

Abstract
Mesenchymal stem cells (MSCs) are mostly found around the vasculature system of the adult bone marrow (BM). They function as immune suppressors, express MHC-II, are phagocytic, and support T-cell cytotoxicity. We hypothesize that these contradictory properties of MSCs are important for BM homeostasis and occur partly through antigen presentation (antigen-presenting cells [APCs]) within a narrow window. Indeed, we have verified APC functions of MSCs to recall antigens, Candida albicans and Tetanus toxoid. The target cells have been identified to be CD4+ T cells. APC assays with IFNγ-knockdown MSCs and with anti–IFNγ receptor confirmed that MHC-II expression requires autocrine stimulation by IFNγ. During APC functions, as IFNγ levels become elevated, there was a concomitant decrease in MHC-II on MSCs. This observation was correlated with flow cytometry studies showing a gradual decrease in MHC-II expression as IFNγ levels were increased. The reduced levels of MHC-II correlated with losses in their allogeneic potential, as indicated in mixed lymphocyte reaction. In summary, endogenous and low levels of IFNγ are required for MHC-II expression on MSCs, and for APC functions. APC functions occur during a narrow window before IFNγ levels are increased. The study has implications for BM protection against infection and exacerbated inflammatory responses.
Introduction
The adult bone marrow (BM) is host to 2 major stem cells: hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs).1,2 While HSCs reside close to the endosteum, MSCs surround the vasculature and trabeculae.3-7 MSCs differentiate along multiple lineages and form cells of mesodermal lineage and can also generate cells of other germ layers through the process of transdifferentiation.8 Studies have shown the generation of functional neurons from MSCs.8 In the adult, MSCs could also be found in the circulation and other tissues.8-10
MSCs exert immune properties that make them desirable for transplantation across allogeneic barriers. These include veto functions and blunting effects on the maturation of professional antigen-presenting cells (APCs).11,12 MSCs, however, appear to be functionally plastic, and, when required, they can modulate their immune-suppressive effects and facilitate T-cell cytotoxic responses to viral infection, albeit at reduced efficiency.13 Recent findings have shown conditional evidence for APC function of MSCs in the presence of interferon-γ (IFNγ).14 These studies are not surprising since MSCs have been reported to express MHC class II (MHC-II), respond to proinflammatory cytokines, and exert phagocytic properties.11,15-18 Despite these immune-enhancing properties of MSCs, they have been reported to suppress immune rejection in different species, including human and nonhuman primates.19-21 However, tolerance of MSCs across the allogeneic barrier might not be absolute since studies by another group report on experimental evidence that questions the status of immune privilege.22
The expression of MHC-II expression on MSCs raises 2 concerns. First, can unstimulated MSCs act as APCs? Second, if MSCs do exert APC function, how can this property explain their role as immune-suppressor cells? These 2 major questions are important and would be relevant to hematologic disorders, especially since the literature reports on dysfunction and abnormal cytogenetics in MSCs from patients with severe aplastic anemia and myelodysplastic syndrome.23-25 Thus, an understanding of the biology of MSCs, in the context of hematopoiesis, could be relevant to hematologic dysfunctions. The studies in this report are based on the overarching hypothesis that MSCs act as a “gatekeeper” in the BM by regulating the types and behavior of cells traversing the BM. We report a role for MSCs as APCs in a model where the readout is based on CD4+ T-cell proliferation. The dual roles of MSCs as APCs and as immune-suppressor cells can be explained by mechanisms that are influenced by the levels of IFNγ levels. The relevance of this report to BM protection is discussed.
Materials and methods
Reagents
Fluoresbrite plain YG 1.0-μm microspheres were purchased from Polysciences (Warrington, PA). Candida albicans was purchased from Greer Laboratories (Lenoir, NC). Tetanus toxoid was obtained from Squibb and distributed by Antigen Supply House (Northridge, CA). Texas Red-Xphalloidin was purchased from Molecular Probes (Eugene, OR). Cell Dissociation Solution was obtained from Sigma (St Louis, MO).
Antibodies
The following antibodies were purchased from BD PharMingen (San Diego, CA): PE HLA-DR mAb, and FITC- and PE-mouse IgG isotypes. FITC anti-CD105 (SH2) was obtained from Cymbus Biotech (Eastleigh, United Kingdom). Goat anti–human IFNγ type I receptor (IFNγR1) and non–immune goat IgG were purchased from R&D systems (Minneapolis, MN). CD4 mAb was generated as ascites with hybridoma.
Culture of MSCs and monocyte isolation
MSCs were cultured from BM aspirates of healthy subjects as described.11 The use of human subjects adhered to guidelines outlined by the institutional review board of UMDNJ (Newark Campus, NJ). Briefly, BM aspirates were cultured in D10 media: DMEM (Sigma) and 10% fetal calf sera (FCS; Hyclone, Logan, UT). Based on the position outlined by the International Society for Cellular Therapy on the nomenclature of MSCs, we have designated the cells cultured for this study as MSCs.26 At passage 5, MSCs were verified by morphology, phenotype, and differentiation.8,11 The cells are adherent to plastic, morphologically symmetrical compared with the asymmetry of fibroblasts, replicated by asymmetry, expressive of telomerase, and able to differentiate into several cell lineages. Based on these indicators, we have achieved purity of more than 99%.
Monocytes were isolated from peripheral blood mononuclear cells (PBMCs) as described.27 Briefly, the adherent population was selected in dishes, and precoated with gelatin and autologous fibronectin. Adherent cells (> 95%) were positive for CD14+ and nonspecific esterase. Macrophages (MΦs) were prepared by culturing monocytes for 1 week in R10 media (RPMI 1640 and 10% FCS).
Phagocytic assay
MSCs or MΦs were incubated at 37°C with fluoresbrite plain YG microspheres at 1 μL/106 cells. After 2 hours, MΦs were washed and then mounted with 1% glycerol. MSCs were permeabilized with 0.1% TritonX-100 and then incubated with Texas Red–X-phalloidin at 28 U/mL for 90 minutes. Both macrophages and MSCs were examined by confocal fluorescence microscopy.
