

Abstract
The bioenergetic response of B lymphocytes is subject to rapid changes following antigen encounter in order to provide ATP and anabolic precursors necessary to support growth. However, the pathways involved in glucose acquisition and metabolism are unknown. We find that B lymphocytes rapidly increase glucose uptake and glycolysis following B-cell antigen receptor (BCR) crosslinking. Inhibition of glycolysis blocks BCR-mediated growth. Prior to S-phase entry, glucose metabolism shifts from primarily glycolytic to include the pentose phosphate pathway. BCR-induced glucose utilization is dependent upon phosphatidylinositol 3-kinase (PI-3K) activity as evidenced by inhibition of glucose uptake and glycolysis with LY294002 treatment of normal B cells and impaired glucose utilization in B cells deficient in the PI-3K regulatory subunit p85α. Activation of Akt is sufficient to increase glucose utilization in B cells. We find that glucose utilization is inhibited by coengagement of the BCR and FcγRIIB, suggesting that limiting glucose metabolism may represent an important mechanism underlying FcγRIIB-mediated growth arrest. Taken together, these findings demonstrate that both growth-promoting BCR signaling and growth-inhibitory FcγRIIB signaling modulate glucose energy metabolism. Manipulation of these pathways may prove to be useful in the treatment of lymphoproliferative disorders, wherein clonal expansion of B lymphocytes plays a role.
Introduction
In response to antigen challenge, resting B lymphocytes exit the G0 phase of the cell cycle and undergo a period of growth before committing to genome replication.1,2 Growth corresponds to an accumulation of cell mass that is accompanied by increased size and is linked to increased de novo macromolecular synthesis.3-5 That mammalian cell growth may be necessary for genome replication underscores its importance in adaptive immunity in that the clonal expansion of antigen-specific B lymphocytes is a prerequisite for humoral immune responses. Most investigations in B cells have focused on the role of genes whose function are important for B-cell antigen receptor (BCR)–induced protein synthesis and increased cell size.4,5 It is recognized, however, that antigen receptor–triggered macromolecular synthesis and gene expression places enormous bioenergetic demands on lymphocytes.5,6 Therefore, one of the fundamental aspects of B-cell responses to antigen challenge that may be critical in vivo is the provision of metabolic substrates to provide ATP and anabolic precursors for cellular growth.
Early studies in lectin-stimulated thymocytes highlighted the importance of glucose uptake and catabolism in providing energy and carbon for macromolecular synthesis.7,8 Further, proliferating thymocytes meet their ATP demand mainly by glycolytic catabolism when sufficient glucose is available.9 It is widely viewed that glucose metabolism is regulated by homeostatic mechanisms wherein mammalian cells respond to a decreased ATP/ADP ratio by adjusting nutrient uptake and catabolism to meet the increased demand for ATP and macromolecular precursors. Recent work has brought new insights into the regulation of energy metabolism by suggesting that the bioenergetic activity of cells is not merely controlled by increased ATP demand but may also be coordinated by signal transduction pathways that act to directly modulate nutrient uptake and metabolism.10-12
Little is currently known about the intracellular signaling and metabolic pathways involved in the acquisition and utilization of glucose to support the bioenergetic demands encountered following BCR engagement.13,14 Upon engagement of the BCR, quiescent B cells undergo a period of growth characterized by de novo RNA and protein synthesis and subsequently enter S phase of the cell cycle within 31 to 48 hours.1,2 Treatment of B cells with inhibitors of phosphatidylinositol 3-kinase (PI-3K) activity in early G1 phase of the cell cycle blunts the ongoing increase in cell size, suggesting that failure of anti-Ig–stimulated B cells to commit to genome replication in the absence of PI-3K activity results from a block at a critical growth checkpoint.15 It has been suggested that in this situation, impaired growth may result from an inability to enact Rel/NF-κB– and c-myc–dependent gene expression programs.4,16,17 Importantly, genomewide analyses have found that Myc increases the expression of specific glycolytic enzyme genes.18-20 It is therefore plausible that impaired growth of B cells in the absence of PI-3K activity may reflect an inability to supply glucose-derived ATP and/or anabolic precursors via glycolysis. With this in mind, we sought to determine if B lymphocytes increase glucose utilization in response to BCR engagement and to identify the signaling and metabolic pathways linking antigen receptors to glucose energy metabolism.
Materials and methods
Antibodies and reagents
Antiphospho(Ser473)Akt and antiphospho(Thr389)p70S6 kinase antibody (Ab) were from Cell Signaling Technology (Beverly, MA). The anti-Glut1 Ab was from Research Diagnostics (Concord, MA). PE-conjugated F(ab′)2 fragments of goat anti–rabbit IgG were obtained from CalTag Laboratories (Burlingame, CA). Anti-CD16/CD32 (FcγIII/II receptor) 2.4G2 monoclonal antibody (mAb) and the Cytofix/Cytoperm Kit were purchased from BD Biosciences (San Jose, CA). F(ab′)2 fragments of goat anti–mouse IgM were obtained from Jackson ImmunoResearch Labs (West Grove, PA). F(ab′)2 fragments of rabbit anti–mouse IgG and rabbit anti–mouse IgG were obtained from ICN Cappel (Aurora, OH). Wortmannin, rapamycin, and LY294002, were obtained from CalBiochem-NovaBiochem (San Diego, CA). Cholera toxin subunit B (CT-B)–Alexa Fluor 488 was obtained from Molecular Probes, Invitrogen (Carlsbad, CA). Protein determinations of whole cell extracts were carried out with a Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories, Hercules, CA).21
Splenic B-lymphocyte isolation
BALB/c mice and p85α-deficient mice (BALB/cAnNTac-Pik3r1 N12) were obtained from Taconic Farms (Germantown, NY) and housed at Boston College. Mice were cared for and handled at all times in accordance with National Institutes of Health and Boston College guidelines. Splenic B cells from mice at 8 to 12 weeks were purified and cultured in RPMI 1640 medium plus 10% FCS as previously described.21 Small dense B cells were then isolated following centrifugation through a discontinuous Percoll gradient as described by Hodgkin et al.22 The A20 cell line expressing a myristoylated Akt–estrogen receptor fusion (Akt-mER) was provided by Dr Michael Gold (Department of Microbiology and Immunology, University of British Columbia).23
Glucose uptake
B cells were resuspended at 3 × 107/mL in uptake buffer (10 mM HEPES [pH 7.4], 136 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, and 1.25 mM MgSO4). Transport was initiated by mixing 200 μL B cells with 200 μL uptake buffer containing 200 μM 2-deoxy-d-glucose (2-DOG) and 2 μCi (7.4 × 104 Bq)/mL [3H]2-DOG (Amersham Biosciences, Piscataway, NJ). At the indicated times, 50 μL of the transport mixture was loaded onto a discontinuous gradient of 200 μL bromododecane per 40 μL 20% perchloric acid, and the gradient was then centrifuged at 14 000g (90 seconds).24 Following centrifugation, the upper aqueous and bromododecane layers were decanted and the perchloric acid phase collected and placed into a vial containing 5 mL scintillation cocktail. The radioactivity was then quantitated by liquid scintillation spectrometry. To assess the contribution of glucose uptake mediated by glucose transporters, uptake of [3H]2-DOG was measured in the presence of 10 μM cytochalasin B, a potent inhibitor of glucose transporter activity.
