

Abstract
As erythroid progenitors differentiate into precursors and finally mature red blood cells, lineage-specific genes are induced, and proliferation declines until cell cycle exit. Cul4A encodes a core subunit of a ubiquitin ligase that targets proteins for ubiquitin-mediated degradation, and Cul4A-haploinsufficient mice display hematopoietic dysregulation with fewer multipotential and erythroid-committed progenitors. In this study, stress induced by 5-fluorouracil or phenylhydrazine revealed a delay in the recovery of erythroid progenitors, early precursors, and normal hematocrits in Cul4A+/– mice. Conversely, overexpression of Cul4A in a growth factor-dependent, proerythroblast cell line increased proliferation and the proportion of cells in S phase. When these proerythroblasts were induced to terminally differentiate, endogenous Cul4A protein expression declined 3.6-fold. Its enforced expression interfered with erythrocyte maturation and cell cycle exit and, instead, promoted proliferation. Furthermore, p27 normally accumulates during erythroid terminal differentiation, but Cul4A-enforced expression destabilized p27 and attenuated its accumulation. Cul4A and p27 proteins coimmunoprecipitate, indicating that a Cul4A ubiquitin ligase targets p27 for degradation. These findings indicate that a Cul4A ubiquitin ligase positively regulates proliferation by targeting p27 for degradation and that Cul4A down-regulation during terminal erythroid differentiation allows p27 to accumulate and signal cell cycle exit.
Introduction
As hematopoietic progenitor cells give rise to more lineage-restricted precursors and finally mature blood cells, they acquire lineage-specific functions, and their proliferative potential declines, usually culminating in G0/G1 arrest. Positive regulators of cell cycle progression are repressed, whereas lineage-specific proteins and regulators that promote cell cycle exit are up-regulated. The coordinated regulation of cell cycle progression and differentiation is critical for normal hematopoiesis, and defects in regulation lead to specific lineage deficiencies, leukemia, or marrow failure. Our studies focus on the role that ubiquitin-mediated protein degradation plays in normal hematopoiesis.
The ubiquitin pathway plays an important role in controlling the turnover of intracellular regulators, including those regulating hematopoiesis. In particular, AML1, the p130 pocket protein, p27, and Jak2 are all regulators of hematopoiesis whose degradation is regulated by the ubiquitin pathway.1-4 First, a ubiquitin polypeptide is attached to a ubiquitin-activating enzyme (E1), then transferred to a ubiquitin-conjugating enzyme (E2), and next passed on to a ubiquitin ligase (E3), which finally transfers ubiquitin to the substrate protein.5 Repetition of these reactions results in the attachment of a polyubiquitin chain to the substrate, which is then degraded by the proteasome. Substrate specificity is largely determined by the E3 ubiquitin ligase, and the Cul4A protein is a core subunit of a multisubunit E3 ligase.
Cul4A encodes a member of the cullin protein family, with 7 members identified in mammals (Cul1, 2, 3, 4A, 4B, 5, and 7).6,7 Each cullin serves as the scaffold around which the other subunits are assembled, including one that recognizes and binds a specific substrate.8 Because each cullin can interact with several different substrate recognition subunits, each cullin is required for degrading multiple substrates and for regulating their corresponding cellular functions. Cul4A ubiquitin ligases target for ubiquitination and degradation Cdt1 (required for DNA replication), DDB2 (DNA repair), c-Jun, HoxA9 (development and differentiation), and p53.9-16 It is also likely there are additional targets.
Studies in nonhematopoietic cells indicate that Cul4A is required for the ubiquitin-mediated degradation of cell cycle regulators. Cul4A is amplified or overexpressed in breast cancer and hepatocellular carcinomas, suggesting a role in regulating the cell cycle.17,18 Also, nuclear Cul4A increases slightly near the G1/S boundary in synchronized HeLa cells, and in a genome-wide analysis of human fibroblast transcripts, Cul4A mRNA was highly expressed at the G1/S transition.12,19
In hematopoietic cells, Cul4A is involved in regulating proliferation and differentiation in maturing granulocytes or monocytes.15,20 We found that Cul4A expression declines during the differentiation of PLB-985 cells (a human, myelomonoblastic cell line) into either granulocytes or monocytes. Enforced Cul4A expression attenuates their differentiation and instead promotes proliferation, indicating that this decline is required for normal differentiation. Our in vivo studies suggest that Cul4A is more broadly required for myeloid and erythroid differentiation and for normal development.21 Cul4A-deficient mice die between 4.5 and 7.5 days after coitus (dpc). However, Cul4A+/– mice are viable, express approximately half the wild-type level of Cul4A, and appear normal, but they have fewer erythroid and multipotential myeloid progenitors, more than 5-fold and 3-fold fewer, respectively.20,21 Because Cul4A haploinsufficiency has a greater effect on erythroid progenitors, we investigated further its role in regulating erythropoiesis.
Here we report that underexpression and overexpression of Cul4A each affect erythroid cell proliferation. Studies with Cul4A+/– mice indicate that Cul4A haploinsufficiency delays the recovery of erythroid progenitors and proerythroblasts after stress induced by 5-fluorouracil or phenylhydrazine. Conversely, overexpression of Cul4A in a proerythroblast cell line increased proliferation. As these cells terminally differentiate, Cul4A declined 3.6-fold, and its enforced expression interfered with erythrocyte maturation and cell cycle exit. Enforced Cul4A expression prevented p27 accumulation during terminal differentiation by reducing p27 protein stability. Furthermore, Cul4A and p27 interact in co-immunoprecipitations. Overall, these findings indicate that Cul4A regulates erythrocyte proliferation by targeting p27 for ubiquitin-mediated degradation and that the decline in Cul4A during terminal differentiation allows p27 levels to increase and signal cell cycle exit.
Materials and methods
Mice and treatments
Cul4A+/– mice and wild-type littermate controls (backcrossed into C57BL/6 11 or more generations) were generated and genotyped as described.21 Sex-matched 6- to 8-week-old mice were used for all experiments. 5-Fluorouracil (5-FU; Faulding Pharmaceutical Elizabeth, NJ), was administered as a single intraperitoneal injection of 150 mg/kg. Phenylhydrazine was administered and blood samples for hematocrits were obtained and analyzed as described.22
Hematopoietic progenitor cell assay
Bone marrow cells were harvested, and hematopoietic progenitors were assayed as described.20,23
Flow cytometry
Freshly isolated bone marrow cells were isolated and stained with phycoerythrin (PE)–conjugated anti-Ter119 (1 μg/mL; PharMingen, San Diego, CA) and fluorescein isothiocyanate (FITC)–conjugated anti-CD71 (1 μg/mL; PharMingen) antibodies and analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA) as described.22 For cell cycle analyses, cells were treated and analyzed as described.20
Cell culture and differentiation
G1E-ER4 cells were a generous gift from M. Weiss (Children's Hospital, Philadelphia, PA) and were maintained and induced to differentiate with 10–7 M β-estradiol (Sigma, St Louis, MO) as described.24,25
Plasmid constructions and transfections
A hemagglutinin antigen (HA) epitope-tagged Cul4A cDNA20 was inserted into the NotI site of MIEG3 (a retroviral vector with a green fluorescent protein [GFP] marker)26 to generate MIEG3-Cul4A-HA. Phoenix ecotropic cells (American Type Culture Collection, Manassas, VA) were transiently transfected with empty MIEG3 or MIEG3-Cul4A-HA via Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Supernatants were collected 48 hours after transfection, and 2 mL virus supernatant supplemented with 8 μg/mL Polybrene was used to transduce 0.5 × 106 G1E-ER4 cells. Transduced GFP+ cells were selected using a fluorescence-activated cell sorter (FACS; Becton Dickinson) and expanded in culture. Independently transduced and selected populations of cells were used for independent experiments.
