

Abstract
Oncogenic RAS expression occurs in up to 40% of multiple myeloma (MM) cases and correlates with aggressive disease. Since activated RAS induces cyclooxygenase-2 (cox-2) expression in other tumor models, we tested a role for cox-2 in mutant RAS–containing MM cells. We used the ANBL-6 isogenic MM cell lines in which the IL-6–dependent parental line becomes cytokine independent following transfection with mutated N-RAS or K-RAS. Both mutated N-RAS– and K-RAS–expressing ANBL-6 cells demonstrated a selective up-regulation of cox-2 expression and enhanced secretion of PGE2, a product of cox-2. Furthermore, in 3 primary marrow specimens, which contained MM cells expressing mutated RAS, 15% to 40% of tumor cells were positive for cox-2 expression by immunohistochemistry. We used cox-2 inhibitors, NS398 and celecoxib, and neutralizing anti-PGE2 antibody to test whether cox-2/PGE2 was involved in the aggressive phenotype of MM ANBL-6 cells containing mutated RAS. Although these interventions had no effect on IL-6–independent growth or adhesion to marrow stromal cells, they significantly inhibited the enhanced binding of mutant RAS– containing MM cells to fibronectin and the enhanced resistance to melphalan. These results indicate a selective induction of cox-2 in MM cells containing RAS mutations, which results in heightened binding to extracellular matrix protein and chemotherapeutic drug resistance.
Introduction
Oncogenic mutations of RAS occur in 30% to 40% of multiple myeloma (MM) patients and are associated with progressive disease, resistance to therapy, and poor survival.1-7 Previous work in other cell models indicates that activated RAS can induce expression of cyclooxygenase-2 (cox-2).8,9 Thus, cox-2 expression may play a role in this aggressive phenotype associated with activating RAS mutations in MM. Furthermore, a recent study10 documented cox-2 expression in malignant plasma cells obtained from a significant percentage of patients, and expression correlated with aggressive disease and poor outcome.
To address the potential role of cox-2 in mutant RAS–containing MM cells, we used the ANBL-6 model, whereby IL-6–dependent MM cells were stably transfected with mutant N-RAS or K-RAS genes or an empty vector.6,7 The RAS-transfected isogenic MM cells demonstrate progressive IL-6–independent growth and resistance to chemotherapeutic agents.6,7,11 Therefore, these cells represent a good model for mechanistic studies on how activated RAS achieves this aggressive phenotype. Our results show a specific induction of cox-2 expression in the RAS-transfected MM cells and an implication of cox-2 expression in the enhanced binding to fibronectin and chemoresistance in these cells.
Materials and methods
Cell lines
Isogenic ANBL-6 cell lines were graciously provided by Dr Brian Van Ness (University of Minnesota). The cells were generated as previously described.6,7 Briefly, the IL-6–dependent ANBL-6 cell line was stably infected with retrovirus expressing either a mutated N-RAS gene (N-RAS cells), a mutated K-RAS gene (K-RAS cells), or an empty vector (wild-type [WT] cells). All 3 lines were maintained in RPMI-1640 media with 10% FBS and 100 U/mL recombinant IL-6. The KM102 bone marrow stromal cell line12 was a generous gift from Dr Michio Kawano (Yamaguchi University, Japan). The KM102 cells were maintained in RPMI-1640 + 10% FBS.
Reagents
NS398 was purchased from Cayman Chemicals (Ann Arbor, MI), dissolved in DMSO, and stored at –20°C until use. Celecoxib was purchased from Searle Pfizer (New York, NY) as 200-mg capsules. The drug was removed from capsules, dissolved in DMSO, and stored at –20°C until use. Purified cox-2 (ovine) electrophoresis standard and PGE2 were purchased from Cayman Chemicals and recombinant IL-6, from R&D Systems (Minneapolis, MN). The following were sources for antibodies: cox-2 and cox-1 (Cayman Chemicals); 2B5 cox-2 neutralizing mAb (Pharmacia, Peapack, NJ); CD44 and α-actin (Santa Cruz Biotechnology, Santa Cruz, CA); α4 and α5 integrin (BD Pharmingen, San Diego, CA); β1 integrin (Chemicon International, Temecula, CA); phospho-Erk1/2 (Thr202/Tyr204) and total Erk (Cell Signaling Technology, Beverly, MA); and α-mouse IgG-HRP (Amersham Biosciences, Piscataway, NJ).
Western analysis
Protein was extracted with SDS lysis buffer containing leupeptin, aprotinin, and PMSF, separated with SDS polyacrylamide, and transferred onto PVDF membrane. After blocking with 5% milk in TBST (Tris-buffered saline, Tween 20), the membrane was probed with diluted primary antibodies overnight at 4°C. The membrane was then washed 3 × 10 minutes with TBST, incubated with HRP-conjugated secondary antibodies for 1 hour at room temperature (RT), washed 3 × 10 minutes with TBST, and developed with enhanced chemiluminescence (ECL) Plus Western Blotting Detection System (Amersham Biosciences).
PGE2 ELISA
Cells were cultured for the indicated time and supernatant was collected. The cells were then counted and total protein was extracted and quantified. Enzyme-linked immunosorbent assay (ELISA) for PGE2 was performed with supernatant samples in duplicate according to the manufacturer's (Cayman Chemicals) protocol. Total PGE2 was normalized to total protein from each sample.
Immunohistochemistry
Immunohistochemical staining was performed on formalin-fixed biopsies using Vector ABC Elite Kit and reagents (Vector Laboratories, Burlingame, CA) as previously described.13 Briefly, tissue sections were stained for one hour with 0.5 μg/mL goat anti–human cox-2 polyclonal IgG (Santa Cruz Biotechnology) followed by horse anti–goat IgG-biotin, avidin–biotin peroxidase complex (ABC), and 3,3′diaminobenzidine (DAB) substrate. Negative controls included incubation with a nonimmune goat anti–human polyclonal IgG (Santa Cruz Biotechnology). Positive controls included previously characterized lung adenocarcinoma samples with high cox-2 expression.13 Images were visualized under a Nikon Microphot-SA microscope equipped with a Plan-Apochromat 40 ×/0.95 objective lens (Nikon, Melville, NY), and were captured with a SPOT-RT digital camera (Diagnostic Instruments, McHenry, IL). MetaMorph 6.1 software (Universal Imaging, West Chester, PA) was used to acquire digital images, and Adobe Photoshop 7.0 software (Adobe Systems, San Jose, CA) was used to prepare them for final publication.