APC assay
Day 1: cell activation. PBMCs (2 × 106/mL) were incubated with 10 μg/mL (optimum in dose-response studies at 5-15 μg/mL) C albicans and T toxoid, at 1:100 final dilution (based on dose-response studies).11 Unactivated cells lacked C albicans.
Day 4: pulsing. MSCs and MΦs (2 × 104/mL) were incubated for 24 hours with 0.1 to 5 μg/mL C albicans or T toxoid. Unpulsed cells lacked antigens. MSCs were either untreated or pretreated with anti-IFNγR1 for 4 hours prior to pulsing.
Day 5: isolation of CD4+ T cells. PBMCs (106/mL) were positively selected by consecutive incubation with 50 μg/mL CD4 mAb and Dynabead goat anti–mouse IgG (Dynal, Oslo, Norway). Immunofluorescence showed more than 95% was positive for CD4.
Day 5: assay. Pulsed MSCs and MΦs were resuspended in D10 media at 106/mL and then subjected to 20 Gy γ-irradiation. Irradiation renders the cells in cycling quiescence, but they remain metabolically active. CD4+ cells (4 × 104/mL) were added to 50, 102, 103, or 104/mL γ-irradiated MSCs or MΦs. Similar cultures contained 0.1 to 5 μg/mL anti-IFNγR1 or isotype control. After 24 hours, cells were pulsed with 1 μCi (0.037 MBq) [methyl-3H]-TdR/well. After 16 hours, cells were harvested and analyzed for radioactive incorporation, and the simulation indices (SIs) were calculated by dividing the dpm of experimental points by dpm of unactivated CD4+ T cells.
Northern analysis
Northern analysis was performed for IFNγ with 10 μg total RNA as described.13 RNA was developed with a cDNA probe, randomly labeled with [α-32P]-dATP, 111 Tbq/mM (Dupont/NEN, Boston, MA), using Prime-IT II random primer kit (Stratagene, La Jolla, CA). Normalization was done with cDNA for 18S rRNA. Bands were scanned after 24 hours using the Typhoon phosphoimager (Amersham Biosciences, Piscataway, NJ). IFNγ cDNA probe was previously described.13
ELISA for IFNγ levels
The media in cultures of MSCs, at 80% confluence in 100 × 17-mm culture dish, were replaced with 2 mL fresh media. After 16 hours, cell-free culture media were collected and placed in siliconized tubes and then studied for IFNγ levels by enzyme-linked immunosorbent assay (ELISA) with a kit purchased from R&D Systems.
Flow cytometry
At 80% confluence, MSCs were incubated with 10 or 100 U/mL IFNγ. At different times, the MSCs were de-adhered with dissociation solution, washed with 1 × PBS (pH 7.4), and then incubated for 30 minutes at room temperature with PE anti–HLA-DR (MHC-II) or indirectly with anti-IFNγRI, each antibody at a final concentration of 1:500. For indirect labeling, the cells were washed and then incubated for 30 minutes with FITC anti–goat IgG at 1:2000 final concentration. Control slides were incubated with secondary antibody alone or isotype control. Cells were fixed with 0.4% paraformaldehyde and then analyzed by FACScan (FACS Caliber; Becton Dickinson, Franklin Lakes, NJ).
Statistical analysis
Data were analyzed using analysis of variance and Tukey-Kramer multiple comparisons test. A P value of less than .05 was considered significant.
Results
Phagocytic properties of MSCs
MSCs have been shown to engulf foreign particles.16-18 To ascertain that the MSCs propagated in our laboratory exhibit similar function, MSCs from 4 different donors were incubated with 1-μm fluorescence particles and then examined by confocal fluorescence microscopy. Autologous macrophages served as positive control. Figure 1 showed representative images for MΦs (Figure 1A) and MSCs (Figure 1B) in 3 planes: upper, middle, and lower. There was relatively bright fluorescence in the middle layers for both MΦs and MSCs (Figure 1A). This section provides evidence to show phagocytic functions of MSCs.
Figure 1.

Phagocytic and APC functions of MSCs. Macrophages and MSCs were treated with fluorescence microspheres for 2 hours and then examined by confocal microscopy. Three planes are shown in representative microscopic studies from 4 experiments, each performed with cells from different donors: (A) macrophages; (B) MSCs. Activated CD4+ cells were immunoselected from PBMCs that were challenged for 5 days with 10 μg/mL C albicans (C) or T toxoid, at 1:100 final dilution (D). Control studies were done in parallel in media alone (Unactivated CD4+). Activated CD4+ cells were added to autologous MSCs or macrophages, and exposed overnight to C albicans or T toxoid (Pulsed), or to media alone (Unpulsed). Cell proliferation was determined by [methyl-3H]-TdR incorporation, and the results were expressed as stimulation indices (SIs); mean ± SD (n = 6). Background disintegrations per minute (unactivated CD4+ and activated CD4+) were 308 ± 8 and 140 ± 4, respectively. *P < .05 versus unactivated CD4+ cells/pulsed and activated/unpulsed cultures. **P < .05 versus pulsed MSCs + activated CD4+ cells.
Antigen-presenting properties of MSCs
MSCs act as immune suppressors in a setting of allogeneic responses, but showed no effect during responses to recall antigens.11 In addition, despite the presence of MSCs, during viral infection, cytotoxic responses were elicited, although at less efficiency.11,13 MSC-derived IFNγ was critical for the development of T-cytotoxic responses to viral infection.13 Another interesting observation was that subsets of MSCs express low density of MHC class II,11 indicating that the immune properties of these cells are complex. This section determined whether MSCs could act as APCs when challenged with 2 different recall antigens, C albicans and T toxoid.
The APC assays are based on the proliferation of antigenspecific CD4+ cells (activated) to autologous MSCs that were preincubated with the antigen (pulsed). Macrophages are professional APCs and therefore served as references to determine the relative efficiency of APC functions by MSCs. Antigen-activated CD4+ T cells were incubated with pulsed or unpulsed MSCs or macrophages. Time-course studies at 24 and 48 hours indicated 48 hours as the optimum time for the proliferation of CD4+ T cells.