Immunoblots
Cells lysates were prepared and fractionated, and immunoblots were prepared as described.20 Loading was normalized with respect to β-actin. Cul4A protein was detected with 1:3000 diluted anti-Cul4A antiserum20 followed by 1:10 000 diluted anti–rabbit immunoglobulin G (IgG) monoclonal antibody (A6154; Sigma). Antiactin monoclonal antibody (H5441; Sigma) diluted 1:4000 was followed by anti–mouse IgG antibody (NA931; Amersham Biosciences, Piscataway, NJ) diluted 1:10 000, the HA epitope tag was detected with horseradish peroxidase (HRP)–conjugated anti-HA antibodies (Roche Biochemicals, Indianapolis, IN) diluted 1:2000, and anti–c-Myc or anti-p27 polyclonal antibody (2 μg/mL; 06-303 or 06-445, respectively; Upstate Biotechnology, Lake Placid, NY) was followed by goat anti–rabbit IgG antibody (Upstate Biotechnology) diluted 1:5000. β-Globin was detected with anti–mouse hemoglobin polyclonal antibody (no. 55447; MP Biomedicals, Aurora, OH) diluted 1:1000, followed by goat anti–rabbit IgG. Rat anti–GATA-1 monoclonal antibody diluted 1:200 was followed by goat anti–rat IgG-HRP diluted 1:10 000 (N1, sc-266 and sc-2032, respectively, Santa Cruz Biotechnology, Santa Cruz, CA).
Proliferation
The proliferation of Cul4A-HA cells and controls was assayed by tritiated thymidine incorporation. Empty vector control or Cul4A-HA cells were plated at 2.5 × 104 cells/well in 96-well plates for 48 hours at 37°C. To each well, 1.0 μCi (0.037 MBq) [3H]thymidine was added, cells were incubated for 8 hours at 37°C and harvested (96-well harvester; Brandel, Gaithersburg, MD), and [3H]thymidine incorporation was measured by scintillation counting.
Immunoprecipitations
Cells were cultured with 10–7 M β-estradiol for 24 hours, collected, washed with PBS, and lysed, and immunoprecipitations were performed as described.14 Rabbit anti-Cul4A antiserum (100-401-A04; Rockland, Gilbertsville, PA), rabbit polyclonal anti-p27 antibody (Santa Cruz Biotechnology, sc-776 or Upstate Biotechnology, 06-445), normal rabbit IgG (Santa Cruz Biotechnology, sc-2027), or rabbit polyclonal anti-HA antibody (Santa Cruz Biotechnology, sc-805) were used. Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto PVDF membranes (Invitrogen), and analyzed by immunoblot as described.20
Results
Cul4A haploinsufficiency does not alter the number of erythroid precursor cells
We previously showed that Cul4A haploinsufficiency causes a 5-fold reduction in the number of primitive erythroid progenitors (erythroid blast-forming units [BFU-Es]).20 We next quantified the number of more mature erythroid colony-forming unit (CFU-E) progenitors in Cul4A+/– mice. Culture of adult Cul4A+/– and wild-type littermate bone marrow hematopoietic progenitors in methylcellulose with recombinant hematopoietic growth factors for 3 days revealed a 2.4-fold reduction in the number of Cul4A+/– CFU-Es (Table 1).
Table 1.
Erythroid progenitors are reduced in Cul4A+/- mice but not total bone marrow and erythroid precursors
|
No. of cells per femur ± SEM
|
||
|---|---|---|
| Wild-type | Cul4A+/- | |
| Total cells/femur, × 106 | 32.25 ± 2.23 | 28.67 ± 2.50 |
| CFU-Es/femur, × 104 | 1.85 ± 0.41 | 0.78 ± 0.15* |
| Erythroid precursors/femur, × 106 | ||
| Proerythroblasts | 0.67 ± 0.11 | 0.33 ± 0.07 |
| Basophilic erythroblasts | 8.11 ± 0.87 | 7.32 ± 0.47 |
| Late basophilic erythroblasts and polychromatophilic erythroblasts | 1.93 ± 0.26 | 1.57 ± 0.10 |
| Orthochromatophilic erythroblasts | 3.59 ± 1.13 | 2.66 ± 0.29 |
Cells were isolated from the bone marrow of wild-type and Cul4A+/- mice. Total bone marrow cells were determined with a Beckman Coulter particle counter. CFU-E colonies were counted 3 days after initiation of the progenitor cell methylcellulose colony assay. Bone marrow cells were incubated with PE-conjugated anti-Ter119 and FITC-conjugated anti-CD71 antibodies. Using flow cytometry, 4 cell populations of erythroid precursors were defined with specific staining characteristics: proerythroblasts as Ter119med CD71high; basophilic erythroblasts as Ter119high CD71high; late basophilic erythroblasts and polychromatophilic erythroblasts as Ter119highCD71med; and orthochromatophilic erythroblasts as Ter119highCD71low. The data presented are from one representative experiment with 3 wild-type and 3 Cul4A+/- mice of 4 independent experiments. The data are the mean ± SEM.
P < .05.
We next quantified the numbers of more mature erythroid cells using the Ter119 and CD71 cell surface markers as previously described.22 The erythroid-specific Ter119 antigen is highly expressed by all erythroid precursors subsequent to proerythroblasts. Conversely, the transferrin receptor (CD71), although not erythroid-specific, is expressed at high levels by proerythroblasts and early basophilic erythroblasts, and its levels decline with erythroid maturation. Simultaneous immunostaining of these 2 antigens and flow cytometry allow the identification of erythroid precursors at 4 stages of maturation: proerythroblasts, basophilic erythroblasts, late basophilic and polychromatophilic erythroblasts, and orthochromatophilic erythroblasts.22 For bone marrow cells isolated from adult Cul4A+/– mice, the frequency of cells in each of these populations was the same as wild-type controls (either with respect to total erythroid precursors or per femur; Table 1). They were also the same for cells isolated from Cul4A+/– or wild-type spleens (data not shown). Therefore, whereas both primitive and more differentiated erythroid progenitors are reduced in Cul4A-haploinsufficient mice, the number of erythroid precursor cells at all stages of maturation examined is normal.
Recovery from 5-FU–induced stress is delayed in Cul4A heterozygous mice
While 10% to 20% of primitive erythroid progenitors (BFU-Es) are in cycle and most (60%-80%) of more mature progenitors (CFU-Es) are proliferating, early erythroid precursor cells are also mitotically active, with DNA synthesis continuing in proerythroblasts, declining in basophilic erythroblasts, and ceasing at the polychromatophilic erythroblast stage.27 Perhaps Cul4A is involved in regulating proliferation and its reduction in Cul4A-haploinsufficient mice affects erythroid progenitors, because these mitotically active cells are more susceptible to a deficiency in proliferation. If so, the stress of killing mitotically active cells with 5-FU might reveal a deficiency in the ability of Cul4A+/– erythroid cells to recover and reestablish normal homeostasis. Cul4A+/– mice and wild-type controls were treated with 150 mg/kg 5-FU, and hematocrits were monitored during the following 20 days to assess the recovery of mature erythrocytes (Figure 1A). Hematocrits for untreated mice of both genotypes are the same, and they declined with similar rates for 8 days after 5-FU. However, at 12 days, wild-type hematocrits increased slightly from their minimum to 28%, whereas those for Cul4A+/– mice lagged behind at 24% (P = .03). At 14 days, hematocrits for Cul4A+/– mice recovered to 28%, but continued to lag behind controls (34%), and this trend continued throughout the remainder of the time course (P ≤ .04).
Figure 1.
