

Abstract
In patients with severe congenital neutropenia (SCN), sepsis mortality is reduced by treatment with granulocyte colony-stimulating factor (G-CSF), but myelodsyplastic syndrome and acute myeloid leukemia (MDS/AML) have been reported. We studied 374 patients with SCN and 29 patients with Shwachman-Diamond syndrome (SDS) on long-term G-CSF enrolled in the Severe Chronic Neutropenia International Registry. In SCN, sepsis mortality was stable at 0.9% per year. The hazard of MDS/AML increased significantly over time, from 2.9% per year after 6 years to 8.0% per year after 12 years on G-CSF. After 10 years, the cumulative incidence was 8% for sepsis mortality and 21% for MDS/AML. A subgroup of SCN patients (29%) received more than the median dose of G-CSF (≥ 8 μg/kg/d), but achieved less than the median absolute neutrophil count (ANC) response (ANC < 2.188 × 109/L [2188/μL] at 6-18 months). In these less-responsive patients, the cumulative incidence of adverse events was highest: after 10 years, 40% developed MDS/AML and 14% died of sepsis, compared with 11% and 4%, respectively, of more responsive patients whose ANC was above the median on doses of G-CSF below the median. Risk of MDS/AML may be similar in SDS and SCN. In less-responsive SCN patients, early hematopoietic stem cell transplantation may be a rational option.
Introduction
Severe congenital neutropenia (SCN) is a heterogeneous disorder of myelopoiesis characterized by an absolute neutrophil count (ANC) persistently below 0.50 × 109/L (500/μL), with maturation arrest of neutrophil precursors in the bone marrow at the promyelocyte/myelocyte stage (reviewed by Zeidler and Welte1). Prior to the introduction of therapy with recombinant human granulocyte colony-stimulating factor (G-CSF), approximately half of affected children died during the first year of life from overwhelming bacterial sepsis. The mortality rate during the second through fourth years was 6% to 7% per year.2 A limited number of case reports from the pre–G-CSF era indicated that patients with SCN3-6 and patients with Shwachman-Diamond syndrome (SDS)7 were also at risk of developing acute myeloid leukemia (AML).
The clinical outlook improved dramatically following the introduction of G-CSF therapy.8 With daily G-CSF dosing at pharmacologic levels, almost all patients with SCN respond with improved ANC values and a reduced incidence of infections and hospitalizations. The Severe Chronic Neutropenia International Registry (SCNIR) was established to monitor the course of SCN and related disorders in patients on G-CSF therapy.9 The SCNIR is the largest prospective cohort study of patients with SCN and related disorders. Through the SCNIR and case reports, it has become clear that patients with SCN and SDS who are receiving G-CSF treatment are at considerable risk of myelodysplastic syndrome or AML (MDS/AML). However, it is not yet clear whether there is any association between the duration and dose of G-CSF therapy and the risk of leukemia in patients with congenital neutropenia. In a previous analysis of data from the SCNIR, no significant association was found between the duration or dose of G-CSF therapy and the risk of MDS/AML.10 In contrast, a report from the French Severe Chronic Neutropenia Study Group did find a positive association with dose in a smaller group of patients.11 However, both of these previous analyses included patients with types of neutropenia other than SCN and SDS who appeared to be at low risk of MDS/AML, possibly obscuring any association.
From a clinical perspective, a key question is whether one can identify early in treatment subgroups of patients with SCN who are at elevated risk of transformation to MDS/AML or death from sepsis.12 In this study, we estimated the cumulative incidence of MDS/AML and death from sepsis over a 12-year follow-up period for patients with SCN and SDS enrolled in the SCNIR. We also examined the associations of risk with daily doses of G-CSF at 6 months and blood counts obtained before and after the start of G-CSF therapy. Our findings have implications for SCN practice guidelines, and may provide new clues about leukemogenesis in these disorders.
Patients, materials, and methods
SCNIR
We studied 374 patients with SCN and 29 patients with SDS who enrolled in long-term clinical G-CSF treatment trials or who directly entered the SCNIR between November 1987 and September 2000 and were followed through February 26, 2001 (Table 1). After February 2001, new regulations affecting long-term observational studies required us to renew the consent of all enrollees, and new privacy regulations interrupted data collection. It also became necessary to transfer the study data from a legacy system to a modern relational database. Therefore, this report includes validated data through February 26, 2001. The study was conducted in accordance with the Declaration of Helsinki, under the auspices of the Human Subjects Committee of the University of Washington, the Hannover Medical School in Hannover, Germany, and other participating institutions.
Table 1.
Follow-up of patients with SCN and SDS in the SCNIR
| No. | Person-y | Median no. (range) of pre-Rx CBC | Median G-CSF dose at 6 mo, μg/kg/d | Total MDS/AML, no. | Total deaths from sepsis, no. | MDS/AML crude rate, % per y* | Death from sepsis crude rate, % per y | |
|---|---|---|---|---|---|---|---|---|
| SCN | ||||||||
| Group 1 | 374 | 2043 | NA | NA | 44 | 19 | 2.2 | 0.9 |
| Group 2 | 365 | 2007 | NA | 6.0 | 44 | 19 | 2.2 | 0.9 |
| Group 3 | 305 | 1769 | 3 (1-76) | 6.2 | 42 | 17 | 2.4 | 1.0 |
| Group 4 | 307 | 1963 | NA | 6.2 | 41 | 15 | 2.1 | 0.8 |
| Group 5 | 238 | 1583 | 9 (1-80) | 8.0 | 34 | 12 | 2.1 | 0.8 |
| Group 6† | 211 | 1445 | 12 (2-116) | 8.0 | 33 | 10 | 2.3 | 0.7 |
| SDS | ||||||||
| Group 1 | 29 | 161 | NA | NA | 2 | 2 | 1.2 | 1.2 |
| Group 2 | 18 | 67 | NA | 4.3 | 2 | 2 | 3.0 | 3.0 |
| Group 3 | 16 | 57 | 3 (1-19) | 4.3 | 1 | 2 | 1.8 | 3.5 |
| Group 4 | 14 | 65 | NA | 4.3 | 1 | 1 | 1.5 | 1.5 |
| Group 5 | 9 | 38 | 3 (2-11) | 4.2 | 1 | 1 | 2.7 | 2.7 |
| Group 6† | 8 | 28 | 8 (4-25) | 4.3 | 1 | 1 | 3.6 | 3.6 |
Dose data indicates G-CSF dose in μg/kg/d at 6 months; Rx, treatment; and NA, not applicable. The pre-Rx CBCs were obtained from 3 years before treatment through the day that treatment was started. Groups were defined as follows: group 1, enrolled; group 2, enrolled with dose data; group 3, enrolled with dose data and CBC data pre-Rx; group 4, enrolled with dose data and followed beyond 18 months; group 5, enrolled with dose data and followed beyond 18 months with CBC data at 6-18 months; and group 6, enrolled with dose data and followed beyond 18 months with CBC data at 6-18 months and pre-Rx.