Flow cytometry
B cells were washed twice in staining buffer (1 mL PBS containing 1% FCS/0.1% NaN3) and then incubated for 20 minutes (4°C) with the anti-CD16/CD32 (2.4G2) mAb Fc block reagent (1:500 vol/vol). Cells were washed twice in 0.1 mL staining buffer and resuspended in 250 μL Cytofix/Cytoperm solution (4°C). After 20 minutes, cells were washed twice in 0.5 mL Perm/Wash solution, resuspended in 0.1 mL Perm/Wash solution, and incubated with 1:500 dilution of anti-Glut1 Ab or isotype control Ab (4°C). After 60 minutes, cells were washed 3 times in 0.1 mL Perm/Wash solution and then incubated with PE-conjugated F(ab′)2 fragments of goat anti–rabbit IgG (1:800) for 1 hour (4°C). Cells were then washed in 0.1 mL Perm/Wash solution, and Glut1 staining was measured by flow cytometry on a BD FACSCanto cytometer and the data analyzed by FACSDiva software (BD Biosciences).
Indirect immunofluorescence
Splenic B cells were centrifuged onto glass slides using a Cytospin (Thermo Electron, Pittsburgh, PA) and incubated with 5 μg/mL CT-B–Alexa Fluor 488 for 30 minutes at 22°C. Slides were washed once with PBS and incubated in 3.7% (vol/vol) formaldehyde for 20 minutes (22°C). Permeabilization was performed in 0.5% (vol/vol) Triton X-100 (5 minutes), and slides were then blocked for 30 minutes in 2% (wt/vol) BSA (22°C). Slides were incubated for 1 hour (22°C) with anti-Glut1 rabbit polyclonal Ab, followed by a TRITC-conjugated goat anti–rabbit IgG (Jackson ImmunoResearch Labs). After 4 washes with PBS, slides were mounted with Aqua Polymount and analyzed by confocal laser scanning microscopy at 488 nm excitation (Leica TCS SP2 confocal microscope; Leica Microsystems, Wetzlar, Germany). A 100 ×/0.53 numeric aperture objective was used to visualize the images. Leica Confocal Software version 2.61 was used to acquire the images.
Glucose utilization measurements
B cells (106 cells/0.5 mL) were incubated with [5-3H]glucose (Amersham Biosciences) for 90 minutes. The initial rate of anti-Ig–induced glycolysis remained linear for 180 minutes. At the indicated times, 100 μL of cells was removed and placed in 1.5 mL microcentrifuge tubes containing 50 μL 0.2 N HCl. The microcentrifuge tubes were then placed in 20 mL scintillation vials containing 0.5 mL water and the vials capped and sealed. 3H2O was separated from unmetabolized [3H]glucose by evaporation diffusion for 48 hours (25°C). The amount of diffused and nondiffused tritium was quantitated by liquid scintillation spectrometry and compared with parallel vials containing [5-3H]glucose only and 3H2O alone.14 Tricarboxylic acid (TCA) cycle flux from glucose catabolism was measured by the rate of 14CO2 production from [6-14C]glucose (Amersham Biosciences).14 Glucose and lactate were measured using assays obtained from Sigma Diagnostics (St Louis, MO) according to the manufacturer's instructions.
Nuclear magnetic resonance (NMR) spectroscopy
B cells (3 × 107) were cultured in RPMI 1640 containing 10 mM [1-13C]glucose or [2-13C]glucose (Cambridge Isotope Laboratories, Andover, MA). B cells were extracted 3 times with 1 mL of 70% (vol/vol) ethanol. Washing of the pelleted cells was avoided because it caused efflux of a large portion of the lactate. The soluble ethanol extracts were pooled, frozen in liquid nitrogen, and lyophilized. The dry material was then resuspended in 0.5 mL D2O for 1H NMR analyses at 28°C. 1H- and 13C-coupled 1H spectra were acquired using a Varian INOVA 500 spectro-photometer (Varian, Palo Alto, CA) equipped with a 5-mm indirect detection probe as described previously.25 In brief, conventional 1H NMR spectra were acquired using a 5006.6 Hz sweep width, 9984 datum points, 90-degree pulse width, 3.0-second recycle delay time, and 128 transients. The 1D heteronuclear multiple quantum coherence (HMQC) spectra (which detect only those protons coupled to 13C) were acquired with a sweep width of 7509.6 Hz, 2048 datum points, 90-degree pulse width, a 1.0-second recycle delay time, and 13C decoupled. For 13C decoupling, the average one-bond 1H-13C J-coupling constant was set to 135 Hz. Spectra were obtained using 4000 transients. Under these acquisition conditions, the lactate methyl doublet was not resolved, but integrated intensities of small molecules at early time points could easily be quantified.
Results
BCR engagement leads to increased glucose uptake and Glut1 expression in a PI-3K–dependent manner
We sought to determine if BCR signaling modulates glucose uptake in B cells. To this end, small dense (quiescent) splenic B cells exhibited a relatively low rate of 2-DOG uptake, whereas stimulation with 10 μg/mL F(ab′)2 fragments of anti–mouse IgM (anti-Ig) for 15 hours resulted in an approximately 10-fold increase in 2-DOG uptake (Figure 1A). The initial rate of anti-Ig–stimulated 2-DOG uptake was linear for approximately 90 seconds and judged specific as cytochalasin B, an inhibitor of facilitated glucose transporters, blocked the increased 2-DOG uptake (Figure 1A, inset). BCR crosslinking induces PI-3K activation within minutes, leading to activation of Akt and p70S6 kinase.15 To determine if PI-3K activity was necessary for the increased glucose uptake following BCR ligation, LY294002 and wortmannin were used to inhibit PI-3K. In the presence of LY294002, anti-Ig–stimulated glucose uptake was significantly reduced in comparison with nontreated B cells (Figure 1B). Pretreatment of B cells with wortmannin also resulted in inhibition of anti-Ig–induced glucose uptake, although not to the same extent as that observed with LY294002 (Figure 1B). The efficacy of the PI-3K inhibitors was demonstrated insofar as pretreatment of B cells with LY294002 or wortmannin blocked BCR-induced Akt phosphorylation on Ser473 (Figure 1C). To determine the contribution of the mammalian homolog mTOR, we tested the effects of the mTOR inhibitor rapamycin on anti-Ig–induced glucose uptake. In several experiments, rapamycin only had a moderate inhibitory effect on anti-Ig–induced glucose uptake (Figure 1B). Importantly, rapamycin inhibited anti-Ig–induced p70S6 kinase phosphorylation on Thr389, which is dependent on mTOR, indicating that the concentration of rapamycin used was sufficient to inhibit mTOR (Figure 1C). These data suggest that BCR ligation acts to increase glucose transport and does so via a pathway dependent on PI-3K activity.
