

Abstract
A robust humoral immune response against human factor VIII (hFVIII) following naked DNA transfer into immunocompetent hemophilia A mice completely inhibits circulating FVIII activity despite initial high-level hFVIII gene expression. To prevent this undesirable response, we compared transient immunomodulation strategies. Eight groups of mice (n = 4-9 per group) were treated with naked DNA transfer of pBS-HCRHPI-hFVIIIA simultaneously with immunosuppressive reagents that included cyclosporine A (CSA), rapamycin (RAP), mycophenylate mofetil (MMF), a combination of CSA and MMF, a combination of RAP and MMF, a monoclonal antibody against murine CD40 ligand (MR1), recombinant murine Ctla4Ig, and a combination of MR1 and Ctla4Ig. All animals except those receiving only CSA exhibited delayed or absent immune responses against hFVIII. The most effective immunosuppressive regimen, the combination of Ctla4Ig and MR1, prevented inhibitor formation in 8 of 9 animals; the ninth had transient low-titer antibodies. All 9 mice of this group produced persistent, therapeutic levels of hFVIII for more than 6 months. When challenged with the T-dependent antigen bacteriophage Φx174, tolerized mice exhibited normal primary and secondary antibody responses, suggesting that transient immunomodulation to disrupt B/T-cell interaction at the time of plasmid injection effectively promoted long-term immune tolerance specific for hFVIII. (Blood. 2006;108:19-27)
Introduction
Hemophilia A is a congenital bleeding disorder caused by a deficiency of coagulation factor VIII (FVIII). Currently, hemophilia patients are treated with repeated infusions of plasma-derived or recombinant FVIII protein concentrates. Gene therapy is expected to provide a more beneficial and cost-effective treatment for this disease. We have demonstrated previously that naked DNA transfer of a liver-specific, high-expressing plasmid (pBS-HCRHPI-hFVIIIA) into Rag2(-/-) severe combined immune deficient (SCID) mice led to persistent highlevel expression of human factor VIII (hFVIII).1 Supraphysiologic expression levels of hFVIII also were achieved in immunocompetent hemophilia A mice, leading to complete but transient phenotypic correction during the first few days after gene therapy treatment. However, a strong humoral immune response against hFVIII subsequently completely inhibited circulating hFVIII activity in immunocompetent hemophilia A mice.2
The formation of inhibitory antibodies to infused hFVIII represents a major complication in the clinical treatment of hemophilia A patients. A similar problem is predicted to occur following gene therapy for hemophilia A. Various strategies have been explored to modulate the immune responses in hFVIII-treated hemophilia patients and in hemophilia A murine models undergoing repetitive hFVIII protein delivery. High-dose tolerance induction protocols are expensive and only partially effective.3,4 Anti-CD40L,5 cyclosporine A (CSA),6 and rituximab7,8 each have been successfully used to treat acquired hemophilia A in individuals who developed anti-FVIII autoantibodies. A partial suppressive response has been achieved in a group of hemophilia A patients using a combination of high-dose hFVIII tolerance induction and cyclophosphamide, with or without concomitant intravenous immunoglobulin (IVIG) therapy.9,10 Notably, both the immune response to hFVIII and the effectiveness of immunomodulation appear to differ significantly in patients with congenital and in patients with acquired hemophilia A, with more frequent responses and better results in the acquired cases. This difference is likely related, at least in part, to the sustained presence of circulating hFVIII antigen in the latter group of patients. In hemophilia A mice, acquisition of inhibitors requires expression of the B7 costimulating molecule, because treatment with Ctla4-Ig transiently blocks the response to repeated hFVIII protein infusion.11 Treatment with anti-CD40L to disrupt signaling via CD40 also delays anti-hFVIII antibody formation in this model.12,13 However, long-term tolerance has not been achieved in any of these animal models.12,13 Recently, several new strategies to prevent the production of inhibitory antibodies also have been reported, including high doses of FVIII to inhibit FVIII-specific memory B cells,14 oral feeding of FVIII-C2 domain,15 lipopolysaccharide (LPS)-activated B-cell blasts transduced with a fusion IgG containing the C2 or A2 domains of FVIII,16 and ex vivo transduction of hematopoietic stem cells.17
In the hFVIII gene transfer mouse model using naked DNA of a liver-specific, high-expressing hFVIII plasmid previously established in our laboratory,2 the expression of hFVIII leads to a predominantly Th2 immune response associated with high-titer anti-hFVIII antibodies that persist for the life of the animals. In this report, we have evaluated and compared multiple candidate immune suppression strategies designed to modulate or prevent the generation of hFVIII-specific T and B cells and the production of high-affinity inhibitory antibodies of the IgG isotype. To accomplish this, we designed protocols to eliminate or reduce key effector populations (antigen-specific B and CD4+ T cells), modulate T-helper-cell activation, alter T/B-cell interactions, or inhibit specific pathways of T- and/or B-lymphocyte activation. Immunomodulation strategies were designed to meet the following criteria: (1) only transient exposure to immunosuppression; (2) inhibition of one or more critical B- and/or T-cell activation pathways; (3) induction of minimal cytoablation and other associated toxicity; and (4) minimal effects on immune competence. These experimental criteria are essential for the translation of such protocols into human gene therapy trials. We demonstrate that suppression of immune responses against FVIII is most effectively achieved by combined immunomodulation therapy at the time of hFVIII plasmid transfer using a combination of murine Ctla4-Ig and anti-murine CD40L (MR1), and most importantly, that transient inhibition of costimulatory molecules can promote long-term immune tolerance that is specific for hFVIII, allowing long-term gene correction in immunocompetent hemophilia A mice.