Cox-2 expression and IL-6
ANBL-6 cells (1-2 × 106) were cultured with or without IL-6 (1000 U/mL) for 0 and 48 hours. Cell lysates were collected at each time point and Western analysis for cox-2 was performed.
In vitro cell proliferation
ANBL-6 isogenic cell lines were first depleted of IL-6 overnight. Cells (2-2.5 × 105 cells in 2 mL) were then cultured in RPM1 + 10% FBS containing either NS398 (10 μM), celecoxib (15 μM), or an equal volume of DMSO. After every 3 days of culture, the media were changed and fresh drugs were added. Live cells were quantified by trypan blue exclusion at each time point and each group was assayed in duplicate.
Cell adhesion to fibronectin (FN)
In a 96-well plate, 50 μLof40 μg/mL of FN or BSA were pipetted into each well and allowed to air-dry overnight at room temperature. The following day, cells were resuspended in serum-free RPMI media and 105 cells in 100 μL were added to each well. Each sample group was run in either triplicate or quadruplicate. After 2 hours in culture at 37°C, nonadherent and weakly adhered cells were removed by inverting the plate to remove media, PBS was added to wells, and this process was repeated 2 more times. The remaining adherent cells were fixed with 3.7% formaldehyde for 20 minutes, washed with PBS, and stained with 0.2% crystal violet solution for 2 hours to 24 hours at RT. Stained cells were then washed with copious amount of distilled water and air-dried overnight at RT. After adding 100 μL of 2% SDS to each well, the plate was rotated for 30 to 120 minutes to dissolve the dye, and optical densities (ODs) were read at 570 nm in a 96-well plate reader.
Cell adhesion to bone marrow stromal cells (BMSCs)
KM102 BMSCs (2 × 104) were added in 100 μL to each well of a 96-well plate and cultured overnight. The media for the KM102 cells were removed and 105 myeloma cells were then resuspended in 100 μL RPMI (serum free) and added to the KM102 cells. Each sample group was performed in triplicate or quadruplicate. After 2 hours of culture at 37°C, the nonadherent myeloma cells were removed by inverting the plate and PBS was added. We repeated the PBS wash 2 more times. Adherent cells were then fixed with 3.7% formaldehyde, washed with PBS, and stained with 0.2% crystal violet for 2 to 24 hours at RT. The crystal violet was then washed off multiple times with distilled water and plates were air-dried overnight at RT. The dye was then solubilized with 2% SDS for 30 to 120 minutes by rotating plate and ODs were measured at 570 nm in a 96-well plate reader. The ODs resulting from KM102 BMSCs cultured alone were subtracted from each group.
Chemotherapeutic drug treatment
Cells (3-5 × 105) were cultured in 2.5 mL in full growth media with 10 μM NS398 overnight at 37°C for 24 hours. Melphalan was then added, and after an additional 24 or 48 hours in culture, nonviable cells were quantified by trypan blue uptake. In the case of using PGE2 neutralizing antibodies, 5 μg/mL PGE2 neutralizing antibodies14 or mouse IgG control was added 24 hours prior to drug treatment. Similarly, wild-type cells were cultured with 10 and 20 μM PGE2 24 hours before melphalan addition.
Identification of RAS mutations in primary myeloma cells
Bone marrow cells were first separated by Ficoll-Hypaque density centrifugation and plasma cells were further isolated by negative selection (> 98% purity). The denaturing gradient gel electrophoresis (DGGE) technique in combination with the polymerase chain reaction (PCR) was used to screen for mutations at codons 12, 13, or 61 in N-RAS or K-RAS genes using the amplification primers described by Nedergaard et al.15 Preliminary experiments with cell lines allowed optimization of urea-formamide concentrations and electrophoresis conditions such that mutations could be identified in heterogenous myeloma specimens containing as little as 5% tumor cells. Any RAS mutations were confirmed by sequencing.
Statistics
The t test was used to determine significance of differences between groups.
Results
Oncogenic RAS induces cox-2 expression and PGE2 secretion
We initially investigated whether expression of activated RAS mutants induces cox-2 expression. For this analysis, we exploited the ANBL-6 MM model of isogenic cell lines where the IL-6–dependent parental line is stably transfected with an empty vector or vectors expressing activated mutated N-RAS or K-RAS genes. All 3 cell lines were cultured in the absence of IL-6 for 48 or 72 hours, and protein extracts were collected and analyzed. Although empty vector–transfected wild-type ANBL-6 cells will ultimately cease growth and die in the absence of IL-6, after this short duration of 48 to 72 hours in IL-6–depleted media, cell viability was still comparable (> 90%) between the 3 isogenic cell lines. Immunoblotting with a cox-2–specific antibody demonstrated a marked increase in cox-2 protein levels in the mutant N-RAS– and K-RAS– expressing cells compared with the wild-type cells (Figure 1A). As a positive cox-2 control, purified cox-2 electrophoresis standard (Cayman Chemicals) was run in parallel. In addition, this increase was specific for cox-2 as comparable cox-1 protein levels were detected in all cell lines (Figure 1B).
Figure 1.