Figure 1C shows the mean SI ± SD (n = 6) of CD4+ T-cell proliferation in APC assays with C albicans. Each experiment was performed with cells from a different donor. The 2 left data sets represent unpulsed macrophages and MSCs cultured with unactivated or activated CD4+ T cells. There were no significant (P > .05) differences in cell proliferation. The 2 right data sets (Figure 1C) show studies in which pulsed macrophages and MSCs were incubated with activated and unactivated CD4+ cells. There was significant (P < .05) proliferation by activated CD4+ T cells compared with unactivated CD4+ T cells. The proliferation observed in cultures with pulsed MSCs and activated CD4+ T cells was significantly less (P < .05) than similar studies with macrophages (Figure 1C). However, there was significant (P < .05) proliferation of pulsed MSCs cocultured with activated CD4+ T cells compared with coculture with unactivated CD4+ T cells (Figure 1C second set of graphs from the right). This suggests that although MSCs showed APC properties, the efficiency was less than macrophages. To verify that the APC function of MSCs exposed to C albicans was not an artifact, the studies were repeated with T toxoid and the results showed a similar response (Figure 1D). MSCs differentiated toward fibroblasts do not show APC functions (data not shown). In summary, studies with 2 different antigens show evidence of MSCs exhibiting APC properties.
Role of endogenous IFNγ on MHC-II expression in MSCs
The next set of studies was aimed to get insights on the mechanism that could reconcile the dual but opposing functions of MSCs: APCs (Figure 1) and immune-suppressor effects.11,20 We used the veto property of MSCs as a basis to formulate a hypothesis. In general, veto function occurs in a setting of graft-versus-host responses where the microenvironment would have high levels of inflammatory mediators, such as IFNγ.28 We proposed that the APC property of MSCs occurred during a narrow window of infection, specifically when the level of the inflammatory mediator IFNγ was low. We further propose that at high levels of IFNγ, MHC-II expression was decreased. Consequently, the MSCs are able to act as veto cells.11 To test our hypothesis, we first studied the timeline expression of MHC-II in MSCs incubated with 2 concentrations of IFNγ: low (10 U/mL) and high (100 U/mL).
MSCs were incubated with IFNγ and, at different times, they were studied for MHC-II expression by flow cytometry using PE anti–HLA-DR. Representative results for 4 different experiments, each with MSCs from a different donor, are shown in Figure 2A. The isotype controls and anti–HLA-DR–labeled cells are presented as solid and open graphs, respectively, in the figure. Each time point was performed as unstimulated and IFNγ-stimulated cultures, and were therefore analyzed by flow cytometry as paired samples. The mean fluorescence intensities (MFIs) ± SD for all studies are shown in the panels. MSCs incubated with 100 U/mL IFNγ showed significantly (P < .05) reduced MFI compared with similar cultures with 10 U/mL IFNγ. The MFI at 100 U/mL IFNγ showed a trend toward increase in MHC-II at 16 hours following IFNγ exposure. Incubation with IFNγ at both concentrations did not result in cell death, as indicated by trypan blue exclusion. The variability in cell numbers at different time points was similar for both stimulated and unstimulated cultures. In summary, there is a decrease in MHC-II expression at high levels of IFNγ, but unchanged at low IFNγ level. The decreased expression of MHC-II showed a reversed effect after prolonged exposure to high IFNγ levels (16-hour point).
Figure 2.

Effects of IFNγ concentrations on the expressions of MHC-II and IFNγRI on MSCs. Cells were stimulated with 10 U/mL or 100 U/mL IFNγ. After 4, 8, 12, and 16 hours, MSCs were studied for (A) MHC-II and (B) IFNγRI expression by flow cytometry. The results represent 4 experiments, each performed with MSCs from a different donor. The MFIs ± SD (n = 4) are shown for MHC-II in the panels. Solid histograms indicate isotype; open histograms, specific antibodies.
Effects of IFNγ on the type I receptor (IFNγRI)
Since MHC-II expression was decreased in MSCs stimulated with 100 U/mL IFNγ (Figure 2A), we next asked whether this could be explained by reduced expression of IFNγRI. In 4 experiments, MSCs were treated with 10 U/mL or 100 U/mL IFNγ, and, at different times, the cells were studied for IFNγRI expression by flow cytometry. Representative results for MSCs untreated (solid histogram) or treated with IFNγ (open histogram) are shown in Figure 2B. The top panel (Figure 2B) showed the histogram of isotype control (open histogram) compared with labeling with anti-IFNγRI. The results showed no change in IFNγRI despite treatment with IFNγ. This indicates that the down-regulation of MHC-II (Figure 2A) could not be due to decreased expression of IFNγRI.
Endogenous IFNγ and MHC-II expression in MSCs
We previously showed endogenous production of IFNγ by MSCs with cytokine protein arrays.13 We now quantitated IFNγ by ELISA and Northern analyses. Northern analyses with MSCs from 3 different donors showed similar band intensities for IFNγ mRNA (Figure 3A upper bands). ELISA determined the production of IFNγ protein in untreated MSCs for 5 different BM donors. The results showed levels ranging between 22 and 25 pg/mL (Figure 3A, lower section).
Figure 3.