Materials and methods
Construction of vectors
The construction of pBS-HCRHPI-hFVIIIA containing a B-domain deleted human factor VIII cDNA18 has been described previously.1
Animal experiments
Animals were kept in accordance with National Institutes of Health guidelines for animal care and the guidelines of the Children's Hospital and Regional Medical Center, Seattle, WA. Hemophilia A mice in 129/SV × C57BL/6 mixed genetic backgrounds were prepared by targeted disruption of exon 16 of the FVIII gene and provided by Drs Rita Sarkar and Haig Kazazian Jr at the University of Pennsylvania.19 All the mice were housed under specific pathogen-free conditions at the vivarium of the Children's Hospital and Regional Medical Center.
Immunosuppressive drugs and antibody
Antibody against murine CD40L (MR1) was a kind gift from Dr Randolph J. Noelle at Dartmouth Medical School, Lebanon, NH. Murine Ctla4-Ig was produced by BD Pharmingen (San Diego, CA), CSA by Bedford Laboratories (Bedford, OH), rapamycin (RAP) by Wyeth-Ayerst Laboratories (Madison, NJ), and mycophenolate mofetil (MMF) by Hoffman-La Roche (Nutley, NJ).
Delivery of plasmid DNA and immunomodulating agents
The methods of plasmid DNA preparation and DNA infusion have been described previously.1,2 Briefly, 50 micrograms of hFVIII plasmids in 2 mL phosphate-buffered saline were injected into the tail vein of 20 g to 24 g FVIII knock-out mice over 6 to 8 seconds.20-22 Scheduled blood samples were taken from the retro-orbital plexus.
Following plasmid injection, mice selected for transient immunosuppression received the immunomodulation components intraperitoneally according to the dosage and schedule listed in Table 1. Combination therapy was given using the same schedule and dosages as the single agent.
Table 1.
Dosage and schedule of delivery for single immunosuppressive agents
| Agent | Dosage and schedule* |
|---|---|
| CSA | 5 mg/kg/d; days 0-14 |
| RPA | 2 mg/kg/d; days 0-14 |
| MMF | 40 mg/kg/d; days 0-14 |
| Ctla4-lg | 5 mg/kg/d; days 0 and 2 |
| MR1 | 10 mg/kg/d; days - 1, 0, 2, 7, and 14 |
Day 0 represents the day of plasmid injection. At day 0, the immunosuppressive agents were given following the plasmid injection.
Via intraperitoneal injection. Combination regimens use the same combined schedule and dosages.
Assays for measuring human factor VIII antigen and its activity
hFVIII antigen levels were examined by enzyme-linked immunosorbent assay (ELISA) using ESH4/ESH8 monoclonal antibodies (American Diagnostica, Greenwich, CT) against hFVIII that are species-specific and do not cross-react with murine factor VIII. The background antigen level of hFVIII in untreated control mice was negligible. Serially diluted normal human plasma was used as standard. hFVIII activity was quantitated by a chromogenic assay (COATEST, measuring factor Xa generation; Chromogenix AB, Mölndal, Sweden), and a modified clotting assay using activated partial thromboplastin time (APTT) reagent and factor VIII-deficient plasma. hFVIII levels were calculated from a standard curve generated by using a series of dilutions of normal human pooled plasma.
Assays for anti-hFVIII antibodies and their subclasses
Inhibitory antibodies were measured by hFVIII Bethesda assays as previously described.23 Total antibodies against hFVIII were detected by an antigen-specific ELISA technique using a plasma-derived, monoclonal antibody-purified commercial factor VIII protein concentrate (Monarc-M, American Red Cross, prepared by Baxter Healthcare Corporation, Glendale, CA) and horseradish peroxidase-conjugated monoclonal antibodies against total murine IgG as described.1
Evaluation of hFVIII-specific IgG subclasses
Antigen-specific IgG subclasses were evaluated by ELISA using hFVIII-coated plates and horseradish peroxidase-conjugated antibodies for different murine IgG subclasses, including IgG1, IgG2a, IgG2b, and IgG3 (Roche Diagnostics, Indianapolis, IN). The microtiter plates (Corning, Corning, NY) were coated with purified human factor VIII protein (20 U/mL [1 U = 100 ng human factor VIII protein], Monarc-M), which had been extensively dialyzed against 50 mM imidazole buffer to remove low-molecular-weight additives. Plasma taken at different time points from plasmid-treated mice was diluted (1:100) and applied to the coated wells. Following incubation and extensive washing, antibodies of different IgG subclasses were added, followed by incubation with o-phenylenediamine dihydrochloride substrate. A standard curve for the different IgG subclasses was established using serially diluted mouse immunoglobulin reference serum (Bethyl Laboratories, Montgomery, TX). The background levels of hFVIII-specific IgG subclasses measured in untreated hemophilia A mice were negligible (< 10 ng/mL).