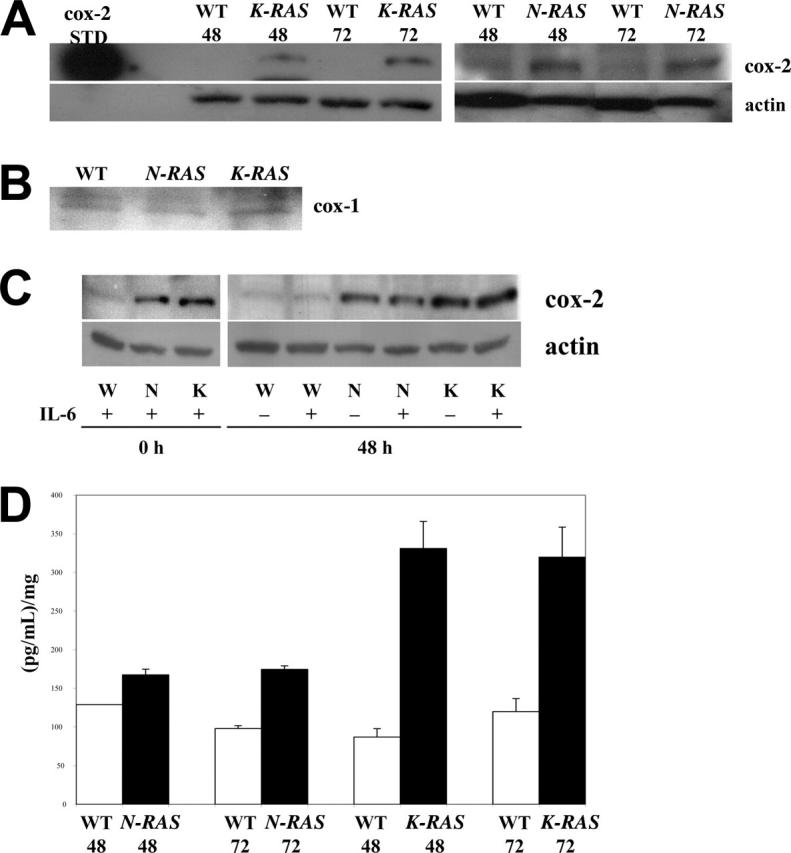
Mutated RAS induces cox-2 expression and activation in MM cells. (A) Western analysis for cox-2 expression in wild-type (WT), N-RAS, and K-RAS MM cells after 48 and 72 hours of IL-6 depletion. Purified cox-2 electrophoresis standard served as positive cox-2 control. (B) cox-1 expression in WT, N-RAS, and K-RAS cells after 48 hours of IL-6 depletion. (C) Western analysis of cox-2 expression in WT, N-RAS, and K-RAS cells cultured for 0 or 48 hours with or without IL-6 (1000 U/mL). (D) Conditioned media were analyzed for PGE2 level. The concentration of PGE2 is reported as picogram of PGE2/mL of media/mg of total protein lysate, mean ± SD.
Although the viability in the IL-6–depleted wild-type cells studied in Figure 1A was comparable with that of the mutated RAS–containing cells, it was possible that cytokine depletion prevented cox-2 expression. Furthermore, prior studies in other cell models16,17 demonstrate that IL-6 is capable of cox-2 induction. Thus, to test a possible role for IL-6 in our ANBL-6 model, the 3 isogenic cell lines were assayed for cox-2 expression in the presence or absence of IL-6 exposure. As shown in Figure 1C, IL-6 had no effect on expression of cox-2 in these cells. Thus, the difference in cox-2 expression between the cell lines is not due to a nonspecific differential effect of cytokine depletion on the health of the cells. The results also rule out the possibility that mutant RAS–containing ANBL-6 cells overexpress cox-2 secondarily to induction of an IL-6 autocrine state. It appears that, at least in the ANBL-6 model, IL-6 does not induce cox-2 expression.
To further support the notion of up-regulated cox-2 expression in mutant RAS–containing MM cells, we assayed conditioned media from the cultures for prostaglandin E2 (PGE2), a product of arachidonic acid metabolism resulting from the enzymatic action of cox-2. The results showed a corresponding increase of PGE2 in the conditioned media obtained from the mutated RAS–containing cells at both 48 and 72 hours after IL-6 depletion (Figure 1D). An increase in PGE2 production was significant (P < .05) for both mutated cell lines at 72 hours, although the increase observed in K-RAS cells was significantly larger than that seen in N-RAS cells. Thus, in the ANBL-6 model, mutant RAS strongly correlates with an increase expression and activity of cox-2.
Activating RAS mutations were also identified in isolated primary myeloma cells from 3 separate patients by the DGGE technique and confirmed by sequencing. These included point mutations at codon 12 of N-RAS in one patient, codon 13 of N-RAS in a second patient, and codon 12 of K-RAS in a third. These mutations were transversions that would have resulted in amino acid changes that have previously been described as activating mutations. To ascertain cox-2 expression in these tumor cells, the bone marrow biopsy sections were stained with a cox-2 antibody. Approximately, 15% to 40% of plasma cells from these marrows stained for cox-2 expression. Figure 2 shows examples of cox-2–positive plasma cells from these 3 patients. In contrast, cox-2 expression was not detected in plasma cells of 4 patients with monoclonal gammopathy of undetermined significance (MGUS). An example of one of these patients is shown in Figure 2D.
Figure 2.
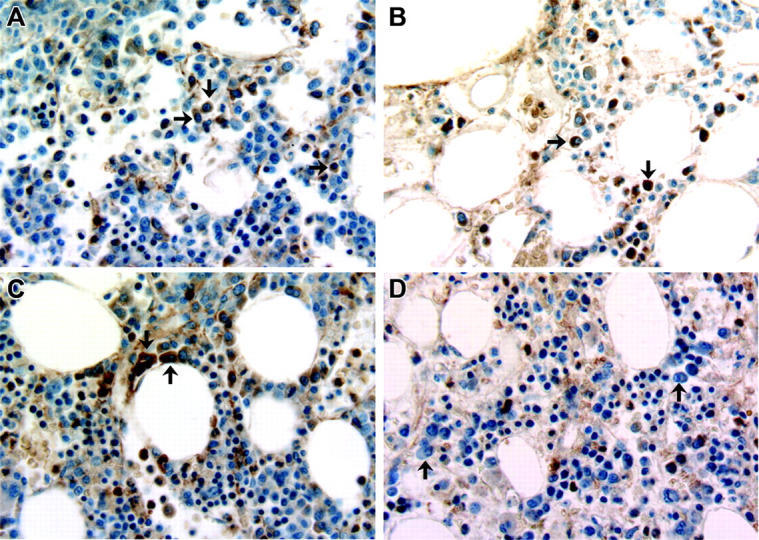
Immunohistochemical detection of cox-2 expression in primary MM cells. Bone marrow samples from 4 patients stained for cox-2 expression. (A-C) Marrow biopsies from 3 patients with myeloma that contains mutated N-RAS or K-RAS. (D) Marrow biopsy from a patient with monoclonal gammopathy of undetermined significance (MGUS). A few plasma cells in each biopsy are depicted with black arrows. Original magnification for all panels, × 40.