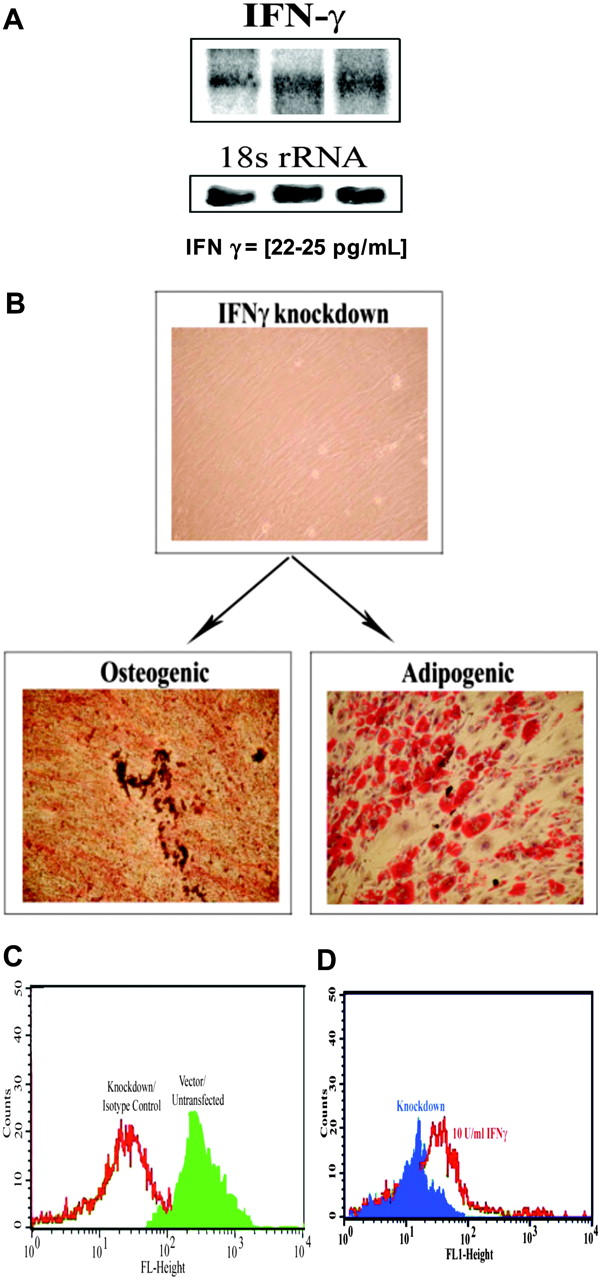
Role of endogenous IFNγ in MHC-II expression. (A) Northern blots for IFNγ using total RNA from MSCs derived from 3 different BM donors. Normalization was done with a cDNA probe for 18S rRNA. (B) Representative osteogenic and chondrogenic differentiation with MSCs, stably knocked down for IFNγ. (C) Representative flow cytometry for MHC-II in MSCs knocked down for IFNγ. (D) IFNγ (10 U/mL) was re-added to cultures of knockdown MSCs. After 24 hours, the cells were studied for MHC-II by flow cytometry. Figure represent 4 experiments, each with a different donor.
Since IFNγ has been shown to regulate the expression of MHC-II in immune cells,29 we asked whether the endogenous low level of IFNγ could be responsible for maintaining MHC-II expression on the MSCs.11 This premise is supported by similar expression of MHC-II in unstimulated MSCs and those exposed to 10 U/mL IFNγ (Figure 2A). The question posed in this section was addressed with IFNγ knockdown MSCs.13 Suppression of IFNγ did not lead to spontaneous differentiation since the knockdown cells were capable of forming osteogenic and adipogenic cells (Figure 3B), and formed NeuN-positive cells following treatment with retinoic acid (not shown).9 Cells were suspended in a 50% PBS/50% glycerol solution and stained with actin phalloidin Alexa Fluor 488. Images were acquired with a Nikon Eclipse TS100 microscope (Nikon, Tokyo, Japan) with a 100 ×/0.3 NA objective, a Nikon PCM 2000, camera, and C. Imaging Series software (Compix Imaging Systems, Orandberry, PA). Flow cytometry for MHC-II in the IFNγ knockdown MSCs showed undetectable fluorescence. A representative study for 4 experiments, each with a different donor, is shown in Figure 3C. We observed overlapping fluorescence for untreated MSCs labeled with isotype control (Figure 3C, open graph) and IFNγ-knockdown MSCs. It should be noted that the cells were stably transfected with the siRNA plasmid. Untreated and MSCs transfected with the siRNA vector alone showed MHC-II expression and overlapping fluorescence (Figure 3C, solid graph).
To further prove that the endogenous levels of IFNγ are responsible for maintaining MHC-II expression in unstimulated MSCs, we re-added 10 U/mL IFNγ to the knockdown MSCs. After 16 hours, the cells were studied for MHC-II by flow cytometry. The MFIs for 4 experiments, each with cells from a different donor, were increased to 750 ± 15, compared with 125 ± 8 for untreated knockdown cells. Representative fluorescence studies, shown in Figure 3D, indicate background fluorescence for knockdown cells (solid graph), compared with increased fluorescence in add-back experiments (open graph). In summary, this section describes studies showing a role for endogenous IFNγ on the expression of MHC-II in MSCs.
Allogeneic functions of IFNγ knockdown MSCs
The significance of reduced MHC-II expression when IFNγ levels were increased was addressed in functional studies. The MLR is a hallmark of allogeneic differences and therefore served as functional readouts to determine the relationship between high levels of IFNγ and MHC-II expression. Furthermore, MSCs have been shown to elicit allogeneic responses.11 We treated MSCs from 7 different donors with 100 U/mL IFNγ. After 8 hours, the cells were washed and then used as stimuli in MLR reactions. MSCs from each donor were tested with PBMCs from 2 different donors (n = 14). Allogeneic differences between the donors of PBMCs and MSCs were verified in one-way MLR with their PBMCs (data not shown). MSCs were pretreated with IFNγ to serve as stimulator cells in MLR. The results showed significantly (P < .05) reduced cell proliferation of responder PBMCs, compared with MLR with untreated MSCs (Figure 4A). Immunofluorescence studies for MHC-II were performed immediately before the treated MSCs were added to the cultures, and the results showed undetectable surface expression.
Figure 4.

IFNγ regulates the allostimulator activity of MSC. MLR with MSC stimulators, pretreated with IFNγ (A), or knockdown for IFNγ (B). (A) MSCs from 7 different donors were incubated with 100 U/mL IFNγ for 8 hours and then used as stimulators in MLR. MSCs from each donor were studied with PBMCs from 2 different donors. (B) MSCs from 5 different donors were stably knocked down for IFNγ using wild-type or mutant siRNA, then used as stimulators in MLR. Control MLR comprised untransfected MSCs or those stably transfected with vector alone (pPMSKH1). *P < .05 versus untreated MSCs. **P < .05 versus other experimental groups. Results are expressed as mean SI ± SE.