Cytokine production and proliferation assay by stimulated spleen cells
Spleens were removed aseptically from hemophilia A mice 180 days after plasmid and Ctla4-Ig and MR1 treatment, and single-cell suspensions prepared in RPMI 1640 medium (Invitrogen, Carlsbad, CA) containing 2 mM glutamine, 50 μM 2-mercaptoethanol (2-ME), 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal calf serum. Naive hemophilia A mice were used as controls. Red blood cells were lysed with sterile 17 mM Tris (tris(hydroxymethyl)aminomethane) and 140 mM NH4Cl buffer, pH 7.4. For cytokine production assays, isolated spleen cells (1 × 105 cells/200 μL/well) were stimulated on day 0 with 10 μg/mL phytohemagglutinin (PHA) or 20 U/mL hFVIII protein (Monarc-M, which had been extensively dialyzed against 50 mM imidazole buffer as described in “Evaluation of hFVIII-specific IgG subclasses”) or 4 μg albumin in imidazole buffer as control. Culture supernatants were collected over a 6-day period and stored at -80°C. Interleukin-2 (IL-2), interferon-γ (IFN-γ, and IL-10 were determined by ELISA using specific antibodies and standards purchased from BD Pharmingen. For assessment of proliferation, triplicates of isolated spleen cells (1 × 105 cells/200 μL/well) from hemophilia A mice 1 year after plasmid injection and Ctla4-Ig and MR1 treatment were stimulated with 10 μg/mL PHA or 1 to 20 U/mL hFVIII protein for 3 days. 3H-thymidine was then added, and cell cultures were harvested 18 hours later. The incorporation of 3H-thymidine was measured, and the results of triplicates expressed as mean counts per minute (cpm). The data presented (▵cpm) were the mean cpm minus background cpm. For both assays, plasmid-treated and naive untreated hemophilia A mice were used as controls.
Immunization with bacteriophage Φx174
Bacteriophage Φx174 was prepared as previously described.24 The stock solution of 1 × 1011 plaque-forming units (PFUs) per mL was diluted and injected intraperitoneally into tolerized and untolerized mice 6 months after plasmid injection at a dose of 1 × 1010 PFU/kg (2 × 108 PFU/mouse). A secondary immunization followed 4 weeks after the primary immunization.
Approximately 200 μL of blood was collected before immunization and 1, 2, and 4 weeks after each immunization. Aliquots of serum were stored at -80°C and subsequently analyzed for phage-neutralizing antibody activity, expressed as the rate of phage inactivation (Kv) using the standard formula24:
Kv = (dilution of serum/time of phage-serum incubation in minutes) × ln (phage assay PFU at time 0 minute/phage assay PFU at time 60 minutes).
Statistical analysis
For comparisons of animals developing inhibitory antibodies with control animals, the unpaired Student t test was performed, and P values less than .05 were considered statistically significant.
Results
We have shown previously that hemophilia A mice mount a T-cell-dependent humoral immune response to exogenous hFVIII following naked gene transfer of pBS-HCRHPI-hFVIIIA and that pretreatment with cyclophosphamide (CTX) induced long-term tolerance in 1 of 6 treated animals.2 The other 5 mice had delayed responses compared to untreated animals but still developed high-titer inhibitory antibodies.
Transient suppression of the inhibitory response with a single immunosuppressive agent
To develop a more effective immunomodulatory regimen, we evaluated the immunosuppressive effects of single agents on the formation of inhibitory antibodies to hFVIII following nonviral gene transfer. After rapid injection of 50 μ gof hFVIII plasmid in 2 mL saline into the tail vein, groups of mice were treated with either CSA, RAP, or MMF, according to the dose and schedule listed in Table 1. The mice treated with CSA produced high levels of hFVIII in the first 2 weeks following gene transfer, but levels then dropped to undetectable within the next few days (Figure 1C), concomitant with the appearance of antibody formation (Figure 1D). This pattern is indistinguishable from that observed in plasmid-treated mice without immunomodulation (Figure 1A,B). Mice treated with RAP had a slightly delayed immune response, with inhibitory antibodies appearing at 3 to 4 weeks after plasmid delivery. Eventually, however, the hFVIII concentrations dropped to undetectable levels, with the appearance of high-titer inhibitory antibodies as shown in Figure 1E,F. Among the 3 single-agent regimens, MMF was most effective in suppressing the immune responses against hFVIII. Of the 4 mice treated with MMF, 2 had a significantly delayed immune response with high-titer antibody appearing 4 to 8 weeks after plasmid delivery, when hFVIII levels dropped to undetectable levels (Figure 1G,H). The other 2 mice developed low-titer inhibitory antibodies 4 weeks after plasmid delivery and had a gradual drop of functional hFVIII levels, which stabilized at approximately 20% of normal levels during the observation period of 180 days following plasmid delivery (Figure 1G,H).
Figure 1.

Naked plasmid transfer of hFVIII plasmids into hemophilia A mice with immunosuppressive agents. hFVIII levels and inhibitory activity were assessed in hemophilia A mice after treatment with pBS-HCRHPI-hFVIIIA beginning on day 1. No transient immunosuppression (A,B), CSA (C,D), RAP (E,F), and MMF (G,H). Fifty μg plasmid in 2 mL saline solution was injected into the tail vein of mice (n = 4) in 5 to 8 seconds. Immunosuppressive drugs were administered intraperitoneally for 14 days starting from the day of the plasmid injection. Mice were then bled at regular intervals. Circulating hFVIII activities in plasma were evaluated by a modified clotting assay (A,C,E,G) and confirmed by a COATEST assay. Inhibitory antibody titers were evaluated by Bethesda assay and are expressed as BU/mL (B,D,F,H). For transient immunosuppression with combined agents, CSA and MMF (I,J) and RAP and MMF (K,L) are shown. Combined immunosuppressive drugs were given at the same schedule as the respective single agent (Table 1). Each symbol represents an individual mouse's results from both assays.