Crude rates are defined as the total number of events, divided by the person-years of follow-up, times 100%.
Includes both follow-up intervals.
Patients were enrolled on referral from cooperating hematologists and other physicians, primarily in North America, Western Europe, and Australia, using standard enrollment criteria and forms as previously described.9 At the time of enrollment, clinical and laboratory information was reviewed by an expert clinician associated with the registry. Follow-up involved semiannual reports that were entered into a standardized database; physicians caring for patients with important changes in their clinical status often contacted the SCNIR offices directly to report new events and to seek management advice based on the growing experience of the registry. All diagnoses of MDS/AML and dates of death were confirmed by review of pathological reports and clinical records as available.
All G-CSF dose data were converted to units of micrograms per kilogram per day (μg/kg/d) using the reported dosing frequency and body weight from the corresponding study visit or the visit closest in time. Initially, patients with SCN were started at a comparatively low dose (5 μg/kg/d) that was increased at approximately 2-week intervals over a several-month period until a satisfactory ANC value was achieved, the goal being an ANC value of 1.5 × 109/L cells (1500/μL cells) or more (US) or 1.0 × 109/L cells (1000 cells/μL) or more (Europe). The dose of G-CSF for the patient at 6 months was regarded as the initial effective dose for all comparisons. This dose was subject to change over time for reasons that might be related to various events and outcomes for each patient. For this reason, we chose to study the dose of G-CSF at 6 months, rather than the “predominant” dose analyzed by Freedman et al,10 or the time-averaged dose analyzed by Donadieu et al.11
The results of complete blood cell counts (CBCs) were also a critical component for this analysis. Because absolute counts for each type of leukocyte in a CBC are quite variable, we computed the average CBC component values using all CBCs that were obtained during follow-up periods of interest, reported to the registry, and accessioned into the registry's computer database. The CBC components included the absolute neutrophil, monocyte, basophil, eosinophil, lymphocyte, and platelet counts (cells × 109/L [cells/μL]), as well as the hemoglobin level (g/L [g/dL]). The interval from 3 years before the start of treatment through the day that treatment was started was chosen for the pretreatment period, and the interval from 6 to 18 months after the start of therapy was chosen to indicate the effective CBC response of each patient after the effective dose of G-CSF had been established.
Statistical methods
Our primary analyses were restricted to patients with SCN, because data for patients with SDS were comparatively limited (Table 1). For both groups, all statistical tests were 2-sided. “CI” denotes confidence interval.
Patients with SCN are at risk of both MDS/AML and death from sepsis, which we analyzed here as competing adverse events. The time scale was years on therapy with G-CSF. We estimated the cumulative incidence of each adverse event using the nonparametric maximum likelihood estimator that accounts for competing risks.13 We obtained flexible and smooth estimates of the absolute cause-specific hazard rates, using spline functions.14 We estimated the effects of the dose of G-CSF and CBC values on the hazard of each adverse event using Cox proportional hazards models.15 In these analyses, each endpoint was considered to be a censoring event for the other, so that a patient was followed for death from sepsis until the last day of follow-up or the diagnosis of MDS/AML, whichever occurred first. For both endpoints, patients were censored at the time of bone marrow transplantation. For analyses including pretreatment but not on-treatment CBC components, we included follow-up beginning from the day that treatment was started. For analyses including CBC components obtained on treatment during the 6- to 18-month follow-up period, we included follow-up only beginning at 18 months, to ensure that these analyses were entirely prospective.
In secondary analyses, we used generalized linear models16 to assess whether the patient-specific mean CBC values differed in subgroups defined by dose of G-CSF and ANC values on treatment. We also studied changes in CBC values (on-treatment values minus pretreatment values). We assessed the association between ANC values on treatment and G-CSF dose using a nonparametric generalized additive model (regression spline17).
We carried out each analysis using all subjects with available data. Patient groups are defined in Table 1 and labeled Group 1 through Group 6. We used Cox models to test whether patients with SCN with CBC data were at increased or decreased risk of each endpoint, compared with patients without CBC data.
We contrasted the median CBC and G-CSF dose values in patients with SDS versus SCN using the Wilcoxon rank sum test.18 We contrasted crude event rates in patients with SDS versus SCN using an exact test.19 We did not fit Cox models to SDS data because there were inadequate numbers of events.
Results
We studied a total of 374 patients with SCN (Table 1, Group 1) and 29 patients with SDS. In patients with SCN, 365 (98%) had G-CSF dose data reported during the first 12 months of follow-up (Group 2). The median age at the start of therapy was 3 years, and the interquartile range was 9 months to 9.7 years. More than half of the patients were male (55%) and 80% were of European ancestry. Of these 365 patients, 305 (84%) had CBCs with differential that were obtained prior to treatment and accessioned into the registry database (Group 3). Of them, 307 (84%) were followed beyond 18 months (Group 4); 238 patients in this group (78%) had CBC data available from the preceding 6- to 18-month follow-up period (Group 5). The majority of this latter group, 211 (89%) of 238, also had pretreatment CBC data (Group 6). In total, we studied 1632 CBCs obtained during the pretreatment window period, and 2335 CBCs obtained during the 6- to 18-month follow-up period. The median patient-specific interval between successive CBCs was 30 days in patients with SCN and 48 days in patients with SDS.
The 374 patients with SCN contributed 2043 person-years, 44 transformations to MDS/AML, and 19 deaths from sepsis (Table 1, Group 1). Using the competing risks definition, the cumulative incidence of MDS/AML was 21% (95% CI = 15%-27%) after 10 years on G-CSF therapy and 36% (95% CI = 22%-50%) after 12 years (Figure 1A). The cumulative incidence of death from sepsis was 8% (95% CI = 4%-12%) after 10 years on G-CSF therapy; no additional deaths from sepsis occurred between years 10 and 12 (Figure 1A). The hazard rates were not significantly different in patients who did and did not have available pretreatment and on-treatment CBC results (Group 2 and 4, respectively; data not shown). The cause-specific hazard of MDS/AML increased significantly over time (P < .001 for trend), rising from 2.9% per year (95% CI = 2.0%-4.1% per year) after 6 years on G-CSF therapy, to 8.0% per year (95% CI = 1.7%-14.0% per year) after 11.7 years (Figure 1B). The cause-specific hazard of death from sepsis was comparatively stable over time (P = .76 for trend; assuming a constant hazard, the mortality rate from sepsis was 0.9% per year; 95% CI = 0.6%-1.4% per year).