We next used similar MLR assays to verify a role for endogenous IFNγ in MHC-II expression by siRNA technology. MSCs, stably knocked down for IFNγ, served as stimulator cells in MLR. Control stimulators were MSCs either stably transfected with vector alone (pPMSKH1), transfected with mutant siRNA, or untransfected. In 5 different experiments, wild-type siRNA resulted in significantly (P < .05) reduced proliferation (SI) of PBMCs (Figure 4B). There were comparable SIs for MSCs untransfected, vector transfectants, and mutant siRNA transfectants (Figure 4B). Together, the results show the reduced allogeneic potential of MSCs that were incubated with 100 U/mL IFNγ.
Role of endogenous IFNγ on the APC function of MSCs
Unpulsed MSCs produced low levels of IFNγ with concomitant expression of MHC class II (Figures 2, 3). Furthermore, in the presence of a relatively higher concentration of IFNγ, the allogeneic responses of MSCs were reduced (Figure 4A). This reduction correlated with a decrease in MHC-II expression (Figure 2A). The question posed is how MSCs could act as APCs in a microenvironment of immune responses when IFNγ levels are expected to be elevated? We therefore propose that the APC function of MSCs is limited to a period before inflammatory responses. We further propose that the APC function of MSCs requires the production of endogenous IFNγ for MHC-II expression (Figure 2A).
The question posed above was addressed by taking advantage of IFNγ receptor type I (IFNγRI) expression on MSCs19 (Figure 2B). APC assays with C albicans and T toxoid were performed with MSCs pretreated with anti-IFNγR1. The antibody was retained in cultures throughout the assay period. The goal was to block the response of endogenously produced IFNγ, thereby preventing the expression of MHC-II. The levels of IFNγR1 antibody used in assays ranged between 0.1 and 5 μg/mL. Parallel cultures contained equivalent concentrations of isotype control. We have determined the optimum concentration of anti-IFNγRI at 1 μg/mL based on the highest MFI in immunofluorescence studies. At 1 μg/mL anti-IFNγRI, we could not activate Stat1 in cells treated with 10 U/mL IFNγ, as determined by gel shift assays (not shown).
The results for studies with T toxoid and C albicans were similar. The results, shown for the optimum level of anti-IFNγRI indicate significantly (P < .05, n = 5) reduced proliferation of activated CD4+ T cells in cultures with pulsed MSCs (Figure 5A right group). Similar reduction was not observed in parallel cultures containing isotype control (Figure 5A second set of bars from the right).
Figure 5.

IFNγ levels in MHC-II expression and APC function of MSCs. (A) APC assays were established as for Figure 1B, in the presence or absence of 1 μg/mL anti-IFNγRI or isotype control. At different times, culture media were determined for IFNγ levels and the CD4+ cells, and flow cytometry was done with the adherent cells colabeled with FITC anti-CD105 (for MSC) and PE anti–HLA-DR. Figure represents 4 different experiments. (B) Studies were set up as for Figure 1B, except that MSCs were pretreated with IFNγ-RI antibody or isotype control. The results are presented as the mean ± SD (n = 5). Background disintegrations per minute (unactivated CD4+ and activated CD4+) were 577 and 653, respectively. *P < .05 versus cultures of pulsed/activated/isotype control. (C) MSCs were incubated for 2 or 12 hours with IFNγ at 10 or 100 U/mL IFNγ. After this, cells were washed and then used in APC assays. *P < .05 versus unstimulated or 10 U/mL IFNγ;**P < .05 versus similar cultures with 100 U/mL IFNγ.
The next set of studies determined whether the blunting effects of anti-IFNγRI correlated with reduced expression of MHC classII. The experiments shown in Figure 5A were repeated, and at 2 hours and 12 hours after the APC assay began, the MSCs were examined for MHC class II by immunofluorescence. The adherent cells in the APC assays were double-labeled with FITC anti-CD105 (MSC marker) and PE anti–HLA-DR. Representative results with C albicans are shown in Figure 5B. The studies represent 10 different experiments, 5 with C albicans and 5 with T toxoid. Control MSCs (not placed in the APC assay), but treated with anti-IFNRI for 12 hours (Figure 5A top panel), showed reduced MFI (solid graph), whereas isotype-treated cells showed increased MFI (open graph). MSCs taken from APC assays at 2 hours showed a gradual reduction in MFI compared with isotype-treated MSCs (Figure 5B middle panel). This decrease in MFI decreased to undetectable levels at 12 hours regardless of whether the cultures consisted of isotype control or anti-IFNγRI (Figure 5B lower panel). The results show a requirement for IFNγRI activation in order to induce the APC function of MSCs. The results also show a gradual reduction in MHC class II expression on MSCs placed in APC assays.
IFNγ levels in APC cultures
The reduced expression of MHC-II in MSCs from APC assays in which anti-IFNγRI was present (Figure 5B lower panel) indicates a situation that makes the MSCs unresponsive to IFNγ with respect to the expression of MHC-II. Thus, these cells would not be able to act as APCs. The MSCs exposed to isotype control for 2 hours in APC assays showed detectable MHC-II (Figure 5B middle panel). This contrasted with similar cultures at 12 hours with isotype control (Figure 5B, bottom panel). We therefore asked whether the time-dependent differences in MHC-II expression correlated with changes in IFNγ levels. The supernatants obtained from APC assays with pulsed MSCs, activated CD4+ cells, and isotype control were quantitated for IFNγ level by ELISA (R&D Systems). In 5 different experiments, we observed 75 ± 6 pg/mL and 402 ± 10 pg/mL IFNγ at 2 and 12 hours, respectively. The IFNγ levels were converted to activity (U/mL) in a bioassay and the results showed 75 to 80 pg/mL and 400 to 450 pg/mL being equivalent to 35 ± 2 U/mL and 130 ± 10 U/mL. The results showed a correlation between IFNγ levels and MHC-II expression, in a time-dependent manner.