Antigen-specific IgG subclasses following single immunosuppressive agent therapy
It has been shown previously that the majority of anti-hFVIII IgG in this model is of the IgG1 isotype; however, small amounts of anti-hFVIII antibodies of the IgG2a, IgG2b, and IgG3 isotype also were detected. These findings suggest that transgene-specific antibodies are predominantly generated in response to Th2-induced activation signals.
To further characterize the nature of the immunosuppressive effect, we examined the anti-hFVIII IgG subclass specificity using an ELISA-based assay in plasmid-treated mice receiving singleagent immunomodulation therapy (Figure 2). Treatment with CSA did not significantly alter the production of any antigen-specific IgG isotype, except for a slight reduction of the IgG3 isotype antibodies. RAP treatment considerably reduced the production of IgG2b and IgG3 but not IgG1 and IgG2a isotypes. The 2 MMF-treated animals that exhibited long-term hFVIII expression and low-titer inhibitory antibodies did not generate detectable levels of inhibitors of any IgG isotype. The other 2 MMF-treated animals, which produced high-titer inhibitors, had a reduction in IgG1 and IgG2b, but not in IgG2a and IgG3 antigen-specific isotypes.
Figure 2.

Subclasses of anti-hFVIII IgG immunoglobulin in hemophilia A mice after naked plasmid transfer of pBS-HCRHPI-hFVIIIA with or without single agent immunosuppression. Titers of subclasses of IgG including IgG1, IgG2a, IgG2b, and IgG3 produced at 2 months following gene transfer were determined by IgG subclass-specific ELISA. All animals treated as described in Figure 1 (A-H) were tested, and their levels were averaged.
Combination immunosuppressive therapy with CSA/MMF or RAP/MMF
To further suppress the inhibitory antibody response to hFVIII, we explored the effects of combining 2 immunosuppressive agents; for example, CSA plus MMF, and RAP plus MMF. While delayed and reduced immune responses were observed in both of the combination therapies, high-titer inhibitory antibodies eventually developed and eliminated functional hFVIII levels in all treated animals (Figure 1I-L). Surprisingly, the combination therapies appeared to be less effective than MMF alone. It is unclear whether these differences reflect heterogeneity in the humoral immune response to hFVIII, as has been observed by others, or another mechanism that reduces the efficacy.12,25-27 Because the hemophilia A mice used in these studies are of mixed genetic background (129sv × C57BL/6), the resulting heterogeneity may have contributed to this variability.
Immunomodulation therapy with Ctla4-Ig, anti-CD40L mAb (MR1), or a combination of both agents
Three groups of plasmid pBS-HCRHPI-hFVIIIA-treated mice were evaluated (Figure 3): group 1 (n = 8) was injected with recombinant murine Ctla4-Ig (Figure 3A); group 2 (n = 9) with MR1, a monoclonal antibody against murine CD40 ligand (Figure 3C); and group 3 (n = 9) with the combination of MR1 and Ctla4-Ig (Figure 3E). When compared with plasmid-injected mice without immunomodulation (Figure 1A,B), treated mice from all groups showed delayed immune responses to hFVIII. Animals treated with Ctla4-Ig alone developed inhibitors 2 to 4 weeks after injection (Figure 3B), and all showed rapid loss of FVIII activity (Figure 3A). Of 9 animals treated with MR1, 6 developed inhibitors 4 to 6 weeks after treatment; among these, one mouse died after developing high-titer inhibitors 4 weeks after treatment. Of the remaining 3, one had a significantly delayed response but eventually developed high-titer inhibitors at 10 weeks; one developed low-titer antibody at 11 weeks after treatment; and the other failed to develop inhibitors and produced supraphysiologic levels of hFVIII throughout the observation period. Most notably, 8 of 9 animals treated with the combination regimen of MR1 and Ctla4-Ig failed to develop detectable inhibitors for at least 20 weeks (Figure 3F). The ninth animal developed transient, low-titer inhibitory antibodies that partially blocked the functional activity of hFVIII in plasma. All animals treated with the combination of MR1 and Ctla4-Ig exhibited persistent, therapeutic, or supraphysiologic levels of circulating hFVIII protein for more than 140 days (Figure 3E), demonstrating that transient immunomodulation given at the time of gene transfer can induce long-term tolerance following gene therapy. In addition, the expression levels of FVIII and inhibitor titers were followed in 3 tolerized mice for up to one year, and no significant changes were observed compared to those obtained at 5 months after treatment.
Figure 3.

Naked plasmid transfer of hFVIII plasmids into hemophilia A mice with or without immunomodulation (costimulation blockade). hFVIII levels and inhibitory antibody formation over time in hemophilia A mice after treatment with pBS-HCRHPI-hFVIIIA beginning on day 1. Transient immunomodulation included Ctla4-Ig on days 1 and 2(n = 8 total; A,B); MR1 on days -1, 1, 2, 7, and 14 (n = 9 total; C,D); and Ctla4-Ig with MR1 (n = 9 total; E,F) using the same combined schedule and dosages as with each individual agent. Fifty μg plasmid in 2 mL saline solution was injected into the tail vein of mice in 5 to 8 seconds. Two separate cohorts of animals (n = 4-5 per group) were used at separate times for each set of immunomodulation experiments; the data from 2 cohorts were combined and presented in the figure. Respective immunosuppressive drugs were administered intraperitoneally at indicated times. #Death of one treated mouse in panel C. Mice were bled at regular intervals. Circulating hFVIII activities and inhibitory antibody titers were evaluated as in Figure 1.