Figure 1.

Cumulative incidence and hazard rates of MDS/AML and death from sepsis in patients with SCN. Results are shown for patients in SCN Group 1 (Table 1). One additional death from sepsis at year 12.4 is not shown on the plot. (A) Cumulative incidence (cumulative proportion experiencing each event as initial cause of failure in subjects at risk of each adverse event), by years on therapy with G-CSF, and 95% CIs at selected years (error bars). Observed cumulative incidence (stair-step curves), and smoothed cumulative incidence (dashed curves) derived from estimated cause-specific hazard functions are shown. (B) Annual hazard rates (incidence rate per year among subjects who are still susceptible) of MDS/AML and death from sepsis, by years on G-CSF therapy, and 95% point-wise confidence envelopes (shaded regions).
The pretreatment ANC value was approximately normally distributed on the log2 scale (Figure 2A; histogram shown in red). The median pretreatment ANC value was 0.129 ×109/L (129/μL), and the absolute interquartile range defining the middle one-half of the data was 0.031-0.285 × 109/L (31-285/μL). In 95% of patients, the ANC value on treatment was greater than the ANC value prior to treatment. The median absolute ANC change in all patients was 2.125 × 109/L (2125/μL), and the interquartile range was 1.019-4.028 × 109/L (1019-4028/μL) (Figure 2B). However, on treatment, 32% had ANC values below 1.5 × 109/L (1500/μL), and 19% had ANC values below 1.0 × 109/L (1000/μL) (Figure 2A; histogram shown in blue).
Figure 2.

Pretreatment ANC, on-treatment ANC, and dose of G-CSF in patients with SCN. Panels A-D show data for SCN Group 6, while panel E shows data for SCN Group 5 (Table 1). (A) Histograms of mean ANC values prior to treatment (red bars), and mean ANC values during the 6-18 month follow-up period (blue bars), shown on a log2 scale. A vertical reference line is shown at 1500 cells/μL, corresponding to the therapeutic target. (B) Box plot of mean ANC values during the 6-18 month follow-up minus pretreatment mean ANC values, displaying the median value, inter-quartile range, 10th and 90th percentiles, and outliers (plus signs). Notches show 95% confidence intervals for the median. (C) Box plots of G-CSF dose values (μg/kg/d), by follow-up period. Box plots are constructed as described for panel B. Two hundred eleven patients contributed 2,440 G-CSF dose values over time; all values for each patient are shown. Starting dose corresponds to 0-3 days of follow-up; 6 m, to between 3 days and 8 months; 1 y, to between 9 and 14 months; 1.5 y, to between 15 and 20 months; 2 y, to between 21 and 26 months; and after 2 y, to between 27 months and 12.44 years. (D) Histogram of G-CSF dose (μg/kg/d) for each patient at or closest to 6 months. The right-most bar represents 4 patients with dose values of 120, 128, 189, and 576 μg/kg/d. (E) Dose of G-CSF at 6 months, and average ANC response over the subsequent year. Dose of G-CSF is plotted on the log2 scale. Curve plots the mean value. Shaded area corresponds to 95% point-wise confidence limits. The nonparametric regression incorporated precision weights equal to the number of CBC values available for each patient during the 6- to 18-month follow-up period.
As expected, the dose of G-CSF at 6 months was significantly higher than the starting dose, and it was comparatively stable after 6 months (Figure 2C). The G-CSF dose at 6 months was highly right-skewed (Figure 2D). The median dose of G-CSF in SCN Groups 5 and 6 was 8.0 μg/kg/d; the minimum dose was 0.2 μg/kg/d, and the maximum dose was 576 μg/kg/d. Therefore, although most patients responded to G-CSF with increased neutrophil counts, the magnitude of the dose at 6 months and the ANC response over the subsequent year varied several-fold.
We assessed the associations between the dose of G-CSF at 6 months and the cause-specific hazards of MDS/AML and death from sepsis. Cox proportional hazards models are summarized in Table 2. Because the dose of G-CSF at 6 months had a skewed distribution (Figure 2D), we considered associations with the dose of G-CSF at or above the median value versus below, and with the log2 dose of G-CSF.
Table 2.
Risk factors for MDS/AML and death from sepsis in patients with SCN in the SCNIR
|
MDS/AML
|
Death from sepsis
|
||||
|---|---|---|---|---|---|
| Factor* | Group | RH (95% CI)† | P‡ | RH (95% CI)† | P |
| G-CSF at 6 mo | |||||
| 6 μg/kg/d or more vs less than 6 μg/kg/d | 2 | 2.5 (1.2-5.2) | .008 | 2.0 (0.7-5.5) | .180 |
| 8 μg/kg/d or more vs less than 8 μg/kg/d | 2 | 1.9 (1.0-3.7) | .036 | 2.9 (1.0-8.1) | .029 |
| Per doubling§ | 2 | 1.22 (1.03-1.44) | .024 | 1.31 (1.02-1.69) | .039 |
| ANC at 6-18 mo | |||||
| Less than 2188/μL vs at least 2188/μL | 5 | 2.3 (1.2-4.7) | .016 | 3.4 (0.9-12.9) | .056 |
| Per halving¶ | 5 | 1.25 (1.00-1.56) | .058 | 1.55 (1.17-2.05) | .006 |
| G-CSF at 6 mo, and ANC at 6-18 mo | |||||
| A: 8 μg/kg/d or more, and ANC less than 2188/μL | 5 | 4.5 (1.5-13.4) | .008 | 3.8 (0.79-18.3) | .094 |
| B: Less than 8 μg/kg/d, and ANC less than 2188/μL | 5 | 1.7 (0.5-6.4) | .422 | 0.6 (0.05-6.9) | .689 |
| C: 8 μg/kg/d or more and ANC of at least 2188/μL | 5 | 1.7 (0.5-5.5) | .402 | 0.5 (0.04-5.1) | .525 |
| D: Less than 8 μg/kg/d, and ANC of at least 2188/μL | 5 | 1.0 (Referent) | NA | 1.0 (Referent) | NA |
| A: 8 μg/kg/d or more and ANC below 2188/μL, vs B-D: other | 5 | 3.1 (1.5-6.1) | .002 | 5.6 (1.7-18.7) | .004 |
| ANC before treatment | |||||
| Less than 138 cells/μL vs 138 cells/μL or more | 3 | 0.9 (0.5-1.6) | .627 | 2.3 (0.75-7.25) | .123 |
| Per halving¶ | 3 | 0.96 (0.79-1.16) | .648 | 1.27 (0.92-1.74) | .145 |
RH indicates relative hazard; NA, not available.