Figure 1.

Engagement of the BCR increases glucose uptake in a PI-3K–dependent pathway. (A) Splenic B cells were cultured in the absence (Med) or presence of 10 μg/mL anti-Ig (Ig) for 15 hours. Uptake of 2-[3H]deoxyglucose was measured for 60 seconds in the presence or absence of 10 μM cytochalasin B. Transport represents the difference between the total 2-[3H]deoxyglucose uptake (cpm/106 cells/sec) minus uptake in the presence of 10 μM cytochalasin B. (Inset) Splenic B cells were cultured in the presence of 10 μg/mL anti-Ig (Ig) for 15 hours. Cells were collected, and the initial uptake of 2-[3H]deoxyglucose was measured for the indicated times (in seconds) in the presence (bottom line) or absence (top line) of 10 μM cytochalasin B as described in “Materials and methods.” The data indicate that uptake of 2-[3H]deoxyglucose in anti-Ig–stimulated B cells is linear for 90 seconds. (B) Parallel B cells were also stimulated with 10 μg/mL anti-Ig (15 hours) in the absence (Ig) or presence of 10 μM LY294002 (Ig+LY), 50 nM wortmannin (Ig+Wort), or 20 nM rapamycin (Ig+Rap). Cells were then harvested, and 2-[3H]deoxyglucose uptake was measured. The inhibitors had no measurable effect on 2-[3H]deoxyglucose uptake in B cells cultured in medium alone (data not shown). Error bars reflect standard deviation from the mean of triplicate measurements, and the data for panels A-B are representative of 3 independent experiments. (C) B cells were pretreated with 10 μM LY294002 (LY), 50 nM wortmannin (Wort), or 20 nM rapamycin (Rap) for 30 minutes and cultured in medium alone (M) or stimulated with 10 μg/mL anti-Ig (αIg) for 15 minutes. Cell lysates were prepared, and phosphorylation of Akt on Ser473 and p70S6K on Thr389 was monitored by Western blot.21 The data are representative of 4 independent experiments.
We next determined if the increased glucose uptake in response to anti-Ig might be attributed to changes in glucose transporter expression. Expression of Glut1 was evaluated, given reports that Glut1 is the primary facilitated glucose transporter expressed on hematopoietic cells.26 Although quiescent B cells expressed Glut1, the expression of Glut1 increased in response to anti-Ig stimulation as determined by Western blot analysis and flow cytometry (Figure 2A-B, respectively). Of note, we also measured Glut1 staining of nonpermeabilized B cells cultured in medium alone or stimulated with anti-Ig for 12 hours; the mean fluorescence intensity of surface Glut1 expression increased 5-fold following anti-Ig stimulation (data not shown). Anti-Ig–induced Glut1 expression in B cells pretreated with LY294002 mirrored that of quiescent B cells, suggesting that increased Glut1 expression requires PI-3K activity (Figure 2B). Consistent with these findings, anti-Ig–induced Glut1 expression was impaired in splenic B cells from p85α-null mice, which exhibit markedly reduced BCR-induced PI-3K activity (Figure 2B).27-29 BCR-induced Glut1 expression in wild-type B cells was not sensitive to rapamycin (data not shown). We also evaluated B cells for expression of Glut1 by immunofluorescence. To monitor surface staining, B cells were incubated with Alexa Fluor 488–conjugated CT-B prior to fixation (Figure 2C, left panels). CT-B binds to glycosphingolipids, with a strong affinity for GM1, and therefore can be used as a plasma membrane marker.30 Quiescent B cells exhibited a relatively low level of anti-Glut1 Ab staining, which was increased in response to BCR engagement (Figure 2C).
Figure 2.
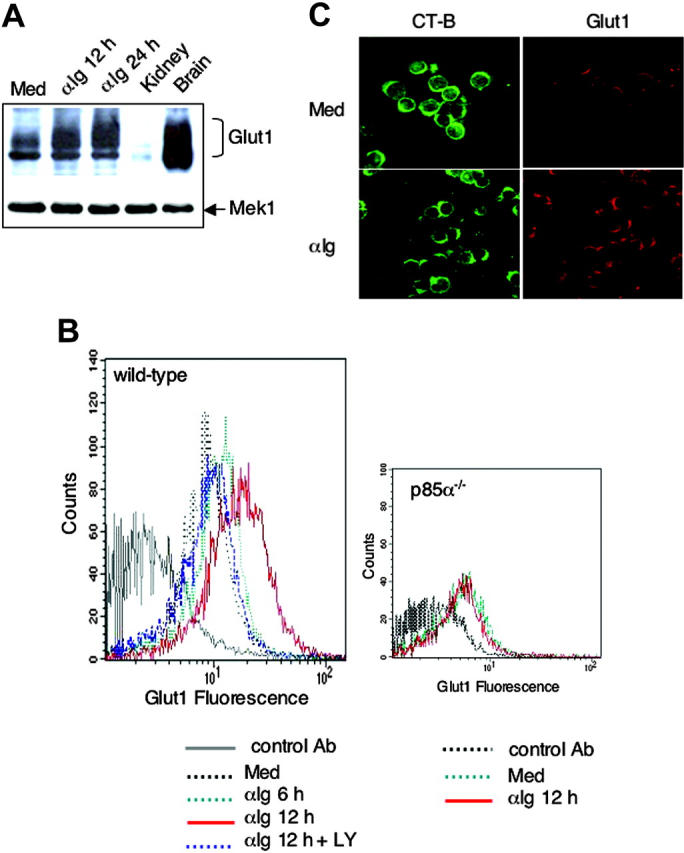
BCR crosslinking increases Glut1 expression. (A) B cells were cultured in the absence (Med) or presence of 10 μg/mL anti-Ig (αIg) for 12 and 24 hours. Cell lysates were prepared, and equivalent amounts of total protein were examined by Western blot for Glut1 and Mek-1 protein levels; the latter serves as a loading control. Brain and kidney lysates serve as tissue sources expressing relatively high and low levels of Glut1, respectively. (B) B cells were cultured in the absence (Med) or presence of 10 μg/mL anti-Ig (αIg) for 6 and 12 hours, and then Glut1 expression was determined by flow cytometry. Parallel B cells were pretreated for 30 minutes with 10 μM LY294002 and then stimulated with anti-Ig for 12 hours (αIg 12 h + LY). B cells were also isolated from p85α-deficient mice, cultured in the absence (Med) or presence of 10 μg/mL anti-Ig (αIg) for 12 hours, and then Glut1 expression was determined by flow cytometry. In both the wild-type and p85α-deficient analyses, control Ab indicates an isotype-matched control staining of B cells stimulated with 10 μg/mL anti-Ig for 12 hours. In data not shown, the staining of B cells with an anti-MHC Ab was similar in all conditions. (C) B cells were cultured in medium alone (Med) or stimulated with 10 μg/mL anti-Ig (αIg) for 12 hours. Cells were stained with Alexa Fluor 488–conjugated CT-B and anti-Glut1 Ab as described in “Materials and methods.” The data are representative of 3 independent experiments.