Antigen-specific IgG subclasses following combination immunomodulation therapy
We similarly evaluated the anti-hFVIII IgG subclasses in the plasmid-treated mice exposed to Ctla4-Ig and/or anti-CD40L mAb (MR1) (Figure 4). Treatment with Ctla4-Ig alone slightly reduced the production of IgG2b and, more significantly, IgG3, but did not alter significantly the production of IgG1 and IgG2a isotypes. MR1 treatment considerably reduced production of IgG2a, IgG2b, and IgG3, and moderately reduced the production of IgG1. As expected, mice treated with the combination of Ctla4-Ig and MR1 had undetectable levels of all 4 IgG isotypes, suggesting a synergistic interaction between these 2 agents.
Figure 4.

Subclasses of anti-hFVIII IgG immunoglobulin in hemophilia A mice after naked plasmid transfer of pBS-HCRHPI-hFVIIIA with or without immunomodulation (costimulation blockade). Titers of subclasses of IgG including IgG1, IgG2a, IgG2b, and IgG3 produced at 2 months following gene transfer were determined by IgG subclass-specific ELISA. All animals treated as described in Figure 3 were tested, and their levels were averaged except that in the MMF-treated group, the IgG levels from only 3 mice that developed inhibitors were averaged.
Cytokine release and proliferation assay following gene transfer and combination immunomodulation therapy
T-cell responses were evaluated by an in vitro proliferation assay in response to a wide range of hFVIII concentrations using splenocytes collected from mice at 1 year after plasmid treatment. Cells isolated from mice treated with hFVIII plasmid had significantly higher proliferation rates compared to those from tolerized mice treated with hFVIII plasmid and combination therapy of Ctla4-Ig and MR1 and untreated mice (Figure 5). The proliferative response is dependent on the hFVIII concentration in the culture medium. T-cell responses initiated by in vitro exposure to hFVIII also were examined using cytokine profiling of splenocytes collected from mice at 6 months after plasmid treatment. Cytokine production by splenic T cells isolated from plasmid-treated and untreated hemophilia A mice are shown in Figure 6. Both groups of animals produced large quantities of lymphokines following PHA stimulation. IL-10 secretion in response to hFVIII was significantly higher in plasmid-treated hemophilia A mice compared with untreated mice, whereas IL-2 and IFN-γ production was increased only slightly. These findings are consistent with a Th2-dominant immune response to hFVIII in this model. These results are comparable to those seen in animals that were challenged with hFVIII protein.28 Most importantly, mice tolerized by the combination therapy with Ctla4-Ig and MR1 showed neither an increase in cytokine production nor enhanced T-cell proliferation when stimulated with hFVIII.
Figure 5.

Proliferation assay following in vitro stimulation of T cells isolated from hemophilia A mice. Two animals per group were untreated (n = 2, ▵), hFVIII plasmid-treated (n = 2, ○), or hFVIII plasmid-treated receiving immunomodulation by combination of Ctla4 and MR1 (n = 2, ▴, animals from the group shown in Figure 3E,F). Splenic T cells were isolated from hemophilia A mice and cultured in the presence of hFVIII for 3 days. T-cell proliferation was measured in triplicate against a range of FVIII concentrations. Each data set represents the mean ▵cpm obtained from 2 mice.
Figure 6.

Cytokine production following in vitro stimulation of T cells isolated from hemophilia A mice. Four animals per group were untreated, hFVIII plasmidtreated, or hFVIII plasmid-treated with immunomodulation by combination of Ctla4 and MR1 (4 animals from the group shown in Figure 3E,F). Splenic T cells were isolated from hemophilia A mice and incubated with hFVIII for 6 days. Production of IL-2, INF-γ, and IL-10 was measured from the culture media using ELISA.
Long-term immune tolerance is specific for hFVIII
To evaluate whether short-term immunomodulation led to longterm defects in host immune function, animals were challenged 6 months after plasmid injection with the T-dependent neoantigen, bacteriophage Φx174.29 Three groups of mice (n = 2 per group) were immunized twice, 4 weeks apart, including tolerized mice treated with plasmid DNA and a combination of Ctla4Ig and MR1, mice treated with plasmid only, and untreated control mice. As shown in Figure 7, animals tolerized to hFVIII exhibited normal primary and secondary responses to bacteriophage Φx174, showing a strong amplification of antibody titers and isotype switching, comparable to antibody responses observed in mice treated with plasmid only and exhibiting high-titer inhibitory antibodies and to those of control untreated mice. These results strongly suggest that transient inhibition of costimulatory molecules in hFVIII plasmidtreated hemophilia A mice can promote long-term immune tolerance that is specific for hFVIII without altering immune responses to other T-cell-dependent antigens.
Figure 7.