Time-dependent effects of IFNγ on APC functions of MSCs
Blocking studies with anti-IFNγRI showed decreases in APC functions (Figure 5A). In addition, there was a decrease in MHC-II expression in MSCs obtained from 12-hour APC assay (Figure 5A-B). We next determined whether MSCs, preincubated with IFNγ at 10 and 100 U/mL for 2 and 12 hours, could affect their ability to act as APCs. Studies with both C albicans and T toxoid showed significantly (P < .05) reduced proliferation of CD4+ cells when the MSCs were incubated with 100 U/mL IFNγ, compared with MSCs incubated with 10 U/mL IFNγ (Figure 5C). The time at which the MSCs were exposed to 100 U/mL IFNγ was important since 12-hour incubation showed significantly (P < .05) less proliferation of CD4+ cells compared with 2-hour incubation (Figure 5C).
IFNγ levels in APC assays with IFNγRI-pretreated MSCs
Four sets of data formed the basis for the studies described in this section: (1) APC assays with anti-IFNγRI showed significant reduction in cellular immune responses (Figure 5A); (2) the presence of IFNγRI and high levels of IFNγ led to decreased expression of MHC-II (Figure 5B); (3) knockdown of IFNγ led to decreased expression of MHC-II (Figure 3C); and (4) exogenous IFNγ restored the expression of MHC-II on MSCs (Figure 3D). Since MSCs could present antigen only if MHC-II was expressed, the aforementioned 4 points indicate that these cells would be able to present antigen only at a time before IFNγ levels were increased. We therefore determined the levels of IFNγ from cultures in which MSCs were pretreated with anti-IFNγRI or isotype control. The data, described in Table 1, showed a significant decrease in IFNγ levels for cultures in which MSCs were pretreated with anti-IFNγRI compared with isotype control, as described in the table's footnotes.
Table 1.
IFNγ levels in APC cultures with MSCs pretreated with anti-IFNγRI
| Cells/pretreated MSCs | IFNγ, pg/mL |
|---|---|
| Unpulsed MSCs | |
| Isotype | 20 ± 4 |
| Anti-IFNγRI | 22 ± 2 |
| Pulsed MSCs | |
| Isotype | 120 ± 8 |
| Anti-IFNγRI | 30 ± 3 |
| Activated CD4+ | |
| None | 88 ± 7 |
| Unactivated CD4+ | |
| None | 5 ± 0.5 |
| Unpulsed MSCs + unactivated CD4+ | |
| Isotype | 18 ± 3 |
| Anti-IFNγRI | 15 ± 2 |
| Unpulsed MSCs + activated CD4+ | |
| Isotype | 92 ± 5 |
| Anti-IFNγRI | 78 ± 5 |
| Pulsed MSCs + unactivated CD4+ | |
| Isotype | 160 ± 5 |
| Anti-IFNγRI | 28 ± 3 |
| Pulsed MSCs + activated CD4+ | |
| Isotype | 385 ± 12* |
| Anti-IFNγRI | 60 ± 6* |
MSCs from 4 different donors were subjected to overnight incubation with 0.1 to 1 μg/mL IFNγRI. Cells were washed and then pulsed with C albicans or T toxoid. After this, the MSCs were washed and then placed in APC cultures in the presence of anti-IFNγRI (0.1-1 μg/mL) or isotype control. After 24 hours, culture supernatants were quantitated for IFNγ by ELISA. The data are shown for values at 1 μg/mL anti-IFNγRI.
These values showed significant (P < .05) differences between anti-IFNγRI and isotype control.
Discussion
The following summarizes the major findings in this report: (1) APC properties of MSCs have been demonstrated for 2 recall antigens, C albicans and T toxoid; (2) MHC-II expression on MSCs requires autocrine stimulation by endogenous, but low, IFNγ levels; and (3) at high IFNγ levels, MHC-II is decreased, causing loss in the ability of MSCs to act as APCs. This third point formed the basis for our conclusion that MSCs could act as APCs during a narrow window of being exposed to at least 2 recall antigens (Figure 6). As proof of principle, the MSCs incubated for 12 hours showed loss of their ability to act as APCs (Figure 5C). Thus, it appears that increased immune response with concomitant elevation of IFNγ levels could lead to reduced expression of MHC-II (Figures 2A, 4A, and 5). Although the significance of reduced MHC-II expression has not been addressed in this study, it may be implied that the MSCs could change their functions from APCs to immune suppressors.
Figure 6.

An image that summarizes the relative functional responses of MSCs in the presence or absence of C albicans or T toxoid. The studies show APC properties heightened during a small window when IFNγ levels are less than 25 pg/mL. As IFNγ levels are increased, the efficiency of APC functions is decreased. Based on previous studies, the reduced efficiency could correlate with the MSCs switching roles as immune-suppressor cells.
There are 2 questions that arise in trying to understand why MSCs exhibit dual roles: (1) Why do MSCs act as immune suppressors during the late stages of the immune response, or at a later time after being exposed to recall antigens? (2) What is the advantage of MSCs to decrease the expression of MHC-II during exacerbated immune response? The premise of immune suppressor during the later stage of an immune response is in line with previous findings showing MSCs acting as veto cells.11 The MSCs would be required to exert veto function during graft-versus-host response, which is a disorder correlating with high IFNγ levels.28 The physiologic significance for halting the APC function of MSCs could be partly explained by the anatomic location of these cells surrounding blood vessels of the BM.6 As the vasculature system controls cells traversing the BM cavity, the vessels' integrity needs to be maintained. Thus, an infection will require quick clearance at this periphery/BM boundary, whereas an exacerbated or prolonged, localized response could be detrimental to the blood vessel. Another reason for blunting the APC functions could be relevant to hematopoietic homeostasis. Inflammation could be suppressive to hematopoietic activities and would therefore be undesirable in the setting of hematopoiesis.30 Recent reports indicate dysfunctions in MSCs from patients with hematologic disorders, known to be linked to immune-mediated mechanisms, such as aplastic anemia.23 Thus, the MSCs might be cells with functions to offset exacerbated inflammatory responses, when cytokines such as IFNγ are increased. Such negative feedback of MSCs on the inflammatory response is particularly important since IFNγ has also been linked to aplastic anemia.31 At high levels, IFNγ acts as a hematopoietic suppressor,32,33 thus it would be undesirable for the BM to have an ongoing inflammatory reaction. Although speculative, these arguments are in line with the recent report of MSCs being significant to hematologic disorders.23-25
Since the MSCs were pulsed with the antigen, and then presented to CD4+ cells that were preactivated with the same antigen, it is deduced that the responses are toward CD4+ T cells that have recently been primed with the recall antigen. Therefore, our studies suggest that MSCs can act as APCs in expanded memory T cells. Future studies are planned to examine the efficiency of MSCs to long-term memory T cells. The APC assays used autologous MSCs and PBMCs. Each donor reported vaccines for tetanus. Since most individuals have been exposed to C albicans, this study was limited to recall antigens. If MSCs were able to efficiently present antigen, then the pulsed MSCs should have been able to elicit the proliferation of unactivated CD4+ cells. We, however, did not observe a marked increase in CD4+ proliferation when the pulsed MSCs were exposed to unactivated CD4+ cells (Figure 1C-D). These cellular-based readouts need further investigation on the efficiency of MSCs as APCs since the IFNγ levels in assays with pulsed MSCs and unactivated CD4+ T cells might suggest slight responses (Table 1). Thus, based on the studies, we could claim only that MSCs act as APCs when they are exposed to CD4+ cells that have been challenged with the recall antigens. Future studies with naive antigens are warranted to address these questions.