Cox-2 activity is not required for progressive IL-6–independent growth of mutated RAS–containing MM cells
Previous results demonstrated that expression of mutated N-RAS and K-RAS in ANBL-6 MM cells allows for progressive IL-6–independent growth.6,7,18 Thus, we first tested whether this growth relies on cox-2 activity by comparing in vitro growth in the presence or absence of the selective cox-2 inhibitors NS398 or celecoxib. In preliminary experiments, we demonstrated that optimal inhibition of cox-2 activity in mutant cells could be achieved with concentrations as low as 10 μM NS398 and 15 μM celecoxib (data not shown). We thus depleted mutant N-RAS and K-RAS cells of IL-6 and cultured them with or without NS398 at 10 μM or celecoxib at 15 μM. Fresh drug was re-added after the first 72 hours of culture. Cell growth and survival were assessed for up to 6 days in culture. As shown in Figure 3, mutant RAS MM cells cultured without inhibitors (DMSO-treated controls) demonstrated a logarithmic growth in the absence of IL-6 as has been previously described.6,7,18 Addition of either NS398 or celecoxib to the culture to inhibit cox-2 had no significant effect on proliferation. Thus, up-regulated cox-2 activity is not required for progressive in vitro growth of oncogenic RAS–expressing ANBL-6 cells.
Figure 3.

Cox-2 does not regulate IL-6–independent growth of mutant RAS–containing MM cells. Cells were cultured in full growth media (without IL-6) with DMSO, 10 μM NS398, or 15 μM celecoxib. Live cells were quantified by trypan blue exclusion, and the results are presented as number of viable cells (× 105) for mutant N-RAS (A) and mutant K-RAS cells (B). This experiment was performed twice (n = 2) with each sample group in duplicate, and these data are representative of both experiments. Error bars indicate standard deviation of duplicate experiments.
Effects of oncogenic RAS on MM-cell adhesion
Since cox-2 has also been reported to affect cell-cell and cell-ECM (extracellular matrix) interactions,19,20 we tested its effects on MM cell adhesion to fibronectin (FN) and bone marrow stromal cells (BMSCs). We first compared adhesion of low cox-2–expressing WT ANBL-6 cells to the high cox-2–expressing mutant cell lines. Cells were first depleted of IL-6 for 48 to 72 hours to negate the potential effect of IL-6 on adhesion. WT, oncogenic N-RAS, or oncogenic K-RAS cells were then resuspended in serum-free media (SFM), added to FN-coated wells versus BSA-coated wells (control) or confluent BMSCs layers (the KM102 stromal cell line), and cultured for 2 hours at 37°C. After removing nonadherent cells, adherent cells were enumerated by staining with crystal violet. As shown in Figure 4A-B, the adhesion of mutant N-RAS and K-RAS MM cells was significantly greater (P < .05) than WT cells when bound to both FN and BMSCs.
Figure 4.

Mutant RAS–containing MM cells demonstrate enhanced binding to fibronectin (FN) and stromal cells. (A) Adhesion to fibronectin (FN) was compared between WT, N-RAS, and K-RAS ANBL-6 cells, and results are presented as OD (570 nm) of crystal violet–stained cells, mean ± SD, n = 3. □ indicates adhesion to BSA, a nonspecific substrate; ▪, adhesion to FN. (B) WT, N-RAS, and K-RAS ANBL-6 cells were adhered to monolayers of the BMSC cell line KM102, and relative cell adhesion to BMSCs was quantified as OD of crystal violet–stained cells, mean ± SD, n = 3.
Cox-2 and PGE2 regulate adhesion to fibronectin
We next examined whether enhanced binding of mutated RAS–containing MM cells to FN is mediated through cox-2. We cultured oncogenic RAS cells in the absence of IL-6 and with or without the cox-2–specific inhibitors NS398 and celecoxib for 72 hours. NS398 was used at concentrations of 5 μM and 10 μM, while celecoxib was added to a final concentration of 5 μM and 15 μM. Equal amounts of DMSO were used as a drug vehicle control. As in the previous experiment, there were no observable effects on growth rate or cell morphology resulting from exposure to NS398 or celecoxib. The cells were then assayed for adhesion to FN. With corresponding adhesion of WT cells arbitrarily defined as 100%, Figure 5A for N-RAS and 5B for K-RAS demonstrate that both cox-2 inhibitors are capable of curtailing adhesion to FN at concentrations as low as 5 μM (Figure 5A-B dark columns). The resulting binding to FN in the presence of the inhibitors was comparable with that of WT cells, which express wild-type RAS and a small amount of cox-2. A similar exposure to the cox-2 inhibitor celecoxib had no effect on the constitutive phosphorylation of Erk (Figure 5C) in mutant cells. Constitutive Erk phosphorylation in these MM cells is a direct downstream effect of mutated RAS.18 These data support the notion that the inhibition of binding to FN induced by the cox-2 inhibitors is due to specific effects on cox-2 function rather than nonspecific effects or inhibitory effects on RAS.
Figure 5.