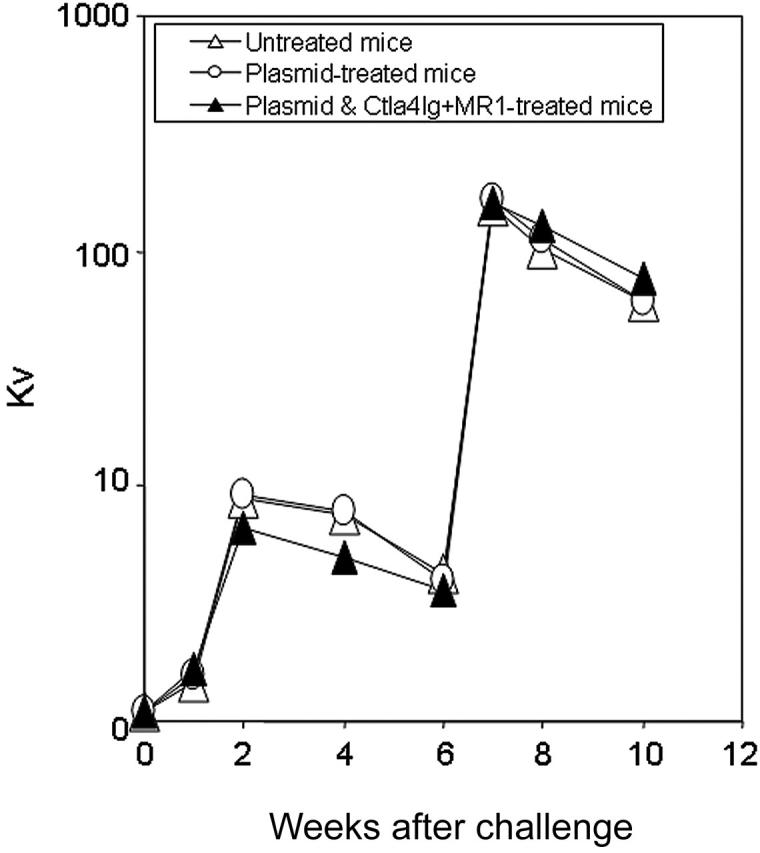
Bacteriophage challenge of hemophilia mice following naked gene transfer and transient immunomodulation. Tolerized hemophilia A mice (n = 2, ▴), selected mice from the group shown in Figure 3E,F) were challenged twice 4 weeks apart with the neoantigen bacteriophage Φx174 (2 × 108 PFU/each challenge). Plasmid-treated (n = 2, ○) and untreated hemophilia A mice (n = 2, ▵) were used as controls. Phage-neutralizing antibody activity was expressed as the rate of phage inactivation (Kv) using a standard formula.24 Mice not receiving bacteriophage did not produce neutralizing antibody (data not shown).
Discussion
Naked gene transfer of hFVIII into the liver of hemophilia A mice is an effective technique to generate a high level of functional hFVIII. However, hFVIII activity declines rapidly within 2 weeks of plasmid delivery. At the same time, sustained hFVIII protein expression can be readily detected in the mouse livers for a minimum of 6 months, as demonstrated by immunostaining using anti-hFVIII antibody.2 The decline of functional hFVIII protein is directly related to the appearance of high-titer inhibitory antihFVIII antibodies of the IgG isotype that appear 2 weeks after plasmid treatment and last for the lifetime of these animals. The immune responses induced in hFVIII plasmid-treated hemophilia A mice are primarily humoral responses and include antigen-specific B and T cells; there is no evidence for the induction of cytotoxic T lymphocytes (CTLs). This antibody response is similar to that in hemophilia mice treated with repeated protein infusions28 and to that observed in hemophilia patients with inhibitors.30 The majority of anti-hFVIII antibody in the plasmid-treated mice is of the IgG1 isotype, but IgG2a, IgG2b, and IgG3 antibodies also are present. These findings suggest that the immune response in these animals is mediated mainly by Th2-induced signals. Most notably, by using naked gene transfer, it is unlikely that these immune responses are driven or potentiated by viral vector components, as we observe little decline in plasmid-containing hepatocytes over time. This model therefore represents an excellent system to study inhibitory antibody formation generated via persistent transgene expression and permits us to investigate potential immunomodulation strategies designed to prevent or eliminate the formation of inhibitory antibodies against hFVIII.
In established animal models of tolerance, the presence of antigen appears to be required to maintain the tolerant state. Previous studies suggest that the minimal number of FVIII required to maintain hyporesponsiveness is in the range of 10-11 Mto 10-10 M per mouse,31,32 a level readily achieved by the sustained hFVIII gene expression (10-9 M-10-8 M) in our animal model. Based upon this concept, high-dose tolerance protocols have been used since the 1970s in efforts to tolerize hemophilia patients to infused hFVIII. As mentioned earlier, this approach also has been combined with the long-term use of immunosuppressive agents. However, such protocols are costly and have been successful only in patients with low-titer inhibitors.
We used the model of long-term expression of hFVIII following naked plasmid delivery to explore the effect of combining the administration of plasmid DNA with transient immunosuppression to induce long-term tolerance in hemophilia A mice. As has been shown previously,2 the administration of the cytotoxic and immunosuppressive agent cyclophosphamide following pBS-HCRHPI-hFVIIIA injection resulted in the prevention (1 of 6 animals) or delay (5 of 6 animals) of inhibitory antibody formation against the transgene. The complete and sustained phenotypic correction of hemophilia A achieved in one animal following transient immunomodulation with cyclophosphamide lasted for more than 6 months, strongly suggesting that consistent tolerance to this “neoantigen” and adequate expression of functional hFVIII might be achievable with an optimized immunosuppressive regimen.