See Table 1 for a summary of follow-up data by group.
Cut-points for dose of G-CSF and ANC values were defined by the group-specific medians.
RHs and 95% CIs estimated using Cox proportional hazards models.
P values for likelihood ratio tests (factors G-CSF at 6 mo; ANC at 6-18 mo; and ANC before treatment) or Wald tests (contrasting levels in factor G-CSF at 6 mo and ANC at 6-18 mo).
A model with log2 G-CSF dose was fitted; the RH per doubling is the RH corresponding to a 1-unit increase of G-CSF dose on the log2 scale or a doubling of a G-CSF dose on the absolute scale.
Models with log2 ANC values were fitted; the RH per halving is the RH corresponding to a 1-unit decrease of G-CSF dose on the log2 scale or a halving of G-CSF dose on the absolute scale.
The dose of G-CSF was significantly and positively associated with the risk of MDS/AML. In patients who required 6 μg/kg/d or more (the median dose in SCN Group 2), compared with patients who required less than 6 μg/kg/d, the hazard of MDS/AML was 2.5-fold (95% CI = 1.2- to 5.2-fold) higher (P = .008). The hazard of MDS/AML was similarly elevated in patients who required 8 μg/kg/d or more (the median dose in SCN Group 5). On a continuous scale, the hazard of MDS/AML increased by 1.22-fold (95% CI = 1.03- to 1.44-fold) per doubling of the dose of G-CSF (P = .024).
In patients who required 6 μg/kg/d or more, compared with patients who required less than 6 μg/kg/d, the hazard of death from sepsis was 2.0-fold higher (95% CI = 0.7- to 5.5-fold), but this association did not reach statistical significance (P = .18). The hazard of death from sepsis was significantly elevated in patients who required 8 μg/kg/d or more. On a continuous scale, the hazard of death from sepsis increased by 1.31-fold (95% CI = 1.02- to 1.69-fold) per doubling of the dose of G-CSF; this association was significant (P = .039).
These associations could reflect confounding or interaction of the dose of G-CSF with other risk factors. We considered both possibilities. For death from sepsis, the ANC value on treatment is a logical risk factor; therefore, we considered its association with both death from sepsis and MDS/AML. Because neutrophil counts had a skewed distribution (Figure 2A), we considered associations with ANC values at or above the median versus below the median, and with log2 ANC.
Regarding potential confounding, as expected, the risk of death from sepsis beyond the 18th month was negatively associated with the ANC value during the 6- to 18-month follow-up period. Patients with ANC values on treatment below the median of 2.188 × 109/L (2188/μL) were at 3.4-fold higher (95% CI = 0.9-12.9; P = .056) risk of death from sepsis than patients with ANCs of 2.188 × 109/L cells (2188/μL cells) or more. On a continuous scale, the hazard of death from sepsis increased by 1.55-fold (95% CI = 1.17- to 2.05-fold; P = .006) per halving of the ANC value on treatment (eg, the patient with 1.0 × 109/L neutrophils [1000/μL neutrophils] was at 55% greater risk of death from sepsis than the patient with 2.0 × 109/L neutrophils [2000/μL neutrophils]). The hazard of MDS/AML was also negatively associated with the ANC value on treatment. Patients with ANCs lower than 2.188 × 109/L (2188/μL) were at 2.3-fold higher (95% CI = 1.2- to 4.7-fold; P = .016) risk of MDS/AML than patients with ANCs of 2.188 × 109/L cells (2188/μL cells) or more. On a continuous scale, the hazard of MDS/AML increased by 1.25-fold (95% CI = 1.00- to 1.56-fold; P = .058) per halving of the ANC value on treatment.
The ANC value on treatment was also associated with the dose of G-CSF at 6 months (Figure 2E). Patients who responded to comparatively low doses, below 8 μg/kg/d (the median value in Group 5), had the highest ANC values on treatment, whereas patients who required higher doses had significantly lower ANC values on treatment (P < .001 for the significance of the model shown versus no association). Therefore, because the ANC value on treatment is associated with the dose of G-CSF, and is also a risk factor for MDS/AML, the association of the dose of G-CSF with the risk of MDS/AML could be the result of confounding. However, for each endpoint, the relative hazards for each factor did not change appreciably when both factors were included in a Cox model (data not shown), indicating that both factors affect the hazards.
Next, we considered Cox models that allowed for interactions between the dose of G-CSF at 6 months and the ANC value on treatment. We categorized each risk factor according to the median values in SCN Group 5 (8 μg/kg/d and 2.188 × 109/L cells [2188/μL cells], respectively). The corresponding subgroups are labeled A to D, as defined in Table 2; the proportion of patients in these subgroups was 29%, 21%, 21%, and 29%, respectively. For both MDS/AML and death from sepsis, the risk was significantly increased in patients who were given more than the median G-CSF dose, but who achieved less than the median ANC response (subgroup A), compared with patients who were below the median for dose but above the median for ANC response (subgroup D). The relative hazards were not significantly increased for patients in subgroups B or C compared with subgroup A. These results are consistent with an interaction. In subgroup A, the risk of MDS/AML was increased by 3.1-fold (95% CI = 1.5- to 6.1-fold; P = .002) and the risk of death from sepsis was increased by 5.6-fold (95% CI = 1.7- to 18.7-fold; P = .004), compared with subgroups B to D combined (Table 2).
The cumulative incidence of MDS/AML and death from sepsis in subgroups A to D is shown in Figure 3 (top panels). After 10 years, the number of patients under follow-up fell to 10 or fewer in subgroups A to C, and the cumulative incidence curves were unstable thereafter. Therefore, we report the cumulative incidence in each subgroup up to year 10.
Figure 3.