Cox-2 regulates adhesion of mutant RAS MM cells to fibronectin. (A) N-RAS–containing and (B) K-RAS–containing ANBL-6 cells were treated with cox-2–specific inhibitors, NS398 or celecoxib, at the concentration shown below the bars (in μM) or DMSO control vehicle for 72 hours and then assayed for adhesion to FN (▪) or BMSCs (□). Adhesion is expressed as binding relative to WT cells where wild-type cells were arbitrarily determined to be 100%. Results reflect average binding ODs of quadruplicate samples. The experiment was repeated once with identical results. (C) Mutant K-RAS cells were cultured with celecoxib for 48 or 72 hours and protein lysates were collected. Western analysis on the lysates was performed using phospho-specific Erk (Thr202/Tyr204) antibody and total Erk antibody. No effect of celecoxib on Erk phosphorylation was observed. (D) Both N-RAS– and K-RAS–containing ANBL-6 cells were cultured in the presence of 5 μg/mL α-PGE2 neutralizing antibodies or mouse IgG isotype control for 72 hours and then assayed for adhesion to FN (▪) or BMSCs (□). The results are reported in percentage as relative adhesion of untreated N-RAS or K-RAS ANBL-6 cells cultured in parallel.
To further confirm that cox-2 regulates RAS-dependent binding to FN, we used a PGE2 neutralizing antibody to test whether inhibiting PGE2, a product of cox-2, can prevent the enhanced binding to FN. We cultured oncogenic N-RAS– and K-RAS–expressing ANBL-6 with α-PGE2 neutralizing antibodies or an equal concentration of IgG isotype control for 72 hours and assayed for adhesion to FN. As shown in Figure 5D, N-RAS and K-RAS cell binding to FN (dark columns) decreased markedly in the presence of the α-PGE2 antibodies, while cells cultured with IgG control antibody were not affected. The results show the percent of control (no antibody) of an experiment performed with quadruplicate samples where the SDs were all less than 5%. The differences between the α-PGE2–treated cells and control IgG–treated cells were significant (P < .05). These results, with 2 separate cox-2 inhibitors and anti-PGE2 antibodies, collectively indicate that the enhanced binding to FN of MM cells containing mutant RAS is mediated by cox-2.
Cox-2 activity has no effect on adhesion to BMSCs
Similar studies were undertaken to determine if cox-2 also regulates adhesion of oncogenic RAS cells to BMSCs. WT, N-RAS, and K-RAS cells were similarly depleted of IL-6 and cells were then challenged with DMSO (control) or cox-2 inhibitors, and, after 72 hours, adherence to BMSCs was assessed. As shown in Figure 5A and 5B (open columns), respectively, adherence of N-RAS and K-RAS cells to BMSCs was unaffected by NS398 or celecoxib used at the same concentrations that significantly inhibit PGE2 production and inhibit binding to FN. Furthermore, treatment with α-PGE2 neutralizing antibodies used in the same concentration that inhibited binding to FN (Figure 5D dark columns) had no effect on N-RAS or K-RAS cells binding to KM102 stromal cells (Figure 5D open columns).
Effect of cox-2 on expression of MM-cell FN receptors
One possible mechanism by which cox-2 mediates adhesion to FN is via its effect on expression of FN receptors. Thus, we performed assays to measure the expression of known FN receptors that are reported to mediate MM cell interactions with the extracellular matrix.21,22 We compared the expression of CD44 and α4, α5, and β1 integrins between WT and mutant RAS–containing cells by Western or flow cytometry analysis. Our result showed by Western analysis that CD44 protein expression was up-regulated in both mutant RAS cells (Figure 6A), but β1 integrin expression was not enhanced in the mutant RAS–expressing cells (Figure 6B). In addition, flow cytometric analysis demonstrated comparable high α4 and low or no α5 protein expression in the WT and mutant RAS cells (Figure 6C).
Figure 6.

Protein expression of FN receptors in ANBL-6 cells. Protein expression was compared by Western analysis between the WT, N-RAS, and K-RAS cells for (A) CD44 and (B) β1. (C) Flow chart for isotype control, and α4 and α5 integrins in WT, N-RAS, and K-RAS cells. Fluorescent intensity is represented by the x-axis, and the percent of positive cells is indicated at the bottom right corner of each chart.
To determine if cox-2 regulates the expression of CD44, we compared CD44 expression in the presence and absence of cox-2 inhibitors. At a dosage of 10 μM cox-2 inhibitors, CD44 protein level was modestly down-regulated in K-RAS cells by NS398 and celecoxib, and in N-RAS by NS398 (data not shown). However NS398 did not have a significant effect on CD44 protein expression in either mutant N-RAS or K-RAS cells when used at 5 μM, which is the same concentration that was able to inhibit both N-RAS and K-RAS cell binding to FN, thus suggesting that cox-2–dependent adhesion to FN cannot be completely explained by an up-regulation of CD44 expression.
Oncogenic RAS–mediated chemotherapeutic drug resistance is partially regulated by cox-2
Previous work has shown that mutant N-RAS– and K-RAS–transfected ANBL-6 cells are resistant to melphalan.11 Since cox-2 can induce chemoresistance in other tumor models,23,24 we tested this notion in our MM cell lines. We first compared drug sensitivity of WT and mutant RAS cells by evaluating the response of each cell line to melphalan-induced cell death. We quantified the percentage of cell death in each cell line challenged with 0 μM, 5 μM, or 20 μM melphalan. Although a high concentration of melphalan (20 μM) exerted comparable amounts of cell death in the WT, N-RAS, and K-RAS cells, mutant RAS–containing MM cells were significantly less sensitive to a concentration of 5 μM (Figure 7A). This is consistent with previous work reporting a RAS-mediated protection against chemotherapeutic drug–induced cell death.11 We next asked whether protection against melphalan-induced cell death in mutant RAS cells is mediated by cox-2. We cultured mutant RAS cells with melphalan alone (5 μM), the cox-2 inhibitor NS398 alone (10 μM), or melphalan + NS398. Although NS398 by itself had little effect on cell death, it significantly sensitized the mutant RAS cells to melphalan-induced cytotoxicity (Figure 7B).