We therefore evaluated multiple immunosuppressive strategies (Table 2) designed to prevent and/or modulate the generation of hFVIII-specific T cells and the production of inhibitory antibodies by hFVIII-specific B cells without induction of nonspecific tolerance. We first evaluated single agents that target the signaling events essential for sustained T-cell activation. CSA inhibits proximal T-cell receptor (TCR)-dependent signals, thereby suppressing the early stages of lymphocyte activation. This agent is used routinely in a broad range of applications, including humans with acquired hFVIII inhibitory antibodies. The bacterial macrolide, RAP, is a potent immunosuppressant and a promising anticancer drug. In forming a complex with its cellular receptor, the FK506-binding protein (FKBP12), RAP binds and inhibits the function of the mammalian target of rapamycin (mTOR). RAP inhibits T-cell proliferation following mitogen (PHA) and/or alloantigen (MLR) stimulation. Currently, no published experience with RAP exists in human hemophilia or related animal models. MMF as well as cyclophosphamide was used for nonspecific inhibition of B- and T-cell proliferation and the production of antibodies. MMF is approved for the prevention of renal transplant rejection and is used to treat a wide range of autoimmune disorders.
Table 2.
List of immunosuppressive agents
|
Published experience in hemophilia A
|
||||
|---|---|---|---|---|
| Agent | Primary mechanism of action | Human patients | Murine models | Reference |
| B- and T-cell lineage | ||||
| CTX* | Nonspecific cytotoxicity | Clinical trials w/ or w/o IVIG; partial response | No report | 9, 10 |
| MMF* | IMPDH inhibition - primarily inhibits B + T proliferation | No report | No report | 49 |
| B-cell lineage | ||||
| Anti-CD20 (rituximab) | Peripheral B-cell depletion | Case report | No report | 7 |
| Anti-CD40L, eg, MR* | Blockade of CD40-dependent immunoglobulin class switch, T-helper-dependent B-cell activation | No report | Efficient blockade of inhibitor formation, transient duration | 12, 13, 50 |
| T-cell lineage | ||||
| Anti-CD3 | T-cell depletion or partial activation leading to anergy | No report | No report | 51-53 |
| CSA*† | Inhibition of TCR-dependent signals | Case reports | No report | 6, 54 |
| CTLA4-lg* | Inhibition of T-cell costimulation | No report | Efficient blockade of inhibitor formation, transient | 55 |
| RAP*† | mTOR inhibition - blocks TCR signaling | No report | No report | 56, 57 |
IVIG indicates intravenous immunoglobulin; IMPDH, inosine monophosphate dehydrogenase; TCR, T-cell receptor; and mTOR, mammalian target of rapamycin.
Used in this study.
Also likely alter B-cell activation.
Our results demonstrate that neither single-agent nor combined therapy with any of these 3 agents was sufficient to prevent antibody formation against hFVIII, although treatment with MMF considerably suppressed the production of antigen-specific IgG1 and IgG2b antibodies; the effect, however, was not potent enough to prevent the induction of long-lasting inhibitory antibodies.
Because an effective antibody response to a protein antigen requires the interaction of antigen-presenting cells (APCs) with T and B cells, we next explored the effects of blocking the T/B-cell interaction by interfering with the known costimulatory pathways CD40L/CD40 and B7/CD28 (CTLA-4). Two specific agents, with comparable compounds currently under evaluation in human clinical trials, were evaluated. CTLA4-immunoglobulin (CTLA4-Ig) is a soluble fusion protein that was created by fusion of the extracellular domain of CTLA4 to the heavy chain constant regions 2 and 3 of IgG1. CTLA4 itself is a high-affinity receptor for both the B7-1 and B7-2 ligands. CTLA4-Ig binds strongly to the B7-1 and B7-2 ligands, altering the interaction of B7 with CD28 of this costimulatory pathway, thus initiating a potent down-modulatory effect. CTLA4-Ig has been used successfully as an immunosuppressive regimen in animal models of autoimmunity33 and transplantation34 and, at least transiently, blocks the inhibitor formation in hemophilia A mice.11 The interaction of CD40L expressed by activated CD4+ T cells with constitutively expressed CD40 on B cells is essential for the generation of high-affinity antibody of different isotypes.35 Anti-mouse CD40L monoclonal antibody (mAb) has been used previously to transiently block the inhibitory antibody formation in hemophilia A mice.12 Anti-mouse CD40L mAb alone or the combination of anti-mouse CD40L mAb and soluble murine Ctla4-Ig have been used to facilitate adenovirusmediated transgene expression after both primary and secondary vector administration.36,37
Based on the limited effects observed with either Ctla4-Ig or anti-CD40L alone, we combined these 2 agents to simultaneously target both effector pathways to induce tolerance. As shown in Figure 3, this combined immunotherapy regimen induced longterm tolerance in all treated animals. Only 1 of 9 mice developed low-titer antibodies that were transient and only partially blocked the functional activity of hFVIII in plasma. All 9 animals, including the one with transient antibodies, produced persistent, therapeutic, or supraphysiologic levels of hFVIII expression. These results demonstrate that short-term immunomodulation by blocking costimulation at the time of gene transfer can induce long-term tolerance against a specific neoantigen introduced by gene therapy. Our findings differ from those reported by Jiang et al,38 who observed that local high-level adenovirus-mediated expression of Ctla4-Ig and CD40-Ig did not induce a state of permanent tolerance toward a foreign transgene, eGFP, in primary skeletal muscles. These differences could be due to the presence of adenovirusrelated proteins, differences in the target organs involved (liver vs muscle), and/or the local versus systemic distribution of Ctla4-Ig and anti-CD40L. Another difference is that GFP is an intracellular protein, while FVIII is a secretory protein.