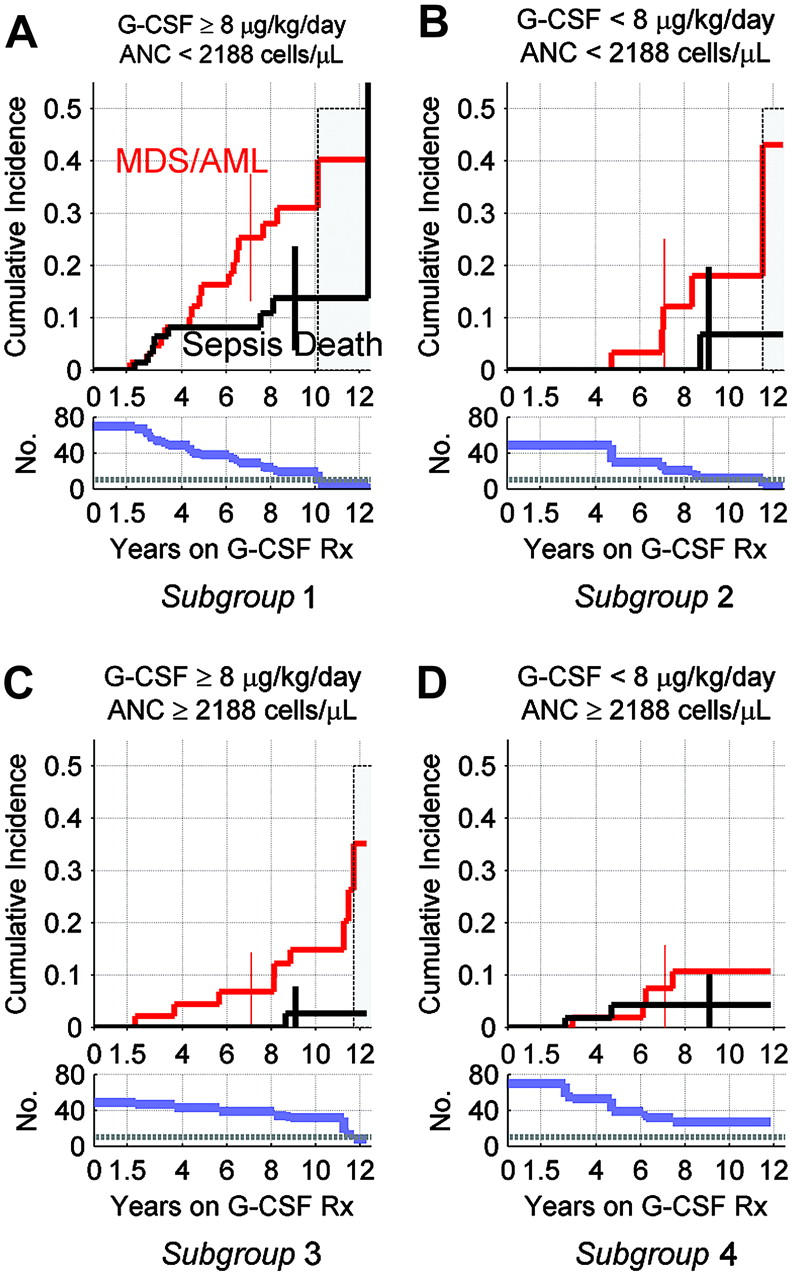
Cumulative incidence of MDS/AML and death from sepsis, by G-CSF dose at 6 months, and average ANC response over the subsequent year, in patients with SCN. Results are shown for patients in SCN Group 5 (Table 1). Top panels show cumulative incidence of MDS/AML and death from sepsis by years on G-CSF therapy; see legend to Figure 1 for details. Bottom panels show corresponding numbers of subjects still at risk and under follow-up, with horizontal reference lines at n = 10 patients. With 10 or fewer subjects, the estimated cumulative incidence curves become unstable, as indicated by gray shaded areas in top panels. Error bars in top panels show 95% confidence limits at years 7 (MDS/AML) and 9 (deaths from sepsis). (A) Cumulative incidence in subjects with mean ANC less than 2.188 × 109/L (2188/μL) cells during the 6- to 18-month follow-up period on G-CSF at 6 month at 8 μg/kg/d or more. (B) Cumulative incidence in subjects with ANC less than 2.188 × 109/L (2188/μL) cells on G-CSF of less than 8 μg/kg/d. (C) Cumulative incidence in subjects with ANC of 2.188 × 109/L (2188/μL) cells or more on G-CSF of 8 μg/kg/d or more. (D) Cumulative incidence in subjects with ANC of 2.188 × 109/L (2188/μL) cells or more on G-CSF of less than 8 μg/kg/d.
Among patients with ANC less than 2.188 × 109/L cells (2188/μL cells) despite G-CSF of 8 μg/kg/d or more (Figure 3A), the cumulative incidence at 10 years was 40% for MDS/AML and 14% for death from sepsis; 54% had one or the other adverse event by the 10-year landmark. In contrast, among patients with ANC of 2.188 × 109/L cells or more (2188/μL cells or more) on G-CSF of less than 8 μg/kg/d (Figure 3D), the cumulative incidence at 10 years was 11% for MDS/AML, 4% for death from sepsis, and 15% for one or the other adverse event. In patients who responded to higher doses of G-CSF (ANC ≥ 2.188 × 109/L cells [2188/μL cells] on G-CSF ≥ 8 μg/kg/d; Figure 3C), the cumulative incidence at 10 years was 15% for MDS/AML, 3% for death from sepsis, and 18% for one or the other adverse event. In patients with ANCs of less than 2.188 × 109/L cells (2188/μL cells) on G-CSF of less than 8 μg/kg/d (Figure 3B), the cumulative incidence at 10 years was 18% for MDS/AML, 7% for death from sepsis, and 25% for one or the other adverse event. We obtained similar estimates of cumulative incidence when the threshold for an ANC response was lowered from 2.188 to 1.5 × 109/L cells (2188 to 1500/μL cells) (data not shown).
In contrast to the significant negative associations seen between the ANC value on treatment and the risks of MDS/AML and death from sepsis, the pretreatment ANC was not significantly associated with the hazard of either endpoint in prospective Cox models (Table 2; SCN Group 3). These analyses included all events and follow up beginning from the day that treatment was started. We evaluated all other pretreatment blood cell counts, and found none that was significantly associated with either endpoint.