Increased catabolism of glucose by the glycolytic pathway in response to BCR crosslinking requires PI-3K activity
To examine the significance of increased glucose uptake, we evaluated whether BCR signaling increased glucose flux into several metabolic pathways involved in glucose catabolism. Generation of [3H]H2O resulting from the dehydration of [5-3H]glucose catalyzed by enolase, the penultimate step in the glycolytic pathway, was monitored as a readout for glycolysis.12 Quiescent B lymphocytes exhibited a measurable, albeit relatively low, rate of glycolysis (Figure 3A). Anti-Ig–stimulated B cells increased glycolysis when measured at 12 hours, which continued to increase for 31 hours, remaining elevated for at least 48 hours (Figure 3A). BCR-induced glycolysis was inhibited more than 90% in the presence of 1 mM 2-DOG (Figure 3A), a potent inhibitor of glycolysis.31 The increase in glycolysis following BCR engagement was blocked by LY294002 or wortmannin and, to a much lesser extent, by rapamycin (Figure 3B). To corroborate these findings, we examined ex vivo splenic B cells isolated from mice deficient in the p85α subunit of PI-3K.21,29 We observed a striking impairment in BCR-induced glycolysis in p85α-null B cells in comparison with wild-type B cells (Figure 3C). To understand the biologic significance of increased glycolysis, we incubated quiescent B cells with 1 mM 2-DOG along with anti-Ig. 2-DOG blocked increases in cell size in response to anti-Ig, as assessed by mean forward scatter (Figure 3D). In addition, 2-DOG blocked increases in protein content in response to anti-Ig (Figure 3D). Notably, 2-DOG reduced cell number by 11% when measured at 18 hours (data not shown). Taken together, these results suggest that glycolytic flux is necessary for BCR-induced B-cell growth.
Figure 3.

BCR engagement increases glycolysis in a PI-3K–dependent manner. (A) Quiescent splenic B cells were cultured in the absence (t = 0 hours) or presence of 10 μg/mL anti-Ig (♦). Anti-Ig–stimulated B cells were also cultured with 1 mM 2-DOG (2DOG) for 12 hours (▵). At 90 minutes prior to the indicated times, B-cell cultures were supplemented with [5-3H]glucose, and glycolysis was then measured as described in “Materials and methods.” The standard deviations for each time point are less than 5% of the mean of triplicate measurements, and the data are representative of 3 independent experiments. (B) Glycolysis was conducted in parallel B cells that were pretreated with 10 μM LY294002 (Ig+LY), 50 nM wortmannin (Ig+Wort), or 20 nM rapamycin (Ig+Rap) for 30 minutes and then stimulated with 10 μg/mL anti-Ig (Ig) for 18 hours. The inhibitors had no measurable affect on glycolysis in B cells cultured in medium (Med) alone (data not shown). (C) Splenic B cells from wild-type (WT) and p85α-deficient (KO) mice were cultured in the absence (Media) or presence of 10 μg/mL anti-Ig (Ig). At 90 minutes prior to the indicated times, glycolysis was measured. The standard deviations for each condition are less than 5% of the mean. (D) B cells were cultured in media alone (Med) or stimulated with 10 μg/mL anti-Ig (Ig) for 18 hours in the absence or presence of 1 mM 2-DOG. Cells were then harvested for flow cytometric analysis for size (mean fwd scatter) and total cellular protein content. Protein determinations from total cellular extracts were carried out using a Bio-Rad Protein Assay Dye Reagent according to the manufacturer's recommendations.21 (E) B cells were cultured in medium alone (Media) or stimulated with 10 μg/mL anti-Ig (Ig) for the indicated times, and the amount of lactate secreted into the tissue culture medium was then measured. The data are represented as millimoles per liter (mmol/L). In panels D-E, error bars reflect standard deviation from the mean of triplicate measurements, and the data are representative of 3 independent experiments.
During the course of these experiments we observed that the increase in glycolysis following BCR crosslinking was accompanied by production of lactate (Figure 3E). We estimate that more than 80% of the glucose consumed in anti-Ig–stimulated B cells, as measured by glucose depletion from the culture medium, was converted to lactate when assayed at 24 hours and 48 hours. An important caveat to this interpretation is that carbon sources other than glucose can contribute to the lactate pool (eg, from incomplete oxidation of glutamine via malate and PEP-carboxykinase enzymes). To more fully assess glucose metabolism, we compared the flux of radiolabeled glucose through the glycolytic pathway and the TCA cycle in anti-Ig–stimulated B cells. Glucose flux through the glycolytic pathway was substantial in comparison with glucose oxidation through TCA cycle flux (Table 1). Because the 14CO2 generated in these assays derives from the complete oxidation of [6-14C]glucose, we cannot entirely rule out the possibility that some glucose may be incompletely oxidized via a “truncated” TCA cycle.
Table 1.
Catabolism of glucose in anti-Ig–stimulated B lymphocytes
| 12 h | 24 h | 48 h | |
|---|---|---|---|
| Glycolysis, nmol/106 cells/min | 0.50 | 1.11 | 1.39 |
| TCA cycle, nmol/106 cells/min | 0.031 | 0.017 | 0.020 |
The rates of metabolic flux of [5-3H]glucose (glycolysis) and [6-13C]glucose (TCA cycle) molecules in anti-Ig—stimulated B cells were determined at the indicated times. The data are represented as mean values of 3 determinations. The standard error is less than 5% of the mean.
NMR spectroscopy reveals changes in glucose catabolism in activated B lymphocytes
We next sought to discern further the role of BCR signaling in controlling glucose carbon flow. Our approach was to use a 1H NMR experiment, which specifically monitors 13C fixation from [1-13C]glucose into metabolites by selecting for only those 1H's coupled to 13C nuclei.25 We favored 1H NMR because it allowed glucose metabolism to be followed immediately in response to BCR engagement, whereas the biochemical assays used in Figure 3A-C did not exhibit the sensitivity necessary to reproducibly monitor changes in glucose utilization early in response to anti-Ig stimulation (ie, 0 to 8 hours). There was little detectable difference in the normal 1H spectrum between quiescent and anti-Ig–stimulated B cells during an initial 8-hour period, quantified by integrating the resonances for the lactate methyl group and the glutamate methylenes as a function of time after anti-Ig stimulation (data not shown). However, use of a 1D-HMQC sequence to select out protons attached to 13C adequately could follow glucose metabolism in quiescent and anti-Ig–stimulated B cells (Figure 4A-B, respectively). Integration of the 13C-coupled 1H resonances revealed that both the lactate methyl group and the glutamate methylenes exhibited increased 13C content as the [1-13C]glucose was metabolized during the initial 8 hours of anti-Ig stimulation (Figure 4C). The specific increase in 13C could be monitored by normalizing the 13C-coupled 1H resonance integral to the total 1H resonance integral (Figure 4D). These data indicate that enhanced glucose utilization, as evidenced by increased 13C incorporation into the methyl group of lactate from [1-13C]glucose, was apparent within 2 hours following BCR engagement. Glutamate pools also increased in 13C content in response to BCR ligation. Notably, incorporation of 13C label into glutamate C-4 from transamination of the TCA cycle intermediate α-ketoglutarate reflects metabolism of [1-13C]glucose by the TCA cycle. C-3 of glutamate also becomes 13C labeled as material repetitively goes through the TCA cycle and the symmetric intermediate succinate. In data not shown, we observed that 13C label into glutamate moderately increased until 12 hours and then declined by 24 hours to a level similar to quiescent B cells.