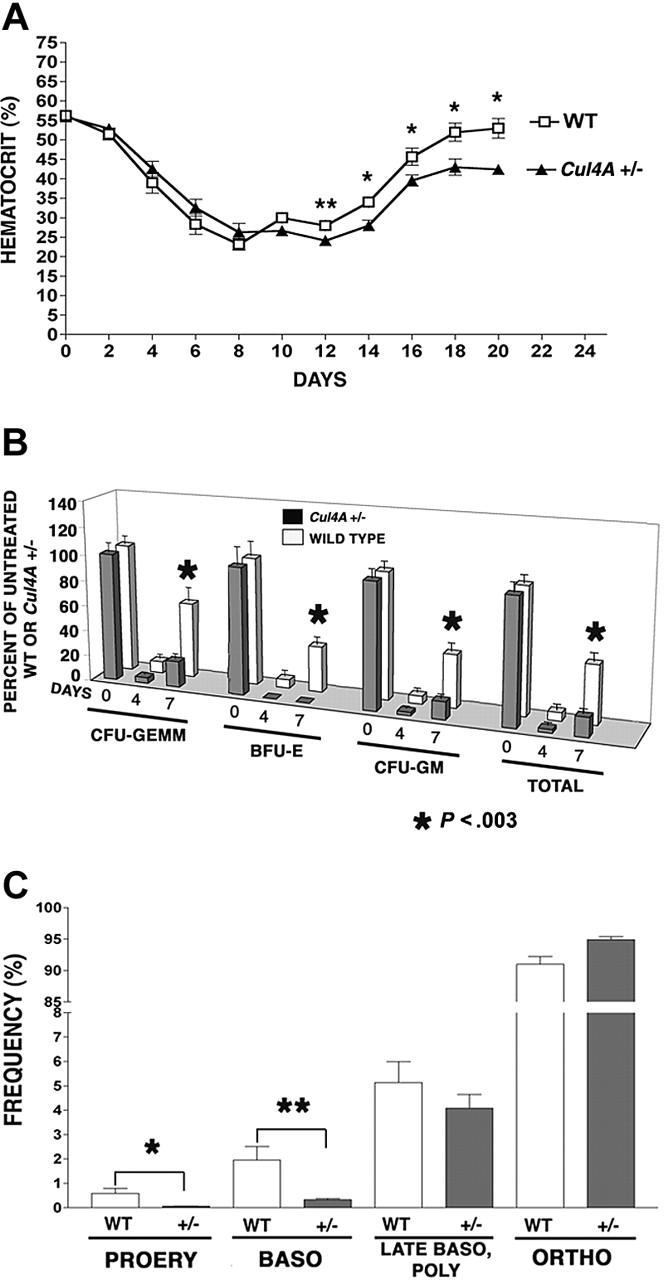
The recovery from 5-FU–induced stress is delayed in Cul4A heterozygous mice. (A) Thirteen Cul4A+/– and 12 wild-type littermates were treated with 150 mg/kg 5-FU and hematocrits were measured 0 to 20 days later. For wild-type (□) and Cul4A+/– (▴) mice, the mean percentages ± SEM are plotted for the results from 3 independent experiments. At day 12, **P = .03, and for days 14 to 20, *P ≤ .04. For days 0 to 16, n = 6-12 mice, and for days 18-20, n = 3-5 mice. (B) Cul4A+/– and wild-type littermates were treated with 5-FU as described in panel A, bone marrow was isolated from femurs 0, 4, or 7 days later and cultured for 7 days. BFU-Es, CFU-GEMMs, and granulocyte-macrophage colony-forming units (CFU-GMs), as well as total colonies, were counted, and the number of progenitors per femur was calculated and normalized with respect to untreated control mice of the same genotype (100%). The means (± SEM) for Cul4A+/– and wild-type animals from 3 independent experiments are shown, where for the 0, 4, and 7 day time points, n = 13, 5, and 9 (respectively) each for wild-type (□) and Cul4A+/– ( ) animals. For day 7, *P < .003. (C) Six Cul4A+/– and 6 wild-type littermates were treated with 5-FU as described, and 7 days later bone marrow was isolated from femurs, stained with anti-Ter119 and anti-CD71 antibodies, and analyzed by flow cytometry to quantify the frequencies of proerythroblasts (PROERY), basophilic erythroblasts (BASO), late basophilic and polychromatophilic erythroblasts (LATE BASO, POLY), and orthochromatophilic erythroblasts (ORTHO) with respect to total erythroid precursors (see “Materials and methods”). The means (percent ± SEM) are graphed for the results from 2 independent experiments. For proerythroblasts (PROERY), *P = .03 and for basophilic erythroblasts (BASO), **P = .01.
) animals. For day 7, *P < .003. (C) Six Cul4A+/– and 6 wild-type littermates were treated with 5-FU as described, and 7 days later bone marrow was isolated from femurs, stained with anti-Ter119 and anti-CD71 antibodies, and analyzed by flow cytometry to quantify the frequencies of proerythroblasts (PROERY), basophilic erythroblasts (BASO), late basophilic and polychromatophilic erythroblasts (LATE BASO, POLY), and orthochromatophilic erythroblasts (ORTHO) with respect to total erythroid precursors (see “Materials and methods”). The means (percent ± SEM) are graphed for the results from 2 independent experiments. For proerythroblasts (PROERY), *P = .03 and for basophilic erythroblasts (BASO), **P = .01.
Bone marrow erythroid (as well as myeloid) progenitors were also assayed 0 to 7 days after 5-FU treatment and their numbers were normalized with respect to those of untreated mice of the same genotype. Four days after 5-FU, the numbers of wild-type erythroid, multipotential myeloid, and granulocyte/monocyte progenitors per femur were each only 6% to 9% of untreated controls, whereas erythroid progenitors in Cul4A+/– mice were not detected, and myeloid progenitors were only 2.5% to 4.4% of that for untreated Cul4A+/– animals (Figure 1B). The largest difference was observed at 7 days, where wild-type animals recovered 35% to 59% of unstressed levels, but Cul4A+/– animals reached only 14% to 20% of the numbers for unstressed Cul4A+/– controls, except for erythroid progenitors which remained undetectable (P < .003).
Seven days after 5-FU, when the recovery of erythroid progenitors in Cul4A+/– mice showed the greatest lag, the frequencies of the more mature erythroid precursors were also examined. There was no significant difference in either the total cellularity or total number of erythroid precursors per femur between Cul4A+/– and wild-type controls (data not shown), so the frequency of each erythroid precursor population was determined with respect to total erythroid precursors as previously described.22 The frequency of wild-type proerythroblasts was 0.59% ± 0.21%, whereas the frequency of Cul4A+/– proerythroblasts was 11-fold lower at 0.055% ± 0.01% (mean ± SEM, P = .03; Figure 1C). The frequency of basophilic erythroblasts was 1.96% ± 0.55% and that for Cul4A+/– was 6-fold less at only 0.33% ± 0.05% (P = .01). There was no significant difference between the frequencies of Cul4A+/– and wild-type late basophilic and polychromatophilic erythroblasts, and the frequency of orthochromatophilic erythroblasts was only slightly greater in Cul4A+/– bone marrow, with 91.00% ± 1.21% for wild-type and 94.90% ± 0.48% for Cul4A+/– (P = .01).