The proliferation of CD4+ cells served as readout of APC functions. Thus, it could be argued that the CD4+ cells were the target population in the APC function of MSCs. In general, CD4+ cells could be subdivided as functionally Th1 and Th2. Although we have quantitated IFNγ as increased in the APC assays, we cannot state that the response is distinctly a particular Th subtype. The reason for this uncertainty is based on a related study that reports lack of absolute characterization for Th-cell subsets based on cytokine production.34 In addition to IFNγ, we have also observed increased levels of IL-2 (not shown). Since MSCs have been reported to produce IL-2 following viral infection,13 further studies are required to link cytokine production and cellular subsets (MSCs vs T-cell subsets) during APC responses. Since we have argued that immune responses in the BM would negatively affect hematopoiesis, we then propose that the activated CD4+ cells would leave the BM into the lymphatic system. Future studies are planned with animal models to trace the path of activated cells.
MSCs placed in culture could be heterogeneous. However, the population of cells that was used in this report shows properties consistent with stem cells and responded to IFNγ in a biphasic manner: MHC-II at low levels and at the baseline, and undetectable to low expression at high levels. In other studies,35,36 100 U/mL IFNγ showed an increase in MHC-II by 48 hours for adult MSCs. This is partly in agreement with our studies since a similar concentration of IFNγ correlated with an increase in MHC-II by 16 hours (Figure 2A). However, the difference between other studies35 and this report lies in the baseline expression of MHC-II and with low levels of IFNγ (Figure 2A). At this time, we have no explanation for this difference although previous studies from our laboratory published low expression of MHC-II on MSCs.11 Perhaps our subset of MSCs might be different from the studies of Le Blanc et al.35 Similar studies by Le Blanc and colleagues (Gotherstrom et al36) using fetal MSCs showed a slow expression of MHC-II, as indicated by a necessary stimulation period of 7 days with 100 U IFNγ/mL. This suggests that MSCs might respond differently to IFNγ. In summary, this report clearly shows an intriguing role for MSCs, but also indicates that further studies are required to dissect their potential as BM gatekeeper cells.
Acknowledgments
We thank Julius Potian for his assistance with the osteogenic and adipogenic studies.
Prepublished online as Blood First Edition Paper, February 21, 2006; DOI 10.1182/blood-2006-01-0057.
Supported by grants from the FM Kirby Foundation, and by grants CA-89868 and AN-918096 awarded by the National Institutes of Health.
J.L.C. and K.C.T. contributed equally to the study.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Deans RJ, Moseley AB. Mesenchymal stem cells: biology and potential clinical uses. Exp Hematol. 2000;28: 875-884. [DOI] [PubMed] [Google Scholar]
- 2.Rosenthal N. Prometheus's vulture and the stem-cell promise. New Engl J Med. 2003;349: 267-274. [DOI] [PubMed] [Google Scholar]
- 3.Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO2 distributions in the bone marrow hematopoietic compartment: I, Krogh's model. Biophysical J. 2001;81: 675-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO2 distributions in the bone marrow hematopoietic compartment: II, modified Kroghian models. Biophysical J. 2001;81: 685-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quesenberry PJ, Colvin G, Abedi M. Perspective: fundamental and clinical concepts on stem cell homing and engraftment: a journey to niches and beyond. Exp Hematol. 2005;33: 9-19. [DOI] [PubMed] [Google Scholar]
- 6.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19: 180-192. [DOI] [PubMed] [Google Scholar]
- 7.Sakaguchi Y, Sekiya I, Yagishita K, Ichinose S, Shinomiya K, Muneta T. Suspended cells from trabecular bone by collagenase digestion become virtually identical to mesenchymal stem cells obtained from marrow aspirates. Blood. 2004;104: 2728-2735. [DOI] [PubMed] [Google Scholar]
- 8.Conget PA, Minguell JJ. Phenotypical and functional properties of human bone marrow mesenchymal progenitor cells. J Cell Physiol. 1999;181: 67-73. [DOI] [PubMed] [Google Scholar]
- 9.Cho KJ, Trzaska KA, Greco SJ, et al. Neurons derived from human mesenchymal stem cells show synaptic transmission and can be induced to produce the neurotransmitter substance P by interleukin-1α. Stem Cells. 2005;23: 383-391. [DOI] [PubMed] [Google Scholar]
- 10.Bensidhoum M, Chapel A, Francois S, et al. Homing of in vitro expanded Stro-1- or Stro-1+ human mesenchymal stem cells into the NOD/SCID mouse and their role in supporting human CD34 cell engraftment. Blood. 2004;103: 3313-3319. [DOI] [PubMed] [Google Scholar]
- 11.Potian JA, Aviv H, Ponzio NM, Harrison JS, Rameshwar P. Veto-like activity of mesenchymal stem cells (MSC): functional discrimination between cellular responses to alloantigen and recall antigens. J Immunol. 2003;171: 3426-3434. [DOI] [PubMed] [Google Scholar]