Figure 4.

1H NMR spectra of glucose metabolites in B cells. B cells were cultured in medium containing 10 mM [1-13C]glucose in the absence (A) or presence (B) of 10 μg/mL anti-Ig for 8 hours. Cells were then analyzed by 1D-HMQC as described in “Materials and methods.” The 13C from the [1-13C]glucose is metabolized to the lactate methyl group and the glutamate methylenes. (C-D) Incorporation of [1-13C]glucose into the lactate methyl group (▪) and the glutamate C4H2 (•) and C3H2 (○) pools in anti-Ig–stimulated B cells. (C) Integrated intensity of protons coupled to 13C selected in a 1D-HMQC experiment. (D) Specific 13C content of lactate and glutamate pools as a function of time after BCR crosslinking. (E) Incorporation of [1-13C]glucose into metabolites in B cells after BCR crosslinking (8 hours) in the absence (control) and presence of pretreatment (30 minutes) with 10 μM LY294002 (+LY), 50 nM wortmannin (+wort), or 20 nM rapamycin (+rap). The increases in 13C content (compared with the same metabolites in quiescent B cells where 13C content is 1.0) are shown for the lactate methyl group (black bars) and glutamate methylenes (open bars) (the average increase in 13C uptake into each glutamate methylene carbon is shown) and the citrate methylenes (gray bars) as a control. (F) 13C glucose incorporation and metabolism into lactate in response to anti-Ig stimulation of B cells at the indicated times (h); [1-13C]glucose labeling of the methyl group (•); [2-13C]glucose labeling of methyl (▪) and methine groups (○). (G) Fraction of lactate generated by the pentose phosphate pathway based on 13C label distribution in the CH3 group as a function of time after anti-Ig stimulation of B cells incubated with [2-13C]glucose. Integration scales in each type of spectrum are arbitrary.
We carried out a similar 1D-HMQC experiment to evaluate the contribution of PI-3K activity in BCR-induced 13C incorporation from [1-13C]glucose into the methyl group of lactate. In B cells stimulated via the BCR for 8 hours, incorporation of 13C label increased approximately 3.5-fold above quiescent B cells (Figure 4E, control). Incorporation of 13C label into the glutamate carbons also increased approximately 2.7- to 2.8-fold. The citrate pool, much of which derived from the tissue culture medium, was not appreciably labeled during the activation period. Pretreatment of B cells with wortmannin or LY294002 blocked 13C incorporation into both lactate and glutamate (Figure 4E), whereas rapamycin had little effect on 13C fixation into either lactate or glutamate (Figure 4E, +rap).
Because several glucose-derived metabolites from the pentose phosphate pathway can enter the glycolytic pathway upstream of enolase, the dehydration reaction of 2-phosphoglycerate used herein may reflect glucose catabolism via both glycolysis and the pentose phosphate pathway. To monitor diversion of glucose into the pentose phosphate pathway, a 1D-HMQC experiment was carried out with [2-13C]glucose. If [2-13C]glucose is used instead of [1-13C]glucose, there will be no 13C incorporation into the lactate methyl group by the glycolytic pathway; however, if the pentose phosphate pathway is operational, 13C will be incorporated into the methyl group of lactate. [1-13C]glucose rapidly labeled the lactate methyl group following BCR engagement (Figure 4F, •). In parallel B cells cultured with [2-13C]glucose, no significant 13C incorporation was detected in the lactate methyl group prior to 24 hours after BCR crosslinking (Figure 4F, ▪). As 13C label from [2-13C]glucose appeared in the lactate methyl group, it was depleted from the methine group of lactate (Figure 4F, ○), a finding consistent with decreased glucose catabolism by the glycolytic pathway and increased catabolism via the pentose phosphate pathway. By 48 hours, [2-13C]glucose flux through the pentose phosphate pathway represented approximately 60% of the total lactate production (Figure 4G). These results suggest that as stimulated B cells progress through the G1/S transition, a shift occurs in the metabolism of glucose from primarily glycolytic to include the pentose phosphate pathway.
Conditional Akt activation increases glucose utilization in B cells
Murine A20 B cells expressing a myristoylated Akt-ER fusion protein (Myr Akt-ER) were used to determine if glucose utilization is contingent upon Akt activation, a downstream target of PI-3K. The Myr Akt-ER is constitutively expressed in A20 cells but remains inactive in the absence of 4-hydroxytamoxifen (4-HT).23 Myr Akt-ER–expressing A20 B cells cultured in medium alone or stimulated with anti-Ig were devoid of detectable Myr Akt-ER Ser473 phosphorylation; addition of 4-HT led to increased Ser473 phosphorylation, a modification that correlates with catalytic activity (Figure 5A). The 4-HT–treated A20 B cells exhibited an increase in glucose uptake in comparison with nontreated A20 B cells (Figure 5B). Activation of the Myr Akt-ER fusion protein also resulted in higher glycolytic flux than nontreated A20 B cells (Figure 5C). In data not shown, A20 B cells expressing an empty vector did not exhibit changes in glucose utilization in response to 4-HT. Exposure to 4-HT did not alter the proliferation of Myr Akt-ER–expressing A20 B cells in comparison with nontreated A20 B cells, indicating that the sum macromolecular synthesis was equivalent (data not shown). These findings suggest that Akt activation is sufficient to increase glucose utilization in A20 B cells.
Figure 5.

Conditional activation of Akt in A20 B cells increases glucose utilization. (A) A20 cells constitutively expressing an Akt-mER fusion protein were cultured in the absence or presence of 4 μM 4-HT or 10 μg/mL anti-Ig (αIg) for 5 and 30 minutes. Cells were then harvested, and phosphorylation of Akt-mER (Myr Akt-ER) on Ser473 was monitored by Western blot. A20 cells constitutively expressing an Akt-mER fusion protein were cultured in the absence or presence of 4 μM 4-HT for 24 hours, and then uptake of 2-[3H]deoxyglucose (B) and glycolysis (C) was measured as described in “Materials and methods.” In panels B-C, error bars reflect standard deviation from the mean of triplicate measurements.