Prior to treatment, high-risk patients in subgroup A had the lowest ANC values, comparatively low hemoglobin levels, comparatively high platelet counts, and elevated counts of all other types of white blood cells (Figure 4). However, the CBC profile for this high-risk subgroup was not specific. Patients in subgroup C who subsequently achieved an ANC above the median on G-CSF of 8 μg/kg/d or more had a similar CBC profile prior to treatment. Hence, the pretreatment CBC could not predict the subsequent outcome, consistent with the Cox models.
Figure 4.

Pretreatment CBC counts in patients with SCN. Data are shown for patients in SCN Group 6 (Table 1). Symbols show mean CBC values for the period from 3 years before the start of treatment through the day that treatment was started (▪ or •) and 95% confidence limits (error bars), for patients in subgroups A to D defined by G-CSF dose at 6 months and ANC response during the subsequent year (for further information about subgroups A to D, see Figure 3 and Table 2). These analyses incorporated precision weights determined by the number of patient-specific CBC values available prior to treatment. Panels show (A) hemoglobin, (B) platelets, (C) eosinophils, (D) basophils, (E) monocytes, (F) lymphocytes, and (G) pretreatment ANC.
On therapy, patients in subgroup A had similar increases in eosinophils, basophils, monocytes, and lymphocytes, and similar decreases in platelets, as other patients maintained on 8 μg/kg/d G-CSF or more (Figure 5). Thus, except for their attenuated increases in neutrophil counts and hemoglobin levels, the CBC response to G-CSF therapy in the high-risk patients was also not remarkable.
Figure 5.

Changes in CBC counts in SCN patients. Data are shown for patients in SCN Group 6 (Table 1). Glyphs show patient-specific changes in mean CBC values during the 6- to 18-month follow-up versus pretreatment mean CBC values (♦ or •), and 95% confidence limits (error bars), for patients in subgroups A to D (see legend to Figure 3 for details). Dotted lines and gray shaded bands show mean values and corresponding 95% confidence limits for changes in all patients combined. These analyses incorporated precision weights equal to nm/(n + m), where m is the number of pretreatment CBC and n is the number of posttreatment CBC. Panels show changes in (A) hemoglobin, (B) platelets, (C) eosinophils, (D) basophils, (E) monocytes, (F) lymphocytes, and (G) ANC.
Finally, we considered the experience of patients with SDS. Prior to treatment, the CBC profile was significantly different between patients with SDS versus SCN. Compared with patients with SCN, patients with SDS had significantly lower median platelet counts (157 versus 384 × 109/L; P < .001), lower median monocyte counts (0.269 versus 0.879 × 109/L cells [269 versus 879/μL cells]; P = .002) and higher median neutrophil counts (0.433 versus 0.138 × 109/L cells [433 versus 138/μL cells]; P < .001). Eosinophil and basophil counts were significantly lower in patients with SDS; half of the patients with SDS had eosinophil and basophil counts that were too low to call (reported counts of zero). The median dose of G-CSF at 6 months was significantly lower (4.3 versus 6.0 μg/kg/d in SDS Group 2 versus SCN Group 2; P = .004). In response to therapy, the median change in neutrophil counts at 6 to 18 months was not significantly different in the 2 disorders (P = .45). The crude rates of MDS/AML and death from sepsis in SDS and SCN shown in Table 1 were not significantly different (P = .88 and .28, respectively). Among all patients with SDS, the 2 subjects who died of sepsis had received the highest doses of G-CSF (11.5 and 16.8 μg/kg/d, respectively). Two patients with SDS developed MDS/AML after 1 month and 2.2 years on G-CSF, at doses of 6.0 and 4.3 μg/kg/d, respectively. These patients were reported previously.10
Discussion
In patients with SCN maintained on long-term therapy with G-CSF, mortality from sepsis remained stable at just under 1% per year over 12 years of follow-up. This rate falls qualitatively below the historical rate of 6% to 7% per year inferred for the precytokine era.2 This secular decline may be attributed to both G-CSF therapy and improvements in supportive care. In contrast, the cause-specific hazard of MDS/AML increased with the duration of therapy, and these increases were statistically significant. Two earlier analyses10,20 that included some of the patients studied here were not able to identify this increase, which is now apparent using exclusively SCN cases, longer follow-up, and more sensitive statistical methods.
Moreover, the cumulative incidence of MDS/AML appears to be higher than previously recognized. A number of patients enrolled in the SCNIR (10%-15%) were reported previously to have developed MDS/AML,9,10 and this crude proportion has often been cited as the “risk” of MDS/AML in SCN. Although this statistic is accurate, it does not account for variable follow-up. The estimates of cumulative incidence we present here may be more appropriate. After 10 years on therapy, the cumulative incidence was 8% for death from sepsis and 21% for MDS/AML. By 12 years, the cumulative incidence of MDS/AML had risen to 36%. This latter value is subject to statistical uncertainty due to the small numbers of subjects under follow-up in the later years; however, it is significantly higher than 15%.
Furthermore, 29% of the patients (subgroup A) had a poorer ANC response despite higher doses of G-CSF. These less-responsive patients were at significantly increased risk of both MDS/AML and death from sepsis: after 10 years, 40% had developed MDS/AML, and 14% had died of sepsis, compared with 11% and 4%, respectively, of patients in subgroup D who had a good ANC response at lower G-CSF doses. Therefore, all patients with SCN appear to be at substantial risk of MDS/AML, including patients maintained on comparatively low doses of G-CSF. However, patients who are less response to G-CSF appear to be at remarkably high risk.
Given the association between the ANC response to G-CSF and the subsequent risk of MDS/AML, we asked whether other features of the CBC could predict the clinical outcome. In clinical subgroups A to D, we observed a trilineage CBC response to therapy with G-CSF: hemoglobin levels increased, platelet counts decreased, and monocyte, eosinophil, basophil, lymphocyte, and neutrophil counts increased from levels observed prior to treatment. However, it was specifically the ANC response to G-CSF therapy that predicted the subsequent clinical outcome, and not the pretreatment CBC values or the changes in other types of blood cell counts.