Recovery from an erythroid-specific stress is delayed in Cul4A heterozygous mice
Because multipotent progenitors (granulocyte, erythrocyte, megakaryocyte, macrophage colony-forming units [CFU-GEMMs]) are reduced in Cul4A+/– mice,20 it was possible that the 5-FU treatment did not directly affect the erythroid lineage and that the delayed recovery of erythroid progenitors and precursors might result from a sluggish recovery of CFU-GEMMs. Therefore, the recovery of Cul4A-haploinsufficient mice from an erythroid-specific stress was examined. Cul4A+/– and wild-type adult mice were subjected to a brief, chemically induced hemolytic anemia by subcutaneous injection with phenylhydrazine on days 0, 1, and 3 (Figure 2A).22 Hematocrits were monitored on days 0 to 7. Hematocrits in Cul4A+/– and wild-type mice declined at similar rates until day 3. By day 5, hematocrits for both had increased slightly, but whereas the hematocrits for wild-type mice increased to 52.7% ± 1.2% by day 7, those for Cul4A+– mice lagged behind at 47.2% ± 1.4% (P = .01). These results demonstrate that Cul4A haploinsufficiency directly affects the erythroid lineage.
Figure 2.

Recovery from an erythroid-specific stress is delayed in Cul4A heterozygous mice. (A) Twelve Cul4A+/– and 12 wild-type littermate controls were treated with 50 mg/kg phenylhydrazine on days 0, 1, and 3 (indicated by arrows), and hematocrits were measured at days 0 to 7. The mean (percent ± SEM) for wild-type (□) and Cul4A+/– (▴) mice are graphed for 2 independent experiments. At day 7, *P = .01. (B) Six Cul4A+/– and 6 wild-type mice were treated with phenylhydrazine as described in panel A, and bone marrow was isolated at day 5. Total cellularity per femur was determined and the number of erythroid precursors per femur for each mouse was determined (see “Materials and methods”). The means (cells per femur ± SEM) are graphed for the results from 2 independent experiments. *P < .001. (C) For the 6 Cul4A+/– and 6 wild-type mice described, the number of cells in each of the 4 erythroid precursor populations (described in “Results” and for Figure 1C) was determined, and these results from 2 independent experiments are graphed (mean ± SEM). For proerythroblasts (PROERY), *P < .001. (D) Spleen colony-forming unit (CFU-S) assays were performed by injecting 3 × 104 low-density mononuclear bone marrow cells isolated from 4 wild-type or 4 Cul4A+/– mice into the tail vein of lethally irradiated adult, female, C57BL/6 recipients (3-4 recipients per wild-type or Cul4A+/– donor). After 8 days, spleens were removed and fixed in Telly fixative (20 parts 70% ethanol, 1 part glacial acetic acid, 1 part formalin), and the numbers of macroscopic colonies were counted. Means ± SEM for the combined results from 2 independent experiments are graphed. When the results for these 4 Cul4A+/– and 4 wild-type donors were compared, *P = .005.
Because there was a significant delay in the recovery of mature erythrocytes in the peripheral blood of Cul4A+/– mice at 7 days, we examined whether there were also fewer bone marrow erythroid precursors at an earlier time point. At 5 days, total cellularity and total erythroid precursors per femur were both significantly lower in Cul4A+/– bone marrow compared to wild-type controls (25% fewer total cells and 26% fewer erythroid precursors; Figure 2B), so the number of cells in each erythroid precursor population per femur was analyzed. There were 4-fold fewer proerythroblasts in Cul4A+/– mice compared to wild-type controls (1.8 × 106 compared to 7.2 × 106 cells, respectively, P < .001) but no significant differences between the more mature erythroid precursors (Figure 2C).
The reduced frequencies of erythroid and myeloid progenitors and precursors could result from an intrinsic property of Cul4A+/– hematopoietic cells or they might require haploinsufficient, nonhematopoietic cells. Therefore, Cul4A+/– or wild-type bone marrow cells were transplanted into wild-type recipients, and the number of spleen colony-forming units were enumerated 8 days later (CFU-S 8). These Cul4A+/– primitive progenitors yielded 3.4-fold fewer colonies than wild-type, consistent with the Cul4A+/– progenitor/precursor defect originating in hematopoietic cells (Figure 2D).
Therefore, reduced Cul4A expression results in fewer erythroid progenitors and a delay in the recovery of these progenitors, proerythroblasts, and basophilic erythroblasts following stress directed at proliferating cells. Proerythroblast recovery is also delayed after an erythroid-specific stress. Overall, multiple in vivo and in vitro assays show consistent and significant, albeit modest for some, reductions in the ability of the erythroid lineage in Cul4A-haploinsufficient mice to recover normal levels of erythroid progenitors and early precursors following stress. These findings suggest that Cul4A plays a positive role in regulating the proliferation of these erythroid populations. To test further this possibility, we examined the effect of overexpressing Cul4A on the proliferation and differentiation of proerythroblasts.
Cul4A overexpression promotes proliferation in proerythroblasts
G1E-ER4 cells are a growth factor-dependent, erythroid cell line-derived from GATA-1 null mice, where GATA-1 function was restored with estrogen-activated GATA-1.25 They proliferate continuously as immature proerythroblasts in culture with supplemented erythropoietin (Epo) and SCF, and 10–7 M β-estradiol activates the GATA-1 fusion, which induces terminal differentiation. Cul4A was stably overexpressed in G1E-ER4 cells (see “Materials and methods”). To distinguish between endogenous and exogenous Cul4A, Cul4A was overexpressed as a Cul4A-HA fusion.20 Using anti-Cul4A antisera to measure total Cul4A (both endogenous and HA tagged), Cul4A-HA cells were found to express 2-fold more Cul4A than G1E-ER4 cells transduced with empty vector (Figure 3A middle panel). The multiple immunoreactive bands correspond to unmodified Cul4A, Cul4A-HA, Cul4A modified by Nedd8 (a previously reported modification), and Cul4A-HA modified by Nedd8.20,28,29
Figure 3.

Cul4A overexpression promotes proliferation in proerythroblasts. (A) Protein lysates were prepared from empty vector control or Cul4A-HA cells, and 10 μg of each was analyzed by immunoblot probed with anti-HA monoclonal antibody (top), anti-Cul4A antiserum (middle), or antiactin monoclonal antibody (bottom). The various forms of Cul4A are indicated with arrows on the right. An immunoblot representative of 3 independent experiments is shown. (B) Cul4A-HA and empty vector control cells were plated in triplicate (2.5 × 104 cells/well) and cultured in 96-well plates in the presence of 2 U/mL Epo and 50 ng/mL SCF for 48 hours, incubated with tritiated thymidine for 8 hours, and incorporated tritiated thymidine was measured. The mean incorporated tritiated thymidine of triplicate samples was determined for each experiment, and the means (cpm ± SEM) of these values from 3 independent experiments are graphed. *P = .04. (C) Cul4A-HA and empty vector control cells were plated at 2.5 × 105 cells/mL, grown for 24 hours, and the proportion of cells in each phase of the cell cycle was determined by propidium iodide staining and flow cytometric analysis. The means (± SEM) of 3 independent experiments are graphed. For cells in G1 and S phase, *P < .05.
To assess the effect of overexpressing Cul4A on proliferation, tritiated thymidine incorporation was measured in Cul4A-HA cells and found to be 34% greater than that for empty vector controls (P = .04; Figure 3B). Because increased proliferation might result from a larger proportion of cells in S phase, the cell cycle distribution of Cul4A-HA cells was analyzed. Cul4A-HA cells had 8% more cells in S phase than controls (65.12% ± 0.86% compared to 60.27% ± 1.96%, respectively, P = .02; Figure 3C). Correspondingly, there were fewer Cul4A-HA cells in G1 phase.25 (25.13% ± 0.32% compared to 28.2% ± 1.5% for controls, P = .04). The difference between the proportion of Cul4A-HA cells and empty vector control cells in G2/M was not significant. Similar results were obtained when Cul4A was overexpressed 2.2-fold in G1E-ER2 cells, another independently derived G1E-ER proerythroblast cell line,24 with 83% more tritiated thymidine incorporation, 20% more cells in S phase, and nearly 24% fewer cells in G1 phase (data not shown). Therefore, Cul4A overexpression consistently causes an increase in proerythroblast proliferation.