Coengagement of the BCR and FcγRIIB blocks glucose utilization
The FcγRIIB is a low-affinity receptor for the Fc portion of Ig, with IgG immune complexes as its physiologic ligand.32 Coclustering of the BCR with FcγRIIB provokes a dominant-inhibitory signal that blocks B-cell growth induced by BCR engagement and ultimately leads to apoptosis.33-35 FcγRIIB inhibits BCR signaling by reducing the accumulation of phosphoinositide 3-kinase products and recruiting SH2-containing inositol-5-phosphatase to FcγRIIB, hydrolyzing PIP3, and inhibiting distal Akt activation.36,37 We therefore tested if coengagement of FcγRIIB inhibited glucose utilization induced by BCR crosslinking. The level of Akt phosphorylation on Ser473 was significantly reduced by intact anti-Ig, which coligates the BCR and FcγRIIB, in comparison with F(ab′)2 fragments of anti-Ig (Figure 6A). Inhibition of Akt phosphorylation required FcγRIIB engagement, because B cells incubated with intact anti-Ig plus mAb 2.4G2 exhibited levels of Ser473 phosphorylation comparable to BCR crosslinking alone. Coclustering of the BCR and FcγRIIB resulted in an approximately 82% reduction in glucose uptake as compared with B cells stimulated via the BCR alone (Figure 6B). Intact anti-Ig also blocked BCR-induced Glut1 expression and glycolysis (Figure 6C-D, respectively). The 1D-HMQC experiments described in Figure 4E also revealed that B cells treated with intact anti-Ig exhibited decreased 13C incorporation into both lactate and glutamate (Figure 6E). These results suggest that limiting glucose utilization may represent an important mechanism underlying the inhibitory action of FcγRIIB on B-cell growth.
Figure 6.

Cocrosslinking the BCR and FcγRIIB inhibits BCR-induced glucose utilization. (A) B cells were cultured in medium alone (Med) or 10 μg/mL F(ab′)2 fragments of anti–mouse IgG (αIg) for 15 minutes. Parallel B cells were cultured with 10 μg/mL intact anti–mouse IgG (wIg) in the presence or absence of 10 μg/mL 2.4G2 mAb. Cells were then evaluated for phosphorylation of Akt on Ser473 and β-actin by Western blot. (B) B cells were cultured in medium alone (Med), 10 μg/mL F(ab′)2 fragments of anti–mouse IgG (IgG), or 10 μg/mL intact anti–mouse IgG (wIg) for 12 hours. Cells were then assayed for 2-[3H]deoxyglucose uptake as described in Figure 1. (C) Glut1 expression was monitored by flow cytometry in B cells cultured in medium alone (Med) and stimulated with 10 μg/mL F(ab′)2 fragments of anti–mouse IgG (anti Ig) for 12 hours or with 10 μg/mL intact anti–mouse Ig (wIg) for 12 hours. Control Ab indicates an isotype-matched control staining of B cells stimulated with 10 μg/mL F(ab′)2 fragments of anti–mouse IgG for 12 hours. In data not shown, the staining of B cells with an anti-MHC Ab was similar in all conditions. (D) Glycolysis was measured in B cells cultured as described in panel B. In panels B and D, error bars reflect standard deviation from the mean of triplicate measurements. The data are representative of 3 independent experiments. (E) Incorporation of [1-13C]glucose into metabolites in B cells stimulated with 10 μg/mL F(ab′)2 fragments of anti–mouse IgG (control) for 8 hours as described in Figure 4E. In parallel, B cells were cultured with 10 μg/mL intact anti–mouse Ig (FcR). The increases in 13C content (compared with the same metabolites in quiescent B cells where the 13C content is 1.0) are shown for the lactate methyl group (black bars), glutamate methylenes (open bars) (the average increase in 13C uptake into each glutamate methylene carbon is shown), and the citrate methylenes (gray bars).
Discussion
The results herein suggest that the BCR regulates glucose utilization in small dense B cells by increasing glucose uptake and expression of the facilitated Glut1 transporter. The importance of this signaling-induced response is underscored in that quiescent B cells do not contain significant glycogen reserves. Therefore, the capacity to signal increased glucose transport in response to antigen challenge provides a mechanism to rapidly acquire glucose-derived energy and biosynthetic precursors necessary to support growth. Our results also indicate that BCR engagement leads to a rapid and sustained increase in glucose metabolism. The results from 1D-HMQC reveal that glycolysis is increased within 2 hours of BCR ligation, preceding the increase in cell size and entry into the cell cycle.1,2,15 Our results also indicate that incubation of B cells with 2-DOG, a potent inhibitor of glycolysis resulting from the competitive inhibition of phosphoglucoisomerase and depletion of hexokinase II,31 prevents BCR-triggered increases in cell size and protein synthesis, suggesting that heightened glycolytic flux is a requirement for B-cell growth.
These results also suggest that prior to S-phase entry, glucose catabolism shifts from primarily glycolysis to include the pentose phosphate pathway. The biologic significance of the shift toward pentose phosphate pathway–mediated glucose catabolism is currently unknown. This pathway has several purposes, including providing reduced coenzyme NADPH for reductive biosynthesis and ribose 5-phosphate for nucleotide biosynthesis. That elevated pentose phosphate pathway activity coincides with the onset of S-phase entry may reflect a demand for ribose 5-phosphate to support nucleic acid synthesis. Notably, however, growth factor stimulation is accompanied by a rise in intracellular H2O238-40 Recent reports provide compelling support for the requirement of NADPH, not ribose 5-phosphate, for cellular proliferation, presumably to maintain the optimal redox status for cellular proliferation.41,42
The rapid increase in glycolysis following BCR engagement is accompanied by an increase in lactate production, with more than 80% of the glucose consumed by B cells being converted to lactate. An important caveat to our calculations is the possibility that nonglucose sources may contribute to lactate production, which might be significant given that glutamine oxidation accounts for 33% of lactate production in activated T cells.43 At this juncture we cannot eliminate the contribution of nonglucose sources to lactate production; however, in a preliminary 1D-HMQC experiment, [13C]glutamine metabolism in anti-Ig–stimulated B cells (15 hours) was not accompanied by a measurable increase in 13C incorporation into lactate (data not shown). These results suggest that anti-Ig–stimulated B cells carry out aerobic glycolysis characterized by increased lactate production from glucose despite the presence of an intact TCA cycle.