There were comparatively limited data to evaluate the experience of patients with SDS. The pretreatment CBC profile of SDS patients was significantly different from SCN patients, consistent with the different genetic etiologies of these disorders.21,22 Patients with SDS had higher baseline ANC counts, and they responded to comparatively lower doses of G-CSF. The cumulative incidence of MDS/AML in SDS could not be well determined. The 2 leukemias that occurred in patients with SDS were diagnosed 1 month and 2.2 years after the start of G-CSF therapy, respectively, making it unlikely that G-CSF initiated at least one of these events. The limited available data do not suggest that the risk of MDS/AML is lower in SDS than in SCN. In future studies, it will be of interest to compare other inherited bone marrow failure syndromes, such as Fanconi anemia, Diamond-Blackfan anemia, and dyskeratosis congenita, to see whether there are similar hazard rates and mechanisms underlying the natural history of AML in each of these premalignant disorders.2
Several limitations of our current study must be noted. First, although the SCNIR includes the largest existing cohort of patients with SCN with long-term prospective follow-up and longitudinal data on dose of G-CSF and CBC response, some patients had limited or no CBC data available electronically, particularly for the pretreatment period. For this report, it was not possible to search all paper records of the registry or ask the referring physicians for additional CBC data. We do not think the data were differentially missing in our computerized files according to prognosis; however, we cannot verify this assumption further than indicated in the “Results” section. Second, we were not able to incorporate follow-up beyond February 26, 2001 (more recent follow-up will be available in the future), and our statistical precision reflects the comparatively limited numbers of events (44 MDS/AML patients and 19 deaths from sepsis). Third, molecular studies have raised a cautionary note about possible interactions between acquired mutations in the G-CSF receptor (GCSFR), G-CSF therapy, and the risk of leukemia.23-30 The prevalence and pathogenicity of these mutations in unselected patients remains unclear;31,32 unfortunately, genetic data on acquired mutations in the GCSFR were not available for this analysis. Fourth, genetic data on germ-line mutations in ELA2 and SBDS (the 2 genes implicated as the primary genetic basis of SCN and SDS) also were not available for this analysis.
In addition, the major limitation of our observational study must be noted: since all patients studied here were treated with G-CSF, we cannot prove that G-CSF does not play a role in leukemogenesis in patients with this disorder. In fact, we observed a statistically significant association between the dose of G-CSF and the risk of MDS/AML in SCN. However, we observed a similar association between the dose of G-CSF and the risk of death from sepsis. The latter association suggests that the former may reflect confounding by indication. Clearly, further studies are needed to elucidate the molecular mechanisms that determine the risk of MDS/AML in SCN, and the role, if any, of G-CSF therapy.
One possible explanation for these associations is that SCN patients with a suboptimal response to higher than the median dose of G-CSF are at increased risk of MDS/AML because an underlying defect in their stem cell population is comparatively more severe than in other patients. This putative genetic defect appears to be correlated with the hyporesponsive phenotype; clinically, pharmacologic G-CSF is unable to overcome the maturation arrest in the neutrophil lineage. If so, it would follow that G-CSF has reduced mortality from sepsis in SCN to the extent that the suspected leukemic predisposition of SCN has become manifest. Future studies are needed to test this particular hypothesis, to identify the sequence of events causing genomic instability and leukemia in SCN and SDS,33 and to assess genotype-phenotype correlations that might further stratify patients according to risk.34
While such studies should be undertaken, the clinical implications of our current data should be emphasized. G-CSF is justifiably the standard of care for neutropenic patients with SCN and SDS; however, it is not a panacea. Our results suggest that less-responsive patients, in particular, should adhere to recommendations for bone marrow examination at least annually.35 Recent protocols incorporating conscious sedation should make these evaluations less trying for patients and their families.
In patients who are less responsive, as defined by our analysis, early hematopoietic stem cell transplantation may be a rational option. Currently, there are very limited data describing the outcomes of transplantation in this group that might be weighed against the risks of sepsis death or MDS/AML described here. From the limited transplantation data, it appears that SCN patients should receive transplants prior to the onset of MDS/AML,35,36 and that results using matched sibling donors may be superior to those from alternative donors.35-37 Until further experience is accumulated, the decision to use this potentially curative modality must be individualized, and preferably performed at a transplantation center with protocols for SCN patients.
Acknowledgments
We thank the patients and doctors for participating in the registry, and the data collection centers in Seattle, WA, and Hannover, Germany, for maintaining the registry. We are grateful to Bin Yao and Steve Dahlberg from Amgen Inc for helpful suggestions about the statistical analysis.
Appendix
Participating institutions in the SCNIR: Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Department of Health and Human Services, Rockville, MD; Department of Medicine, University of Washington, Seattle, WA; Department of Pediatric Hematology Oncology, St Joseph's Children's Hospital, Paterson, NJ; Department of Pediatrics, University of Michigan Medical Center, Ann Arbor, MI; CancerCare Manitoba, Winnipeg, MB, Canada; Amgen, Inc, Boulder, CO; Hospital for Sick Children, Toronto, ON, Canada; Ballarat Oncology and Haematology Services, Wendouree, Australia; St James's University Hospital, Leeds, United Kingdom; and Kinderklinik, Medizinische Hochschule, Hannover, Germany.
Prepublished online as Blood First Edition Paper, February 23, 2006; DOI 10.1182/blood-2005-11-4370.
A list of the institutions participating in the Severe Chronic Neutropenia International Registry appears in the “Appendix.”
Supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, Division of Cancer Epidemiology and Genetics; NIH Grant 1R24AI049392; and a gift from the Amgen Foundation, Thousand Oaks, CA.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Zeidler C, Welte K. Kostmann syndrome and severe congenital neutropenia. Semin Hematol. 2002;39: 82-88. [DOI] [PubMed] [Google Scholar]
- 2.Alter BP. Inherited bone marrow failure syndromes. In: Nathan DG, Orkin SH, Look AT, Ginsburg D, eds. Hematology of Infancy and Childhood. Philadelphia: WB Saunders Inc; 2003: 280-365.