Cul4A regulates erythroid terminal differentiation
We next investigated the role of Cul4A during erythroid terminal differentiation. G1E-ER4 proerythroblasts were induced to differentiate, and Cul4A protein was measured by immunoblot. Cul4A declined 3.6-fold within 48 hours of induction (Figure 4A). Cul4A mRNA declined at a similar rate (data not shown). To demonstrate that these cells differentiated into erythrocytes, β-globin expression was also measured and confirmed to have increased.30 In addition, the proportion of cells in S phase decreased from 60% to 6%, whereas that in G0/G1 phase increased from 28% to 81%, in agreement with previous findings.24,25
Figure 4.
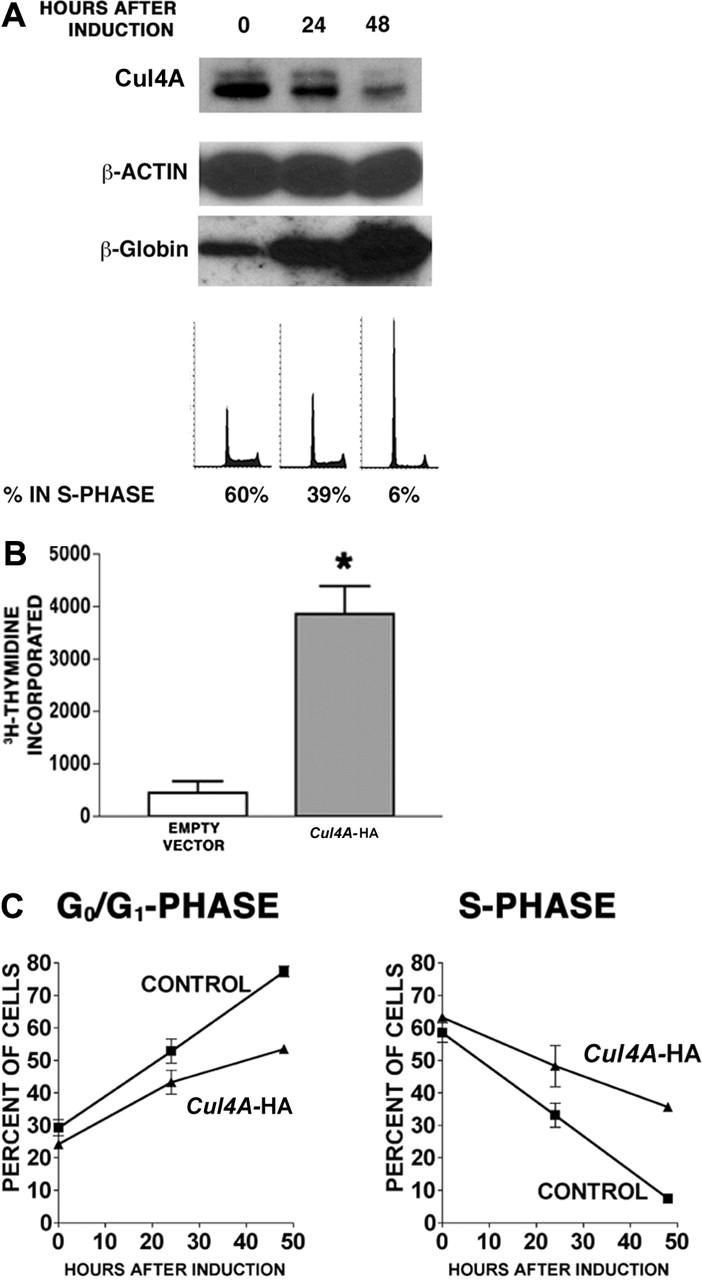
Enforced expression of Cul4A promotes proliferation in proerythroblasts induced to differentiate. G1E-ER4 cells were induced to differentiate into erythrocytes, and Cul4A and β-globin protein levels were measured by immunoblot of 10 μg total lysate. The amounts of immunoreactive protein were quantified and normalized with respect to β-actin. DNA content was determined by propidium iodide staining and flow cytometry, and the relative number of cells in S phase was determined. Representative results from 3 independent experiments are shown. (B) Cul4A-HA and empty vector control cells were plated at 2.5 × 104 cells/well in triplicate in 96-well plates, induced to differentiate, and cultured for 48 hours. Then tritiated thymidine incorporation during 8 hours was quantified. The means (cpm ± SEM) of 2 independent experiments are graphed. *P = .01. (C) Cul4A-HA (▴) and empty vector control cells (▪) were induced to differentiate, and aliquots of cells at 0, 24, and 48 hours after induction were stained with propidium iodide and analyzed by flow cytometry to determine the proportion of cells in each phase of the cell cycle. The mean (percent ± SEM) in either G0/G1 or S phase is graphed with respect to hours after induction for 3 independent experiments.
If the down-regulation of Cul4A expression is required for cell cycle exit and erythrocyte maturation, its enforced expression would interfere with these functions. In Cul4A-HA cells, the exogenous Cul4A-HA fusion is expressed from a constitutive promoter, so to verify its enforced expression in Cul4A-HA cells during erythroid differentiation, protein lysates were prepared from empty vector control and Cul4A-HA cells 0, 24, and 48 hours after induction to differentiate, and total Cul4A protein (native and Cul4A-HA) was measured by immunoblot. Whereas native Cul4A declined in the empty vector control cells, total Cul4A remained constant in Cul4A-HA cells (Figure 5).
Figure 5.

Cul4A-enforced expression attenuates differentiation into erythrocytes and reduces the accumulation of p27 protein. Cul4A-HA and empty vector control cells were induced to differentiate and aliquots of cells were taken 0, 24, and 48 hours after induction. Protein lysates were prepared, and 10 to 20 μg total protein for each sample was analyzed by immunoblot probed with antibodies that recognize HA, Cul4A, β-globin, c-Myc, GATA-1, p27, or actin. Actin was used as a loading control. Representative results of 3 independent experiments are shown.
Cul4A-HA and empty vector control cells were then induced to differentiate for 48 hours, and tritiated thymidine incorporation was measured. Whereas control cells incorporated only 460 ± 220 cpm, Cul4A-HA cells incorporated 8.4-fold more, with 3860 ± 540 cpm (Figure 4B). Next, cell cycle distribution was measured 0 to 48 hours after induction. As expected, the proportion of empty vector control cells in S phase declined 8-fold from 58% to 7%, but that for Cul4A-HA cells only dropped 1.8-fold from 63% to 36% (Figure 4C). Correspondingly, the proportion of control cells in G0/G1 increased from 29% to 77%, whereas that for Cul4A-HA cells increased from 24% to only 54%.
We next examined the effect of enforced Cul4A expression on erythrocyte maturation, as indicated by β-globin induction. Cul4A-HA and empty vector control cells were induced to differentiate, protein lysates were prepared at 0, 24, and 48 hours, and the amounts of β-globin and c-Myc were quantified by immunoblot. Whereas β-globin increased dramatically in controls, its induction was severely reduced in Cul4A-HA cells (Figure 5). Also, c-Myc expression normally declines during erythrocyte terminal differentiation, but Cul4A–enforced expression greatly attenuated this decrease.
Because Cul4A is an E3 ligase subunit, our findings suggest that a Cul4A-containing ubiquitin ligase targets for degradation one or more regulators that promote cell cycle exit and/or erythrocyte maturation. GATA-1 triggers both of these functions.25 Also, during the terminal differentiation of erythroid cell lines, p27 protein increases coincident with G1 arrest.31,32 In addition, Rylski and coworkers showed that in G1E-ER4 cells induced to differentiate, GATA-1 activation results in cell cycle arrest and p27 accumulation.25 Therefore, we examined the effect of Cul4A-enforced expression on the up-regulation of GATA-1 and p27 during terminal erythroid differentiation.