The high rate of aerobic glycolysis in response to anti-Ig stimulation of primary B lymphocytes observed herein mirrors the situation in tumor cells. This might reflect a situation in which the capacity to carry out oxidative degradation in the mitochondria is limited in normal B cells. Of interest, Fitzpatrick and coworkers14 reported that B-cell hybridomas exhibit low levels of pyruvate dehydrogenase, which in turn might represent a barrier for glycolytic intermediates entering the TCA cycle. We note that in response to limiting extracellular glucose, B cells compensate by increasing glycolysis but not oxygen consumption (data not shown). Alternatively, B cells that have encountered antigen may rapidly transition to a high rate of glycolysis to allow for precise control over the time during which ATP and anabolic precursors are needed to support growth, clonal expansion, and Ig synthesis.44,45 This would provide B cells the capacity to redirect glucose carbon flow into the pentose phosphate pathway to provide NADPH and ribose 5-phosphate or redirect the glycolytic end product, pyruvate, into a truncated TCA cycle. It has been demonstrated that citrate can be exported to the cytosol, thereby serving as a substrate for ATP citrate lyase, to produce acetyl-CoA and oxaloacetate46; the former is a precursor for fatty acid and cholesterol synthesis and functions, at least in part, to promote membrane biogenesis.47
Our results also suggest that increased glucose utilization results from a primary PI-3K signaling pathway originating from the BCR. This conclusion is supported by the findings that LY294002 and wortmannin block BCR-induced glucose uptake and glycolysis. In addition, splenic B cells from mice deficient in the class 1A PI-3K regulatory subunit p85α (Pik3r1), which exhibit impaired BCR-coupled PI-3K activation,28,29 fail to increase Glut1 expression and glycolysis. We cannot rule out that a homeostatic response to decreased ATP and/or glycolytic metabolites associated with growth contributes, along with PI-3K signaling input, to increased glycolysis; however, we find that conditional activation of Akt is sufficient to stimulate glucose utilization in A20 B cells in the absence of increased growth. Taken together, these findings suggest that in B lymphocytes PI-3K/Akt signaling functions to regulate glucose utilization, thereby providing B cells the capability to meet the bioenergetic demands associated with growth.
In terms of downstream Akt effector signaling components, our results fail to conclusively implicate mTOR in BCR-induced glucose metabolism, because rapamycin treatment does not significantly abate the increase in glucose transport and glycolysis in B cells. This is surprising given the important role of Akt and mTOR signaling in regulating nutrient uptake in hematopoietic cells.12,24 Interestingly, the growth of splenic B cells was reported to be only partially inhibited by rapamycin, whereas LY294002 completely blocked B-cell growth.16 In this study NF-κB–dependent c-myc transcription was shown to be critical for anti-Ig–stimulated B-cell growth. The potential significance of this pathway to the findings herein is underscored by the observation that many glycolytic genes are directly regulated by c-Myc.18-20 Further, recent gene expression profiling in ex vivo splenic B cells revealed that BCR engagement up-regulates several genes encoding enzymes of the glycolytic pathway.48 These findings taken together raise the possibility that PI-3K may link the BCR to increased glycolysis via c-Myc–dependent transcription of genes encoding glycolytic enzymes. In addition, increased glucose utilization may be regulated via posttranslational mechanisms. The key rate-limiting step in glycolysis is the phosphorylation of fructose-6-phosphate catalyzed by 1-phosphofructokinase (PFK).49,50 Akt has been shown to phosphorylate PFK-2, an enzyme that generates fructose-2,6 bisphosphate, an allosteric regulator of PFK.49 Along these lines, the atypical protein kinase C (PKC) isoforms, PKCλ and PKCζ, together with Akt, have been implicated in insulin-stimulated Glut4 translocation to the plasma membrane.50-51
In summary, our results provide evidence for the first time that both growth-promoting BCR signaling and growth-inhibitory FcγRIIB signaling directly modulate glucose energy metabolism. These results also raise the possibility that manipulation of the BCR-linked pathways involved in glucose acquisition and catabolism may prove effective in the treatment of lymphoproliferative disorders that arise from the clonal expansion of normal B lymphocytes.
Acknowledgments
We thank Dr David Fruman (Center for Immunology, Department of Molecular Biology and Biochemistry, University of California–Irvine) for helpful suggestions.
Prepublished online as Blood First Edition Paper, January 31, 2006; DOI 10.1182/blood-2005-12-4788.
Supported by United States Public Health Service (USPHS) grant AI-49994 (T.C.C.).
M.F.R. and T.C.C. designed research and wrote the manuscript, and all of the authors performed research.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.DeFranco AL, Raveche ES, Paul WE. Separate control of B lymphocyte early activation and proliferation in response to anti-IgM antibodies. J Immunol. 1985;135: 87-94. [PubMed] [Google Scholar]
- 2.Sieckmann DG. The use of anti-immunoglobulins to induce a signal for cell division in B lymphocytes via their membrane IgM and IgD. Immunol Rev. 1980;52: 181-210. [DOI] [PubMed] [Google Scholar]
- 3.Schuhmacher M, Staege MS, Pajic A, et al. Control of cell growth by c-Myc in the absence of cell division. Curr Biol. 1999;9: 1255-1258. [DOI] [PubMed] [Google Scholar]
- 4.Iritani BM, Eisenman RN. c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci U S A. 1999;96: 13180-13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buttgereit F, Burmester GR, Brand MD. Bioenergetics of immune functions: fundamental and therapeutic aspects. Immunol Today. 2000;21: 192-199. [DOI] [PubMed] [Google Scholar]
- 6.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5: 844-852. [DOI] [PubMed] [Google Scholar]
- 7.Cooper EH, Barkhan P, Hale AJ. Observations on the proliferation of human leucocytes cultured with phytohaemagglutinin. Br J Haematol. 1963; 9: 101-111. [DOI] [PubMed] [Google Scholar]
- 8.Roos D, Loos JA. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res. 1973;77: 127-135. [DOI] [PubMed] [Google Scholar]
- 9.Krauss S, Brand MD, Buttgereit F. Signaling takes a breath—new quantitative perspectives on bioenergetics and signal transduction. Immunity. 2001;15: 497-502. [DOI] [PubMed] [Google Scholar]
- 10.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271: 31372-31378. [DOI] [PubMed] [Google Scholar]
- 11.Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23: 7315-7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16: 769-777. [DOI] [PubMed] [Google Scholar]
- 13.Brand K, Leibold W, Luppa P, Schoerner, C, Schulz A. Metabolic alterations associated with proliferation of mitogen-activated lymphocytes and of lymphoblastoid cell lines: evaluation of glucose and glutamine metabolism. Immunobiology.1986;173: 23-34. [DOI] [PubMed] [Google Scholar]