Figure 7.

Resistance to melphalan in mutant RAS MM cells is partially regulated by cox-2. (A) WT, N-RAS–, and K-RAS–containing ANBL-6 cells were cultured with 0μM, 5 μM, and 20 μM melphalan for 48 hours and nonviable cells were then quantified. The results are reported as percent of nonviable cells in each population, mean ± SE of 3 experiments (except the 20-μM treated group, which was performed once). Results from WT cells challenged with 5 μM melphalan are significantly different from N-RAS and K-RAS cells (P < .05). (B) Oncogenic N-RAS– and K-RAS–containing ANBL-6 cells were cultured with or without NS398 (10 μM) for 24 hours and then challenged with melphalan (5 μM). After an additional 48 hours in culture, nonviable cells were quantified. Results are presented as percent cell death, mean ± SE of 4 separate experiments. *Significantly different from corresponding groups without added cox-2 inhibitor (P < .05). (C) Mutant N-RAS and K-RAS cells were cultured without antibody or with 5 μg/mL mouse IgG or 5 μg/mL PGE2 mouse neutralizing antibodies for 24 hours and melphalan was then added at 5 μM. The cells were maintained for an additional 48 hours and the percent of cell death was determined. The data are reported as the percent cell death in each population above untreated group. (D) WT cells were cultured without IL-6 and with or without PGE2 (μM) for 24 hours and 5 μM melphalan was then added. After 48 hours of melphalan treatment, nonviable cells were quantified with trypan blue uptake, mean ± SD, n = 2.
We also tested the notion that cox-2 participates in drug resistance by using the anti-PGE2 neutralizing antibodies. After exposure to anti-PGE2 or IgG isotype control antibody for 24 hours, melphalan was added for an additional 48 hours and cell death was quantified. As shown in Figure 7C, inhibition of PGE2 with neutralizing antibodies significantly reversed melphalan drug resistance in both mutant N-RAS and K-RAS cells. In these experiments, α-PGE2 alone had a small but reproducible adverse effect on N-RAS cells for unknown reasons. However, the increase in cell death induced by the combination of α-PGE2 and melphalan was significantly greater than the sum of the 2 agents used alone. These data indicate that N-RAS– and K-RAS–induced melphalan drug resistance in ANBL-6 is, at least in part, regulated by cox-2 and PGE2.
To further support a role of cox-2 in drug resistance, we tested whether exogenous PGE2 would protect wild-type ANBL-6 cells against melphalan cytotoxicity. Melphalan was added to WT cells after 24 hours in culture with PGE2, and, after 48 hours of melphalan exposure, nonviable cells were quantified by trypan blue uptake. As shown in Figure 7D, PGE2 was able to partially protect wild-type cells against melphalan-induced cell death, further supporting a protective effect of cox-2 and PGE2 against melphalan cytotoxicity in ANBL-6 cells.
Discussion
Our results indicate that oncogenic RAS induces cox-2 expression and secretion of PGE2 in MM cells. Cox-2 was selectively induced in the ANBL-6 MM cell line following stable transfection of both mutated N-RAS and K-RAS alleles. In addition, immunohistochemistry identified cox-2 expression in a significant percentage of malignant plasma cells from 3 patients that also expressed mutated RAS. It is unclear why cox-2 expression in these primary myeloma cells was so heterogenous with many plasma cells negative for immunostaining. It is possible that oncogenic RAS expression was also heterogenous in the marrow as has been suggested by others,25 or, just as likely, other mechanisms also regulate cox-2 expression within these bone marrows. It is of interest that when cox-2 expression was previously identified in a large cohort of patients,10 it was also expressed in a heterogenous manner. Although the percentage of myeloma samples that expressed cox-2 in this previous study (31% at diagnosis and 47% with relapsed/refractory disease) approximates the incidence of RAS mutation in myeloma,1 it is yet unknown whether additional RAS-independent conditions can also mediate cox-2 expression.
Although the ANBL-6 model did not indicate an ability of IL-6 to induce cox-2 expression in our experiments, prior work in other models16,17 did indicate a potential regulatory role of IL-6. Thus, IL-6, or other myeloma growth factors such as IGF-1, could provide a RAS-independent mechanism of cox-2 up-regulation.
There are several possible pathways by which oncogenic RAS may induce cox-2 in MM cells. In Rat-1 fibroblasts, oncogenic H-RAS induced cox-2 via its activation of the RAF-MEK-Erk pathway.8 In a second study9 in rat intestinal epithelial cells (IEC-6), mutated K-RAS induced cox-2 transcription and mRNA stabilization. The up-regulated transcription was due to RAS signaling through Erk, and RNA stabilization was due to Erk as well as AKT activity. In our previous work,18 activated mutations of RAS in myeloma cells induced constitutive activity of both Erk and Akt. These effects may, thus, explain the resulting up-regulated cox-2 expression. Although our data clearly demonstrate an enhanced binding of mutated RAS–expressing cells to FN in a cox-2–dependent manner, the mechanism of this effect remains unclear. In other cell models, cox-2 modulates cell adhesion to ECM by regulating expression of β120 and α526 integrins or CD44.27 To address this, we tested expression of these molecules in our isogenic ANBL-6 cell lines. Only CD44 demonstrated up-regulated expression in mutated RAS MM cells and modest inhibition when cox-2 inhibitors were used. However, concentrations of cox-2 inhibitors that depressed binding to FN (5 μM) were not capable of curtailing CD44 expression. Furthermore, we have not been able to confirm a direct role of CD44 in binding of mutant MM cells to FN (data not presented). In addition, although blocking antibodies to α4 and β1 integrins significantly inhibited binding to FN of mutant RAS MM cells (data not shown), their expression levels were not higher than that of WT cells. Thus, the cox-2–dependent enhanced binding of mutant RAS MM cells to FN is complex. It is possible that cox-2 modulates α4/β1 avidity or affinity, and CD44 may be involved by interacting with this integrin.