After induction to differentiate, GATA-1 expression increased in both control and Cul4A-HA cells, indicating that Cul4A does not target GATA-1 for degradation. Although p27 levels increased during erythrocyte differentiation, enforced Cul4A expression severely reduced its accumulation, suggesting that Cul4A targets p27 for degradation and that during terminal erythroid differentiation, down-regulation of Cul4A allows the accumulation of p27, which signals cell cycle exit.
Cul4A promotes the proteasome-dependent degradation of p27
As part of a ubiquitin ligase, Cul4A targets proteins for degradation by the proteasome.9-16 To show that p27 is regulated by the proteasome in proerythroblasts, G1E-ER4 cells were treated for 4 hours with 5 or 10 μM of the 26S proteasome inhibitor, MG132, and p27 protein was measured by immunoblot. Inhibiting proteasome function caused the accumulation of p27 protein, indicating that proteasome function is required to establish the steady-state level of p27 (Figure 6A).
Figure 6.

Cul4A promotes the proteasome-dependent degradation of p27. (A) Proliferating G1E-ER4 cells were treated with proteasome inhibitor (5 or 10 μM MG132) for 4 hours. Cells were harvested, protein lysates prepared, and 10 μg total protein for each sample was analyzed by immunoblot probed with antibodies that recognize p27 or actin. Actin was used as a loading control. Representative results of 2 independent experiments are shown. (B) Cul4A-HA and empty vector control cells were induced to differentiate for 24 hours, 3 × 106 cells/100-mm dish were treated with 10 μg/mL cycloheximide for the indicated times, cells were harvested, lysates were prepared, and 10 to 30 μg total lysate for each sample was analyzed by immunoblot and probed with anti-p27 or anti–β-actin antibodies. Actin was used as a loading control. For Cul4A-HA (▴) and empty vector control cells (▪), the amount of p27 at each time point was normalized to the amount of p27 at 0 hours (100%). Because significantly less p27 is present in Cul4A-HA cells compared to the control, 2 different exposures of the same anti-p27 immunoblot are shown. Representative results from 2 independent experiments are shown. (C) For lanes 1-4, Cul4A-HA cells were induced to differentiate for 24 hours, cells were harvested, protein lysate was prepared, and for each sample, 1 mg total protein lysate was precleared with protein A and protein G Sepharose beads, followed by immunoprecipitation with anti-p27, anti-Cul4A, or anti-HA antisera (all from rabbit) and incubation with protein A and protein G Sepharose beads. Immunoprecipitated proteins were eluted with sample buffer and analyzed by immunoblot probed with antibodies that recognize HA, Cul4A, or p27. Immunoprecipitation with protein A and protein G Sepharose beads alone was used as a negative control. For lanes 5-9, G1E-ER4 cells were induced to differentiate for 24 hours, protein lysate was prepared, immunoprecipitations with anti-HA, anti-p27, or anti-Cul4A antisera were performed as described for lanes 1-4. Immunoprecipitated proteins were eluted with sample buffer and analyzed by immunoblot probed with antibodies that recognize Cul4A or p27. Immunoprecipitations with either normal rabbit IgG, protein A and protein G Sepharose beads alone, or rabbit anti-HA antiserum (antiserum against an unrelated protein in these cells) were used as negative controls, all of which failed to immunoprecipitate either p27 or Cul4A, indicating that the anti-p27 and anti-Cul4A antibodies interact specifically with their respective antigens. Representative results from 3 independent experiments are shown.
If p27 is a Cul4A ubiquitin ligase substrate, then p27 should be destabilized when Cul4A is overexpressed. Because p27 protein levels are relatively low in proliferating proerythroblasts, and because we observe the largest effect of Cul4A overexpression after induction to differentiate, we analyzed p27 stability 24 hours after induction to differentiate. Cycloheximide (10 μg/mL) was added to stop protein synthesis, and p27 protein levels were quantified by immunoblot. When Cul4A was overexpressed, the decay rate of p27 protein increased 6.7-fold, with a half-time of only 15 minutes for Cul4A-overexpressing cells and 103 minutes for controls (Figure 6B).
Cul4A might act indirectly to promote p27 degradation, so we determined whether Cul4A and p27 physically interact. Again, because p27 protein levels are low in proliferating G1E-ER4 proerythroblasts, Cul4A-HA cells were induced to differentiate for 24 hours and then either anti-p27, anti-Cul4A, or anti-HA antibodies were used to immunoprecipitate p27, both Cul4A and Cul4A-HA, or Cul4A-HA, respectively, and their interactions were tested by immunoblot (Figure 6C lanes 1-4). Immunoprecipitated Cul4A-HA copurified with p27, and the reciprocal occurred as well (lanes 2 and 4). Therefore, overexpressed Cul4A (as epitope-tagged Cul4A–HA) physically interacts with p27. Based on known cullin-containing ubiquitin ligase structures, it is likely a substrate-recognition subunit bridges their interaction.8 This result also indicates that the overexpressed Cul4A-HA acts as part of a ubiquitin ligase to target p27 degradation, increase proliferation, and interfere with terminal erythroid differentiation and cell cycle exit.
Although Cul4A is overexpressed only 2-fold, it was possible that the excess Cul4A results in an interaction with p27 that does not normally occur. Therefore, anti-Cul4A and anti-p27 antibodies were each used to immunoprecipitate their corresponding proteins from G1E-ER4 lysates, where Cul4A is not overexpressed, and their interactions were tested by immunoblot (Figure 6C lanes 5-9). The interaction between Cul4A and p27 persists (Figure 6C lanes 8 and 9), demonstrating that this interaction is not an artifact of Cul4A overexpression and that it occurs under native conditions.
To assess whether Cul4A regulates p27 in vivo, p27 protein was measured in proerythroblasts and basophilic erythroblasts from Cul4A+/– and wild-type mice. In unstressed mice, the low frequency of these cells made it difficult to detect Cul4A and p27, so the frequency of these cells was increased by treatment with phenylhydrazine. Under these conditions, p27 levels were 2.7-fold higher in Cul4A+/– cells, consistent with Cul4A regulating p27 in vivo in these early erythroid precursors (Figure 7). Cul4A protein expression in the Cul4A+/– samples was approximately one-half that in wild-type controls, in agreement with our previous findings.21
Figure 7.

After phenylhydrazine-induced stress, the expression of p27 protein in early erythroid precursors from Cul4A+/– mice is elevated compared to wild-type controls. Wild-type and Cul4A+/– mice were treated with phenylhydrazine as described for Figure 2. On day 4, bone marrow cells were isolated separately from each mouse, and proerythroblasts and basophilic erythroblasts were identified and isolated with a fluorescence-activated cell sorter, as described in Figure 1. Cul4A, p27, and β-actin protein levels were determined by immunoblot. Actin was used as a loading control. A representative immunoblot from 2 independent experiments and their combined results are shown (mean ± SEM for 5 wild-type and 6 Cul4A+/– mice, *P = .03).
Discussion
We investigated the role of Cul4A, which encodes a ubiquitin ligase core subunit, in regulating erythropoiesis, erythrocyte maturation, and cell cycle exit. We hypothesized that Cul4A positively regulates proliferation, so in Cul4A–haploinsufficient mice, proliferating populations of erythroid cells would be expected to exhibit a deficiency in their ability to increase proliferation. The delayed recovery of Cul4A+/– progenitors after 5-FU–induced stress supports this model. The accompanying reduction in proerythroblasts and basophilic erythroblasts might result because Cul4A haploinsufficiency also reduces their capacity to increase proliferation. Alternatively, their reduced numbers might indirectly result from the reduction in progenitors. However, the findings that Cul4A overexpression in G1E-ER4 proerythroblasts increases their proliferation and interferes with maturation and cell cycle exit are consistent with a direct effect and further support the model that Cul4A is a positive cell cycle regulator.