- 3.Miller RW. Childhood cancer and congenital defects: a study of U.S. death certificates during the period 1960-1966. Pediatr Res. 1969;3: 389-397. [DOI] [PubMed] [Google Scholar]
- 4.Gilman PA, Jackson DP, Guild HG. Congenital agranulocytosis: prolonged survival and terminal acute leukemia. Blood. 1970;36: 576-585. [PubMed] [Google Scholar]
- 5.Rosen RB, Kang SJ. Congenital agranulocytosis terminating in acute myelomonocytic leukemia. J Pediatr. 1979;94: 406-408. [DOI] [PubMed] [Google Scholar]
- 6.Wong WY, Williams D, Slovak ML, et al. Terminal acute myelogenous leukemia in a patient with congenital agranulocytosis. Am J Hematol. 1993;43: 133-138. [DOI] [PubMed] [Google Scholar]
- 7.Mack DR, Forstner GG, Wilschanski M, Freedman MH, Durie PR. Shwachman syndrome: exocrine pancreatic dysfunction and variable phenotypic expression. Gastroenterology. 1996;111: 1593-1602. [DOI] [PubMed] [Google Scholar]
- 8.Dale DC, Bonilla MA, Davis MW, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81: 2496-2502. [PMC free article] [PubMed] [Google Scholar]
- 9.Dale DC, Cottle TE, Fier CJ, et al. Severe chronic neutropenia: treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am J Hematol. 2003;72: 82-93. [DOI] [PubMed] [Google Scholar]
- 10.Freedman MH, Bonilla MA, Fier C, et al. Myelodysplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood. 2000;96: 429-436. [PubMed] [Google Scholar]
- 11.Donadieu J, Leblanc T, Bader MB, et al. Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia: experience of the French Severe Chronic Neutropenia Study Group. Haematologica. 2005;90: 45-53. [PubMed] [Google Scholar]
- 12.Ancliff PJ. Congenital neutropenia. Blood Rev. 2003;17: 209-216. [DOI] [PubMed] [Google Scholar]
- 13.Gaynor JJ, Feuer EJ, Tan CC, et al. On the use of cause-specific failure and conditional failure probabilities: examples from clinical oncology data. J Am Stat Assoc. 1993;88: 400-409. [Google Scholar]
- 14.Rosenberg PS. Hazard function estimation using B-splines. Biometrics. 1995;51: 874-887. [PubMed] [Google Scholar]
- 15.Cox DR. Regression models and life-tables. J R Stat Soc Ser B Methol. 1972;34: 187-220. [Google Scholar]
- 16.McCullagh P, Nelder JA. Generalized Linear Models. London: Chapman and Hall; 1983.
- 17.Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. New York, NY: Springer-Verlag; 2002.
- 18.Wilcoxon F. Individual comparisons by ranking methods. Biometrics. 1945;1: 80-83. [PubMed] [Google Scholar]
- 19.Liddell FD. Simple exact analysis of the standardised mortality ratio. J Epidemiol Community Health. 1984;38: 85-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee A, Shannon KM. Leukemic transformation in patients with severe congenital neutropenia. J Pediatr Hematol Oncol. 2001;23: 487-495. [DOI] [PubMed] [Google Scholar]
- 21.Dale DC, Person RE, Bolyard AA, et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96: 2317-2322. [PubMed] [Google Scholar]
- 22.Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33: 97-101. [DOI] [PubMed] [Google Scholar]
- 23.Dong F, Brynes RK, Tidow N, et al. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med. 1995;333: 487-493. [DOI] [PubMed] [Google Scholar]
- 24.Tidow N, Pilz C, Kasper B, Welte K. Frequency of point mutations in the gene for the G-CSF receptor in patients with chronic neutropenia undergoing G-CSF therapy. Stem Cells. 1997;15: 113-119. [DOI] [PubMed] [Google Scholar]
- 25.Tidow N, Kasper B, Welte K. Clinical implications of G-CSF receptor mutations. Crit Rev Oncol Hematol. 1998;28: 1-6. [DOI] [PubMed] [Google Scholar]
- 26.Ward AC, van Aesch YM, Schelen AM, Touw IP. Defective internalization and sustained activation of truncated granulocyte colony-stimulating factor receptor found in severe congenital neutropenia/acute myeloid leukemia. Blood. 1999;93: 447-458. [PubMed] [Google Scholar]
- 27.Hunter MG, Avalos BR. Deletion of a critical internalization domain in the G-CSFR in acute myelogenous leukemia preceded by severe congenital neutropenia. Blood. 1999;93: 440-446. [PubMed] [Google Scholar]
- 28.Hunter MG, Avalos BR. Granulocyte colony-stimulating factor receptor mutations in severe congenital neutropenia transforming to acute myelogenous leukemia confer resistance to apoptosis and enhance cell survival. Blood. 2000;95: 2132-2137. [PubMed] [Google Scholar]
- 29.Hermans MH, Ward AC, Antonissen C, et al. Perturbed granulopoiesis in mice with a targeted mutation in the granulocyte colony-stimulating factor receptor gene associated with severe chronic neutropenia. Blood. 1998;92: 32-39. [PubMed] [Google Scholar]
- 30.Hermans MH, Antonissen C, Ward AC, et al. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999;189: 683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernard T, Gale RE, Evans JP, Linch DC. Mutations of the granulocyte-colony stimulating factor receptor in patients with severe congenital neutropenia are not required for transformation to acute myeloid leukaemia and may be a bystander phenomenon. Br J Haematol. 1998;101: 141-149. [DOI] [PubMed] [Google Scholar]
- 32.Ancliff PJ, Gale RE, Liesner R, Hann I, Linch DC. Long-term follow-up of granulocyte colony-stimulating factor receptor mutations in patients with severe congenital neutropenia: implications for leukaemogenesis and therapy. Br J Haematol. 2003;120: 685-690. [DOI] [PubMed] [Google Scholar]
- 33.Tschan CA, Pilz C, Zeidler C, Welte K, Germeshausen M. Time course of increasing numbers of mutations in the granulocyte colony-stimulating factor receptor gene in a patient with congenital neutropenia who developed leukemia. Blood. 2001;97: 1882-1884. [DOI] [PubMed] [Google Scholar]
- 34.Bellanne-Chantelot C, Clauin S, Leblanc T, et al. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood. 2004;103: 4119-4125. [DOI] [PubMed] [Google Scholar]
- 35.Choi SW, Boxer LA, Pulsipher MA, et al. Stem cell transplantation in patients with severe congenital neutropenia with evidence of leukemic transformation. Bone Marrow Transplant. 2005;35: 473-477. [DOI] [PubMed] [Google Scholar]
- 36.Zeidler C, Welte K, Barak Y, et al. Stem cell transplantation in patients with severe congenital neutropenia without evidence of leukemic transformation. Blood. 2000;95: 1195-1198. [PubMed] [Google Scholar]
- 37.Ferry C, Ouachee M, Leblanc T, et al. Hematopoietic stem cell transplantation in severe congenital neutropenia: experience of the French SCN register. Bone Marrow Transplant. 2005;35: 45-50. [DOI] [PubMed] [Google Scholar]