In contrast to results from FN-binding assays, cox-2 did not regulate the enhanced binding of mutated RAS–containing MM cells to BMSCs. A series of interactions between multiple receptors and counterreceptors, such as CD44, integrin, cadherin, and others, can facilitate adhesion of MM cells to BMSCs.28-31 Mutated RAS may activate many of these regulatory pathways, some of which are cox-2 independent. Thus, inhibition of cox-2 in MM cells may prevent some cell-cell interactions, but this may be insufficient to suppress overall binding to BMSCs.
Melphalan resistance mediated by oncogenic RAS is partially regulated by cox-2. NS398, used at a nontoxic dosage, and anti-PGE2 neutralizing antibody significantly enhanced the killing of mutant RAS–containing cells by melphalan. Furthermore, addition of PGE2 to wild-type ANBL-6 MM cells resulted in significant resistance. These results are similar to those in other tumor models where cox-2 inhibition significantly increased the cytoreductive effects of chemotherapeutic drugs.23,24,32 A recent report also supports this contention in myeloma where the combinatory treatment with the cox-2 inhibitor NS398 and either dexamethasone or thalidomide caused a synergistic effect in RPMI8226 MM cells.33 These data suggest the clinical potential of combining cox-2 inhibitors with chemotherapeutic agents in MM, and a recent phase 2 trial34 suggests potential efficacy. Although a recent study suggests some cox-2 inhibitors may inhibit myeloma growth via cox-2–independent mechanisms,35 the ability of anti-PGE2–blocking antibody to comparably sensitize our MM cells to melphalan confirms a cox-2 effect.
In summary, our results indicate a selective up-regulated expression and activity of cox-2 in MM cells containing activating RAS mutations. Heightened cox-2 activity may play a role in the enhanced ability of these cells to bind to extracellular matrix, migrate and disseminate, and resist chemotherapeutic agents. A recent study confirming the involvement of cox-2 and PGE2 in osteoclastogenesis36 suggests that up-regulated activity in mutant RAS myeloma could also contribute to progressive lytic bone disease. cox-2 inhibitors may, thus, be worthwhile agents for future therapy.
Acknowledgments
We thank Myrna Fisher (Greater Los Angeles VA Healthcare System) for performing the flow cytometry analysis.
Prepublished online as Blood First Edition Paper, February 23, 2006; DOI 10.1182/blood-2005-09-3926.
Supported by funds from the Multiple Myeloma Research Foundation, the Department of Veterans Affairs, and the following grants from the National Institutes of Health: Early Detection Research Network National Cancer Institute CA-86366, CA 111851, CA 96920, and CA 111448.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Neri A, Murphy JP, Cro L, et al. Ras oncogene mutation in multiple myeloma. J Exp Med. 1989; 170: 1715-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corradini P, Ladetto M, Voena C, et al. Mutational activation of N- and K-ras oncogenes in plasma cell dyscrasias. Blood. 1993;81: 2708-2713. [PubMed] [Google Scholar]
- 3.Paquette RL, Berenson J, Lichtenstein A, McCormick F, Koeffler HP. Oncogenes in multiple myeloma: point mutation of N-ras. Oncogene. 1990; 5: 1659-1663. [PubMed] [Google Scholar]
- 4.Portier M, Moles JP, Mazars GR, et al. p53 and RAS gene mutations in multiple myeloma. Oncogene. 1992;7: 2539-2543. [PubMed] [Google Scholar]
- 5.Liu P, Leong T, Quam L, et al. Activating mutations of N- and K-ras in multiple myeloma show different clinical associations: analysis of the Eastern Cooperative Oncology Group Phase III Trial. Blood. 1996;88: 2699-2706. [PubMed] [Google Scholar]
- 6.Billadeau D, Jelinek DF, Shah N, LeBien TW, Van Ness B. Introduction of an activated N-ras oncogene alters the growth characteristics of the interleukin 6-dependent myeloma cell line ANBL6. Cancer Res. 1995;55: 3640-3646. [PubMed] [Google Scholar]
- 7.Billadeau D, Liu P, Jelinek D, Shah N, LeBien TW, Van Ness B. Activating mutations in the N- and K-ras oncogenes differentially affect the growth properties of the IL-6-dependent myeloma cell line ANBL6. Cancer Res. 1997;57: 2268-2275. [PubMed] [Google Scholar]
- 8.Sheng H, Williams CS, Shao J, Liang P, DuBois RN, Beauchamp RD. Induction of cyclooxygenase-2 by activated Ha-ras oncogene in Rat-1 fibroblasts and the role of mitogen-activated protein kinase pathway. J Biol Chem. 1998;273: 22120-22127. [DOI] [PubMed] [Google Scholar]
- 9.Sheng H, Shao J, Dubois RN. K-Ras-mediated increase in cyclooxygenase 2 mRNA stability involves activation of the protein kinase B1. Cancer Res. 2001;61: 2670-2675. [PubMed] [Google Scholar]
- 10.Ladetto M, Vallet S, Trojan A, et al. Cyclooxygenase-2 (COX-2) is frequently expressed in multiple myeloma and is an independent predictor of poor outcome. Blood. 2005;105: 4784-4791. [DOI] [PubMed] [Google Scholar]
- 11.Rowley M, Liu P, Van Ness B. Heterogeneity in therapeutic response of genetically altered myeloma cell lines to interleukin 6, dexamethasone, doxorubicin, and melphalan. Blood. 2000;96: 3175-3180. [PubMed] [Google Scholar]