Cul4A is a core subunit of a ubiquitin ligase, and our findings that Cul4A interacts with and destabilizes p27 show that a Cul4A ubiquitin ligase targets p27 for degradation. We propose a model where this Cul4A ubiquitin ligase targets p27 for degradation and promotes proliferation in erythroid progenitors and proerythroblasts. Supporting this model, p27 protein is elevated nearly 3-fold in early erythroid precursors isolated from the bone marrow of Cul4A–haploinsufficient mice. Furthermore, Cul4A expression and consequently p27 degradation are down-regulated during erythroid terminal differentiation, and p27 accumulates to signal cell cycle arrest. Fewer erythroid progenitors in Cul4A-haploinsufficient mice could result from lowered Cul4A-mediated ubiquitination, elevated p27 levels, and fewer proliferating progenitors. Overexpression of Cul4A would lower p27 levels and promote proliferation. We observe modest increases in the proliferation of G1E-ER4 cells overexpressing Cul4A and in the proportion of these cells in S phase (34% and 8%, respectively, Figure 3). Because the expression of p27 in these cells is already relatively low compared to after the induction of differentiation (Figure 5), modest Cul4A overexpression might have a fairly small impact on p27 protein level and proerythroblast proliferation. However, the persistence of Cul4A expression during terminal differentiation impedes p27 up-regulation and interferes with cell cycle exit and erythrocyte maturation (Figures 4, 5).
In vitro studies demonstrate that p27 levels increase as erythroid cells terminally differentiate.25,31-34 Coupled with its established function as a cyclin-dependent kinase inhibitor,35,36 the model that p27 is involved in directing cell cycle exit during erythroid terminal differentiation is well supported. Our findings indicate that a Cul4A ubiquitin ligase targets p27 for degradation in erythroid cells. However, the SCFSkp2 ubiquitin ligase (containing the Cul1 cullin) was also shown to target p27 for degradation in nonhematopoietic cells,37-39 and 2 reports show indirectly that SCFSkp2 regulates p27 in erythroid cells. Bouscary et al found that blocking phosphoinositide 3-kinase activity in erythroid progenitors with LY294002 prevented Epo-stimulated proliferation and increased p27.40 A protein associated with SCFSkp2 increased in response to Epo, and LY294002 prevented this increase. Myeloid leukemia factor 1 (MLF1) is part of a translocation associated with acute myeloid leukemia.41 Winteringham et al42 induced murine erythroleukemic cells to differentiate with Epo and found that overexpressing MLF1 prevented cell cycle exit and p27 induction. The expression of SCFSkp2 subunits (Cul1, Skp1, and Skp2) were down-regulated during differentiation, and MLF1 overexpression prevented this decline. Therefore, p27 appears to be regulated by both Cul4A and Cul1 ubiquitin ligases.
Given the complex role p27 plays in regulating cell cycle progression, it is not surprising that its regulation involves multiple pathways, including 2 different ubiquitin ligases. Both cyclin E and c-Jun are also regulated by multiple, distinct ubiquitin ligases. Phosphorylated cyclin E bound to Cdk2 is targeted for degradation by the Cul1-containing SCFCdc4 ubiquitin ligase,43-45 whereas unphosphorylated cyclin E is targeted by a different Cul1 ligase, SCFSkp2,46 and a Cul3 ubiquitin ligase.47 Likewise, unphosphorylated c-Jun can be ubiquitinated by a Cul4A-based ligase, DCXDET1-COP1,14 and phosphorylated c-Jun is targeted by SCFCdc4.48
The transcription of p27 is repressed by c-Myc in lymphoid cells,49 and c-Myc expression is not repressed when Cul4A-overexpressing proerythroblasts are induced to differentiate (Figure 5), so it is possible that Cul4A acts indirectly through c-Myc to repress p27 expression. However, our results that Cul4A and p27 coimmunoprecipitate and that p27 protein is destabilized when Cul4A is overexpressed (Figure 6) clearly show that Cul4A acts directly to promote p27 degradation.
In G1E-ER4 cells, enforced c-Myc expression prevents p27 mRNA induction and cell cycle arrest.25 However, erythrocyte maturation proceeds in these c-Myc–overexpressing cells, indicating that c-Myc repression is required for cell cycle exit but not for erythrocyte maturation and that these 2 functions can be separated. Additionally, enforced p27 expression causes the accumulation of cells in G1 and a decline in cells in S phase but not terminal erythrocyte differentiation, indicating that erythrocyte maturation does not necessarily follow from cell cycle exit.32,34 Because enforced Cul4A expression affects the repression of c-Myc, the induction of p27, erythrocyte maturation, and cell cycle exit, it appears that like GATA-1, Cul4A functions upstream of regulators that direct cell cycle exit or erythrocyte maturation or both. However, because GATA-1 levels during differentiation are not affected by enforced Cul4A expression, Cul4A does not appear to regulate GATA-1. A Cul4A ubiquitin ligase might target for degradation a protein that directs erythrocyte maturation, but none of the previously identified Cul4A ligase substrates (Cdt1, DDB2, c-Jun, HoxA9, and p53) are known to be involved in this function. In 32D myeloid cells induced with G-CSF to differentiate into granulocytes, a Cul4A ubiquitin ligase targets for degradation the HOXA9 homeodomain protein, which plays important roles in regulating granulocyte/monocyte differentiation.15 Enforced HOXA9 expression interfered with granulocyte differentiation, which was restored by Cul4A overexpression. A 60% reduction in Cul4A reduced differentiation. These findings indicate Cul4A promotes differentiation, in apparent contrast to our findings for PLB-985 cells induced to differentiate into granulocytes or monocytes with dimethylformamide or phorbol-myristate acetate, respectively.20 These differing findings might result because different pathways were being induced, with Cul4A targeting different regulators of differentiation. However, our findings for the erythroid lineage are consistent with our prior results for myeloid cells: Cul4A promotes proliferation, and its down-regulation is required for cell cycle exit and terminal differentiation.
Ubiquitin ligases regulate cell proliferation, differentiation, and apoptosis, and the ubiquitin pathway is often the target of cancer-related deregulation.50 Bortezomib (PS-341 or Velcade) inhibits proteasome function, and in a large clinical trial, one third of patients with relapsed and refractory multiple myeloma who were treated with bortezomib showed significant clinical benefit.51 A greater understanding of these ligases and their targets will promote the refinement of novel therapies.
Acknowledgments
We thank Merv Yoder and Dave Skalnik for many insightful suggestions and stimulating discussions and Veerendra Munugalavadla for helpful technical discussions and suggestions.
Prepublished online as Blood First Edition Paper, February 7, 2006; DOI 10.1182/blood-2005-08-3349.