- 14.Fitzpatrick L, Jenkins HA, Butler M. Glucose and glutamine metabolism of a murine B-lymphocyte hybridoma grown in batch culture. Appl Biochem Biotechnol. 1993;43: 93-116. [DOI] [PubMed] [Google Scholar]
- 15.Donahue AC, Fruman DA. Proliferation and survival of activated B cells requires sustained antigen receptor engagement and phosphoinositide 3-kinase activation. J Immunol. 2003;170: 5851-5860. [DOI] [PubMed] [Google Scholar]
- 16.Grumont RJ, Strasser A, Gerondakis S. B cell growth is controlled by phosphatidylinosotol 3-kinase-dependent induction of Rel/NF-kappaB regulated c-myc transcription. Mol Cell. 2002;10: 1283-1294. [DOI] [PubMed] [Google Scholar]
- 17.Bone H, Williams NA. Antigen-receptor cross-linking and lipopolysaccharide trigger distinct phosphoinositide 3-kinase-dependent pathways to NF-kappa B activation in primary B cells. Int Immunol. 2001;13: 807-816. [DOI] [PubMed] [Google Scholar]
- 18.Kim JW, Zeller KI, Wang Y, et al. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol. 2004;24: 5923-5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuhmacher M, Kohlhuber F, Holzel M, et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001;29: 397-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osthus RC, Shim H, Kim S, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275: 21797-21800. [DOI] [PubMed] [Google Scholar]
- 21.Piatelli MJ, Wardle C, Blois J, et al. Phosphatidylinositol 3-kinase-dependent mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 and NF-kappa B signaling pathways are required for B cell antigen receptor-mediated cyclin D2 induction in mature B cells. J Immunol. 2004;172: 2753-2762. [DOI] [PubMed] [Google Scholar]
- 22.Hodgkin PD, Yamashita LC, Coffman RL, Kehry MR. Separation of events mediating B cell proliferation and Ig production by using T cell membranes and lymphokines. J Immunol. 1990;145: 2025-2034. [PubMed] [Google Scholar]
- 23.Christian SL, Sims PV, Gold MR. The B cell antigen receptor regulates the transcriptional activator beta-catenin via protein kinase C-mediated inhibition of glycogen synthase kinase-3. J Immunol. 2002;169: 758-769. [DOI] [PubMed] [Google Scholar]
- 24.Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002;13: 2276-2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciulla RA, Roberts MF. Effects of osmotic stress on Methanococcus thermolithotrophicus: 13C-edited 1H-NMR studies of osmolyte turnover. Biochim Biophys Acta. 1999;1427: 193-204. [DOI] [PubMed] [Google Scholar]
- 26.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain cell size or viability. Mol Cell. 2000;6: 683-692. [DOI] [PubMed] [Google Scholar]
- 27.Fruman DA, Cantley LC. Phosphoinositide 3-kinase in immunological systems. Semin Immunol. 2002;14: 7-18. [DOI] [PubMed] [Google Scholar]
- 28.Fruman DA, Snapper SB, Yballe CM, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 1999;283: 393-397. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki H, Terauchi Y, Fujiwara M, et al. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science. 1999;283: 390-392. [DOI] [PubMed] [Google Scholar]
- 30.Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol. 1998;141: 929-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geschwind JF, Georgiades CS, Ko YH, Pedersen PL. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev Anticancer Ther. 2004;4: 449-457. [DOI] [PubMed] [Google Scholar]
- 32.Ravetch JV, Bolland S. IgG Fc receptors. Ann Rev Immunol. 2001;19: 275-290. [DOI] [PubMed] [Google Scholar]
- 33.Ashman RF, Peckham D, Stunz LL. Fc receptor off-signal in the B cell involves apoptosis. J Immunol. 1996;157: 5-11. [PubMed] [Google Scholar]
- 34.Klaus GG, Hawrylowicz CM, Holman M, Keeler KD. Activation and proliferation signals in mouse B cells. III. Intact (IGG) anti-immunoglobulin antibodies activate B cells but inhibit induction of DNA synthesis. Immunology. 1984;53: 693-701. [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips NE, Parker DC. Cross-linking of B lymphocyte Fc gamma receptors and membrane immunoglobulin inhibits anti-immunoglobulin-induced blastogenesis. J Immunol. 1984;132: 627-632. [PubMed] [Google Scholar]
- 36.Carver DJ, Aman MJ, Ravichandran KS. SHIP inhibits Akt activation in B cells through regulation of Akt membrane localization. Blood. 2000;96: 1449-1456. [PubMed] [Google Scholar]
- 37.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90: 293-301. [DOI] [PubMed] [Google Scholar]
- 38.Slekar KH, Kosman DJ, Culotta VC. The yeast copper/zinc superoxide dismutase and the pentose phosphate pathway play overlapping roles in oxidative stress protection. J Biol Chem. 1996; 271: 28831-28836. [DOI] [PubMed] [Google Scholar]
- 39.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270: 296-299. [DOI] [PubMed] [Google Scholar]
- 40.Le Goffe C, Vallette G, Charrier L, et al. Metabolic control of resistance of human epithelial cells to H2O2 and NO stresses. Biochem J. 2002;364: 349-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995;14: 5209-5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian WN, Braunstein LD, Pang J, et al. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem. 1998;273: 10609-10617. [DOI] [PubMed] [Google Scholar]
- 43.Bental M, Deutsch C. Metabolic changes in activated T cells: an NMR study of human peripheral blood lymphocytes. Magn Reson Med. 1993;29: 317-326. [DOI] [PubMed] [Google Scholar]
- 44.Hunt TK, Zederfeldt B, Goldstick TK. Oxygen and healing. Am J Surg. 1969;118: 521-525. [DOI] [PubMed] [Google Scholar]
- 45.Newsholme EA, Board M. Application of metabolic-control logic to fuel utilization and its significance in tumor cells. Adv Enzyme Regul. 1991; 31: 225-246. [DOI] [PubMed] [Google Scholar]
- 46.Baggetto LG. Deviant energetic metabolism of glycolytic cancer cells. Biochimie. 1992;74: 959-974. [DOI] [PubMed] [Google Scholar]
- 47.Zambell KL, Fitch MD, Fleming SE. Acetate and butyrate are the major substrates for de novo lipogenesis in rat colonic epithelial cells. J Nutr. 2003;133: 3509-3515. [DOI] [PubMed] [Google Scholar]
- 48.Zhu X, Hart R, Chang MS, et al. Analysis of the major patterns of B cell gene expression changes in response to short-term stimulation with 33 single ligands. J Immunol. 2004;173: 7141-7149. [DOI] [PubMed] [Google Scholar]
- 49.Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. 1997;272: 17269-17275. [DOI] [PubMed] [Google Scholar]
- 50.Taha C, Liu, Z, Jin J, Al-Hasani H, Sonenberg N, Klip A. Opposite translational control of GLUT1 and GLUT4 glucose transporter mRNAs in response to insulin. Role of mammalian target of rapamycin, protein kinase B, and phosphatidylinositol 3-kinase in GLUT1 mRNA translation. J Biol Chem. 1999;274: 33085-33091. [DOI] [PubMed] [Google Scholar]
- 51.Ueki K, Yamamoto-Honda R, Kaburagi Y, et al. Potential role of protein kinase B in insulin-induced glucose transport, glycogen synthesis, and protein synthesis. J Biol Chem. 1998;273: 5315-5322. [DOI] [PubMed] [Google Scholar]
- 52.Watson RT, Kanzaki M, Pessin JE. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev. 2004;25: 177-204. [DOI] [PubMed] [Google Scholar]