- 12.Harigaya K, Handa H. Generation of functional clonal cell lines from human bone marrow stroma. Proc Natl Acad Sci U S A. 1985;82: 3477-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krysan K, Merchant FH, Zhu L, et al. COX-2-dependent stabilization of survivin in non-small cell lung cancer. Faseb J. 2004;18: 206-208. [DOI] [PubMed] [Google Scholar]
- 14.Sharma S, Stolina M, Yang SC, et al. Tumor cyclooxygenase 2-dependent suppression of dendritic cell function. Clin Cancer Res. 2003;9: 961-968. [PubMed] [Google Scholar]
- 15.Nedergaard T, Guldberg P, Ralfkiaer E, Zeuthen J. A one-step DGGE scanning method for detection of mutations in the K-, N-, and H-ras oncogenes: mutations at codons 12, 13 and 61 are rare in B-cell non-Hodgkin's lymphoma. Int J Cancer. 1997;71: 364-369. [DOI] [PubMed] [Google Scholar]
- 16.Osuka K, Suzuki Y, Watanabe Y, Takayasu M, Yoshida J. Inducible cyclooxygenase expression in canine basilar artery after experimental subarachnoid hemorrhage. Stroke. 1998;29: 1219-1222. [DOI] [PubMed] [Google Scholar]
- 17.Tai H, Miyaura C, Pilbeam CC, et al. Transcriptional induction of cyclooxygenase-2 in osteoblasts is involved in interleukin-6-induced osteoclast formation. Endocrinology. 1997;138: 2372-2379. [DOI] [PubMed] [Google Scholar]
- 18.Hu L, Shi Y, Hsu JH, Gera J, Van Ness B, Lichtenstein A. Downstream effectors of oncogenic ras in multiple myeloma cells. Blood. 2003;101: 3126-3135. [DOI] [PubMed] [Google Scholar]
- 19.Dormond O, Bezzi M, Mariotti A, Ruegg C. Prostaglandin E2 promotes integrin alpha Vbeta 3-dependent endothelial cell adhesion, rac-activation, and spreading through cAMP/PKA-dependent signaling. J Biol Chem. 2002;277: 45838-45846. [DOI] [PubMed] [Google Scholar]
- 20.Yazawa K, Tsuno NH, Kitayama J, et al. Selective inhibition of cyclooxygenase-2 inhibits colon cancer cell adhesion to extracellular matrix by decreased expression of beta1 integrin. Cancer Sci. 2005;96: 93-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93: 1658-1667. [PMC free article] [PubMed] [Google Scholar]
- 22.Jalkanen S, Jalkanen M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J Cell Biol. 1992;116: 817-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin J, Hsiao PW, Chiu TH, Chao JI. Combination of cyclooxygenase-2 inhibitors and oxaliplatin increases the growth inhibition and death in human colon cancer cells. Biochem Pharmacol. 2005;70: 658-667. [DOI] [PubMed] [Google Scholar]
- 24.Zatelli MC, Luchin A, Piccin D, et al. Cyclooxigenase-2 inhibitors reverse chemoresistance phenotype in medullary thyroid carcinoma by a P-gp mediated mechanism. J Clin Endocrinol Metab. 2005;90: 5754-5760. [DOI] [PubMed] [Google Scholar]
- 25.Kalakonda N, Rothwell DG, Scarffe JH, Norton JD. Detection of N-Ras codon 61 mutations in subpopulations of tumor cells in multiple myeloma at presentation. Blood. 2001;98: 1555-1560. [DOI] [PubMed] [Google Scholar]
- 26.Han S, Roman J. COX-2 inhibitors suppress integrin alpha5 expression in human lung carcinoma cells through activation of Erk: involvement of Sp1 and AP-1 sites. Int J Cancer. 2005;116: 536-546. [DOI] [PubMed] [Google Scholar]
- 27.Dohadwala M, Luo J, Zhu L, et al. Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. J Biol Chem. 2001; 276: 20809-20812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Driel M, Gunthert U, van Kessel AC, et al. CD44 variant isoforms are involved in plasma cell adhesion to bone marrow stromal cells. Leukemia. 2002;16: 135-143. [DOI] [PubMed] [Google Scholar]
- 29.Masellis-Smith A, Belch AR, Mant MJ, Pilarski LM. Adhesion of multiple myeloma peripheral blood B cells to bone marrow fibroblasts: a requirement for CD44 and alpha4beta7. Cancer Res. 1997;57: 930-936. [PubMed] [Google Scholar]
- 30.Hurt EM, Wiestner A, Rosenwald A, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5: 191-199. [DOI] [PubMed] [Google Scholar]
- 31.Michigami T, Shimizu N, Williams PJ, et al. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and alpha(4)beta(1)-integrin enhances production of osteoclast-stimulating activity. Blood. 2000;96: 1953-1960. [PubMed] [Google Scholar]
- 32.Dandekar DS, Lopez M, Carey RI, Lokeshwar BL. Cyclooxygenase-2 inhibitor celecoxib augments chemotherapeutic drug-induced apoptosis by enhancing activation of caspase-3 and -9 in prostate cancer cells. Int J Cancer. 2005;115: 484-492. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, Abe Y, Matsushima T, Nishimura J, Nawata H, Muta K. Selective cyclooxygenase 2 inhibitor NS-398 induces apoptosis in myeloma cells via a Bcl-2 independent pathway. Leuk Lymphoma. 2005;46: 425-433. [DOI] [PubMed] [Google Scholar]
- 34.Prince HM, Mileshkin L, Roberts A, et al. A multicenter phase II trial of thalidomide and celecoxib for patients with relapsed and refractory multiple myeloma. Clin Cancer Res. 2005;11: 5504-5514. [DOI] [PubMed] [Google Scholar]
- 35.Kardosh A, Soriano N, Liu Y-T, et al. Multitarget inhibition of drug-resistant multiple myeloma cell lines by dimethyl-celecoxib (DMC), a non-COX-2 inhibitory analog of celecoxib. Blood. 2005;106: 4330-4338. [DOI] [PubMed] [Google Scholar]
- 36.Han SY, Lee NK, Kim KH, et al. Transcriptional induction of cyclooxygenase-2 in osteoclast precursors is involved in RANKL-induced osteoclastogenesis. Blood. 2005;106: 1240-1245. [DOI] [PubMed] [Google Scholar]