Supported by grant DK 66603 from the National Institutes of Health (NIH), an Indiana University School of Medicine Biomedical Research Grant, and the Riley Children's Fund (K.T.C.). An additional grant from the NIH (HL 75816) also provided support (R.K.).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 2001;20: 723-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tedesco D, Lukas J, Reed SI. The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev. 2002;16: 2946-2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tyers M, Jorgensen P. Proteolysis and the cell cycle: with this RING I do thee destroy. Curr Opin Genet Dev. 2000;10: 54-64. [DOI] [PubMed] [Google Scholar]
- 4.Kamizono S, Hanada T, Yasukawa H, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem. 2001;276: 12530-12538. [DOI] [PubMed] [Google Scholar]
- 5.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70: 503-533. [DOI] [PubMed] [Google Scholar]
- 6.Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. cul1–1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;85: 829-839 [DOI] [PubMed] [Google Scholar]
- 7.Dias DC, Dolios G, Wang R, Pan ZQ. CUL7: a DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc Natl Acad Sci U S A. 2002;99: 16601-16606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6: 9-20. [DOI] [PubMed] [Google Scholar]
- 9.Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol. 2004;6: 1003-1009. [DOI] [PubMed] [Google Scholar]
- 10.Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol. 2003;5: 1008-1015. [DOI] [PubMed] [Google Scholar]
- 11.Zhong W, Feng H, Santiago FE, Kipreos ET. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature. 2003;423: 885-889. [DOI] [PubMed] [Google Scholar]
- 12.Nag A, Bondar T, Shiv S, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol Cell Biol. 2001;21: 6738-6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuda N, Azuma K, Saijo M, et al. DDB2, the xeroderma pigmentosum group E gene product, is directly ubiquitylated by Cullin 4A-based ubiquitin ligase complex. DNA Repair (Amst). 2005;4: 537-545. [DOI] [PubMed] [Google Scholar]
- 14.Wertz IE, O'Rourke KM, Zhang Z, et al. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science. 2004;303: 1371-1374. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Morrone G, Zhang J, et al. CUL-4A stimulates ubiquitylation and degradation of the HOXA9 homeodomain protein. EMBO J. 2003; 22: 6057-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nag A, Bagchi S, Raychaudhuri P. Cul4A physically associates with MDM2 and participates in the proteolysis of p53. Cancer Res. 2004;64: 8152-8155. [DOI] [PubMed] [Google Scholar]
- 17.Chen L-C, Manjeshwar S, Lu Y, et al. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer Res. 1998;58: 3677-3683. [PubMed] [Google Scholar]
- 18.Yasui K, Arii S, Zhao C, et al. TFDP1, CUL4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology. 2002;35: 1476-1484. [DOI] [PubMed] [Google Scholar]
- 19.Cho RJ, Huang M, Campbell MJ, et al. Transcriptional regulation and function during the human cell cycle. Nat Genet. 2001;27: 48-54. [DOI] [PubMed] [Google Scholar]
- 20.Li B, Yang FC, Clapp DW, Chun KT. Enforced expression of CUL-4A interferes with granulocytic differentiation and exit from the cell cycle. Blood. 2003;101: 1769-1776. [DOI] [PubMed] [Google Scholar]
- 21.Li B, Ruiz JC, Chun KT. CUL-4A is critical for early embryonic development. Mol Cell Biol. 2002;22: 4997-5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(–/–)5b(–/–) mice due to decreased survival of early erythroblasts. Blood. 2001;98: 3261-3273. [DOI] [PubMed] [Google Scholar]
- 23.Nakahata T, Gross AJ, Ogawa M. A stochastic model of self-renewal and commitment to differentiation of the primitive hematopoietic stem cells in culture. J. Cell Physiol. 1982;113: 455-458. [DOI] [PubMed] [Google Scholar]
- 24.Gregory T, Yu C, Ma A, Orkin SH, Blobel GA, Weiss MJ. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood. 1999;94: 87-96. [PubMed] [Google Scholar]
- 25.Rylski M, Welch JJ, Chen YY, et al. GATA-1-mediated proliferation arrest during erythroid maturation. Mol Cell Biol. 2003;23: 5031-5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams DA, Tao W, Yang F, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96: 1646-1654. [PubMed] [Google Scholar]
- 27.Papayannpoulou T, Abkowitz JL. Biology of erythropoiesis, erythroid differentiation, and maturation. In: Hoffman R, Benz EJJ, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, eds. Hematology: Basic Principles and Practice. 2nd ed. New York, NY: Churchill Livingstone; 1995: 242-254.
- 28.Osaka F, Kawasaki H, Aida N, et al. A new NEDD8-ligating system for cullin-4A. Genes Dev. 1998;12: 2263-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hori T, Osaka F, Chiba T, et al. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene. 1999;18: 6829-6834. [DOI] [PubMed] [Google Scholar]
- 30.Minie M, Clark D, Trainor C, et al. Developmental regulation of globin gene expression. J Cell Sci Suppl. 1992;16: 15-20. [DOI] [PubMed] [Google Scholar]
- 31.Hsieh FF, Barnett LA, Green WF, et al. Cell cycle exit during terminal erythroid differentiation is associated with accumulation of p27(Kip1) and inactivation of cdk2 kinase. Blood. 2000;96: 2746-2754. [PubMed] [Google Scholar]
- 32.Tamir A, Petrocelli T, Stetler K, et al. Stem cell factor inhibits erythroid differentiation by modulating the activity of G1-cyclin-dependent kinase complexes: a role for p27 in erythroid differentiation coupled G1 arrest. Cell Growth Differ. 2000; 11: 269-277. [PubMed] [Google Scholar]
- 33.Panzenbock B, Bartunek P, Mapara MY, Zenke M. Growth and differentiation of human stem cell factor/erythropoietin-dependent erythroid progenitor cells in vitro. Blood. 1998;92: 3658-3668. [PubMed] [Google Scholar]
- 34.Matushansky I, Radparvar F, Skoultchi AI. Manipulating the onset of cell cycle withdrawal in differentiated erythroid cells with cyclin-dependent kinases and inhibitors. Blood. 2000;96: 2755-2764. [PubMed] [Google Scholar]
- 35.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13: 1501-1512. [DOI] [PubMed] [Google Scholar]
- 36.Reed SI. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003;4: 855-864. [DOI] [PubMed] [Google Scholar]
- 37.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999; 1: 193-199. [DOI] [PubMed] [Google Scholar]
- 38.Sutterluty H, Chatelain E, Marti A, et al. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1: 207-214. [DOI] [PubMed] [Google Scholar]
- 39.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9: 661-664. [DOI] [PubMed] [Google Scholar]
- 40.Bouscary D, Pene F, Claessens YE, et al. Critical role for PI 3-kinase in the control of erythropoietin-induced erythroid progenitor proliferation. Blood. 2003;101: 3436-3443. [DOI] [PubMed] [Google Scholar]
- 41.Yoneda-Kato N, Look AT, Kirstein MN, et al. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. 1996;12: 265-275. [PubMed] [Google Scholar]
- 42.Winteringham LN, Kobelke S, Williams JH, Ingley E, Klinken SP. Myeloid leukemia factor 1 inhibits erythropoietin-induced differentiation, cell cycle exit and p27Kip1 accumulation. Oncogene. 2004; 23: 5105-5109. [DOI] [PubMed] [Google Scholar]
- 43.Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. Archipelago regulates cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature. 2001;413: 311-316. [DOI] [PubMed] [Google Scholar]
- 44.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413: 316-322. [DOI] [PubMed] [Google Scholar]
- 45.Koepp DM, Schaefer LK, Ye X, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294: 173-177. [DOI] [PubMed] [Google Scholar]
- 46.Nakayama K, Nagahama H, Minamishima YA, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000;19: 2069-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999;13: 2375-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303: 1374-1378. [DOI] [PubMed] [Google Scholar]
- 49.Yang W, Shen J, Wu M, et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001; 20: 1688-1702. [DOI] [PubMed] [Google Scholar]
- 50.Pagano M, Benmaamar R. When protein destruction runs amok, malignancy is on the loose. Cancer Cell. 2003;4: 251-256. [DOI] [PubMed] [Google Scholar]
- 51.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23: 630-639. [DOI] [PubMed] [Google Scholar]