Malley R, Henneke P, Morse SC, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A. 2003;100: 1966-1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JM, Ng VH, Maeda S, Rest RF, Karin M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J Exp Med. 2004; 200: 1647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull. 2005;28: 886-892. [DOI] [PubMed] [Google Scholar]
- 34.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276: 7811-7819. [DOI] [PubMed] [Google Scholar]
- 35.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101: 2461-2463. [DOI] [PubMed] [Google Scholar]
- 36.Roy CN, Custodio AO, de Graaf J, et al. An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide-induced inflammation in mice. Nat Genet. 2004;36: 481-485. [DOI] [PubMed] [Google Scholar]
- 37.Lee P, Peng H, Gelbart T, Beutler E. The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci U S A. 2004;101: 9263-9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu XB, Nguyen NB, Marquess KD, Yang F, Haile DJ. Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis. 2005;35: 47-56. [DOI] [PubMed] [Google Scholar]
- 39.Yang F, Liu XB, Quinones M, Melby PC, Ghio A, Haile DJ. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J Biol Chem. 2002;277: 39786-39791. [DOI] [PubMed] [Google Scholar]
- 40.Montosi G, Corradini E, Garuti C, et al. Kupffer cells and macrophages are not required for hepatic hepcidin activation during iron overload. Hepatology. 2005;41: 545-552. [DOI] [PubMed] [Google Scholar]
- 41.Lou DQ, Lesbordes JC, Nicolas G, et al. Iron- and inflammation-induced hepcidin gene expression in mice is not mediated by Kupffer cells in vivo. Hepatology. 2005;41: 1056-1064. [DOI] [PubMed] [Google Scholar]