- 12.Beyth S, Borovsky Z, Mevorach D, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105: 2214-2219. [DOI] [PubMed] [Google Scholar]
- 13.Kang HS, Habib M, Chan J, et al. A paradoxical role for IFNγ in the immune properties of mesenchymal stem cells during viral challenge. Exp Hematol. 2005;33: 796-803. [DOI] [PubMed] [Google Scholar]
- 14.Stagg J, Pommey S, Eliopoulos N, Galipeau J. Interferon-γ-stimulated marrow stromal cells: a new type of non-hematopoietic antigen-presenting cell. Prepublished on November 17, 2005, as DOI 10.1182/blood-2005-07-2793. (Now available as Blood. 2006;107:2570-2577.) [DOI] [PubMed]
- 15.Silva WA, Covas DT, Panepucci RA, et al. The profile of gene expression of human marrow mesenchymal stem cells. Stem Cells. 2003;21: 661-669. [DOI] [PubMed] [Google Scholar]
- 16.Hill JM, Dick AJ, Raman VK, et al. Serial cardiac magnetic resonance imaging of injected mesenchymal stem cells. Circulation. 2003;108: 1009-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinds KA, Hill JM, Shapiro EM, et al. Highly efficient endosomal labeling of progenitor and stem cells with large magnetic particles allows magnetic resonance imaging of single cells. Blood. 2003;102: 867-872. [DOI] [PubMed] [Google Scholar]
- 18.Chan J, O'Donoghue K, de la Fuente J, et al. Human fetal mesenchymal stem cells as vehicles for gene delivery. Stem Cells. 2005;23: 93-102. [DOI] [PubMed] [Google Scholar]
- 19.Rasulov MF, Vasichenkow AV, Onishchenko NA, et al. First experience of the use bone marrow mesenchymal stem cells for the treatment of a patient with deep skin burns. Bull Exp Biol Med. 2005;139: 141-144. [DOI] [PubMed] [Google Scholar]
- 20.Bartholomew A, Sturgeon C, Siatskas M, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30: 42-48. [DOI] [PubMed] [Google Scholar]
- 21.Devine SM, Bartholomew AM, Mahmud N, et al. Mesenchymal stem cells are capable of homing to the bone marrow of non-human primates following systemic infusion. Exp Hematol. 2001;29: 244-255. [DOI] [PubMed] [Google Scholar]
- 22.Eliopoulos N, Stagg J, Lejeune L, Pommey S, Glipeau J. Allogeneic marrow stromal cells are immune rejected by MHC class I- and class II-mismatched recipient mice. Blood. 2005;106: 4057-4065. [DOI] [PubMed] [Google Scholar]
- 23.Bacigalupo A, Valle M, Podesta M, et al. T-cell suppression mediated by mesenchymal stem cells is deficient in patients with severe aplastic anemia. Exp Hematol. 2005;33: 819-827. [DOI] [PubMed] [Google Scholar]
- 24.Flores-Figueroa E, Arana-Trejo RM, GutierrezEspindola G, Perez-Cabrera A, Mayani H. Mesenchymal stem cells in myelodysplastic syndromes: phenotypic and cytogenetic characterization. Leuk Res. 2005;29: 215-224. [DOI] [PubMed] [Google Scholar]
- 25.Soenen-Cornu V, Tourino C, Bonnet M-L, et al. Mesenchymal cells generated from patients with myelodysplastic syndromes are devoid of chromosomal clonal markers and support short- and long-term hematopoiesis in vitro. Oncogene. 2005;24: 2441-2448. [DOI] [PubMed] [Google Scholar]
- 26.Horwitz EM, Le Blanc K, Dominici M, et al. Clarification of the nomenclature for MSC: the International Society for Cellular Therapy position statement. Cytotherapy. 2005;7: 393-395. [DOI] [PubMed] [Google Scholar]
- 27.Rameshwar P, Denny TN, Gascón P. Enhanced HIV-1 activity in bone marrow can lead to myelopoietic suppression partially contributed by gag p24. J Immunol. 1996;157: 4244-4250. [PubMed] [Google Scholar]
- 28.Ritchie D, Seconi J, Wood C, Walton J, Watt V. Prospective monitoring of tumor necrosis factor alpha and interferon gamma to predict the onset of acute and chronic graft-versus-host disease after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11: 706-712. [DOI] [PubMed] [Google Scholar]
- 29.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to IFN-γ. Annu Rev Immunol. 1997;15: 749-795. [DOI] [PubMed] [Google Scholar]
- 30.Nishikawa SI, Hashi H, Honda K, Fraser S, Yoshida H. Inflammation, a prototype for organogenesis of the lymphopoietic/hematopoietic system. Cur Opin Immunol. 2000;12: 342-345. [DOI] [PubMed] [Google Scholar]
- 31.Zeng W, Miyazato A, Chen G, Kajigaya S, Young NS, Maciejewski JP. Interferon-γ-induced gene expression in CD34 cells: identification of pathologic cytokine-specific signature profiles. Blood. 2006;107: 167-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Dybedal I, Bryder D, et al. IFN-γ negatively modulates self-renewal of repopulating human hemopoietic stem cells. J Immunol. 2005;174: 752-757. [DOI] [PubMed] [Google Scholar]
- 33.Kato K, Kamezaki K, Shimoda K, et al. Intracellular signal transduction of interferon on the suppression of haematopoietic progenitor cell growth. Br J Haematol. 2003;123: 528-535. [DOI] [PubMed] [Google Scholar]
- 34.Rosloniec EF, Latham K, Guedez YB. Paradoxical roles of IFN-γ in models of Th1-mediated autoimmunity. Arthritis Res. 2002;4: 333-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31: 890-896. [DOI] [PubMed] [Google Scholar]
- 36.Gotherstrom C, Ringden O, Tammik C, Zetterberg E, Westgren M, Le Blanc K. Immunologic properties of human fetal mesenchymal stem cells. Am J Obstet Gynecol. 2004;190: 239-245. [DOI] [PubMed] [Google Scholar]