Several hypotheses have been proposed to explain the strong combined inhibitory effects of Ctla4-Ig and anti-CD40L. Engagement of the T-cell receptor by antigen-major histocompatibility complex in the absence of costimulatory signals commonly produces T-cell anergy or prolonged unresponsiveness39 and may result in prolonged and perhaps indefinite survival of allografted organs.40,41 Patients with X-linked hyper IgM syndrome due to mutations of CD40L and mice treated with anti-CD40L mAb (MR1) are unable to signal via CD40 and as a result lack class switch recombination and somatic hypermutation.42,43 During antigen challenge, the dominant effect of Ctla4Ig consists of blockade of CD28 binding by B7-1 or B7-2, therefore inhibiting further activation of T cells by activated B cells. An alternative hypothesis may postulate that the induction and maintenance of tolerance by costimulatory blockade is the result of active suppression mediated by individual subsets of regulatory T cells (Treg) or cytokines. In Ctla4-Ig and MR1-treated hemophilia mice, the “tolerance” was maintained along with high-level constitutive hFVIII gene expression. To systematically test and delineate the mechanisms for tolerance induction in the mice treated with a combination of Ctla4-Ig and MR1, T cells isolated from tolerized mice have to be adoptively transferred into naive hemophilia A mice. The recipient mice will subsequently be challenged by hFVIII plasmid injection to test for active suppression of the transgene-specific immune response. If the tolerance observed in the treated mice is induced by elimination of antibody production due to T-cell anergy or prolonged unresponsiveness, adoptive transfer of T cells will not induce tolerance in the recipient mice. On the other hand, if the tolerance is the result of immunosuppression by regulatory T cells, adoptive transfer is expected to facilitate tolerance induction in the recipient mice.
Interestingly, neither Ctla4-Ig nor anti-CD40L treatment alone achieved a significant inhibitory effect, whereas the combined therapy induced long-term tolerance. This strongly suggests that the 2 compounds act synergistically in blocking distinct activation pathways. Furthermore, T cells from tolerized mice remained nonresponsive to hFVIII exposure in vitro, whereas plasmidtreated hemophilia A mice not receiving combination therapy responded with strong IL-10 production (Figure 6).
In our experiments, Ctla4-Ig was less effective than MR1 when used alone in reducing the titer of anti-hFVIII antibodies. This result is different from those obtained from immunomodulation of adenoviral gene transfer of factor IX, where Ctla4Ig was more effective than MR1.36,37 In the latter case, the inhibitory response was induced primarily against viral vectors to generate predominantly CTLs that led to destruction of transduced cells. Therefore, blocking the CD28 pathway is likely more effective to block T-cell-dependent responses, whereas the blocking of antibody production through the CD40L pathway is less effective. In contrast, in the plasmid-treated hemophilia A mice, the inhibitory response is predominantly a humoral response against the transgene product, hFVIII, and MR1, which blocks T-help function, is apparently more effective than Ctla4Ig alone for blockade of the antibody response. CD40L also enhances APC function by inducing expression of the B7 proteins. Thus, the synergistic effect of dual blockade of both CD28 and CD40L costimulatory pathways with Ctla4-Ig and MR1 induced long-term antigen-specific tolerance but not a general immunodeficiency, fulfilling the requirements listed in the introduction section.
A clinical trial to induce tolerance is a logical next step to test the efficacy of combined immunomodulation therapy using CTLA4-Ig and anti-CD40L in humans. CTLA4-Ig does not seem to induce permanent immune compromise in humans44 and has been approved recently by the Food and Drug Administration for clinical use. Clinical trials to test efficacy and safety of anti-CD40L have yielded controversial results. In particular, some trials induced thrombotic events or increased thrombotic risks in patients,45,46 whereas some did not.47 In another study,48 administration of heparin in conjunction with anti-CD40L also reduced the frequency of thromboembolic complications. To use this agent in humans, further studies are needed to delineate functional and safety profiles of different monoclonal antibodies targeting different CD40L epitopes and functions. The minimum effective dose of anti-CD40L can be tested first in animals such as our mouse model.
It will be important to compare our findings with other immunomodulation regimens, including a nonactivating, nondepleting antimouse CD3 antibody to eliminate and/or tolerize specific T-cell subsets, and a murine B-cell-depleting antibody that affects mature and transitional murine B cells. Our goal is to develop the most effective immune modulating therapy with the least toxicity. It will be important to design an immunomodulation regimen that induces only a transient immunodeficiency in the host, as was the case in our combined immunomodulation treatment. Additional experiments are required to explore whether transient immunosuppressive regimens can down-regulate or eliminate a pre-existing immune response such as in mice that have already developed inhibitors.
Acknowledgments
We would like to thank Dr Randolph J. Noelle for kindly providing us with the anti-murine CD40L antibody (MR1) and Drs Rita Sarkar and Haig Kazazian Jr for providing us with the hemophilia A mice. We thank Dr Christopher Wilson for his advice and discussions. We also would like to thank Ms Marge Young for assisting us with the Bacteriophage immunization experiments, and Ms Laura Stewart for assisting us with the factor VIII clotting assays.
Prepublished online as Blood First Edition Paper, February 28, 2006; DOI 10.1182/blood-2005-11-4532.
Supported by a Dr Mario Bisordi Research Award from the Hemophilia Association of New York (C.H.M.), a Career Development grant from the National Hemophilia Foundation (C.H.M.), and an R01 grant from the National Institutes of Health/National Heart, Lung, and Blood Institute (HL069049-01).
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Miao CH, Ye X, Thompson AR. High-level factor VIII gene expression in vivo achieved by nonviral liver-specific gene therapy vectors. Hum Gene Ther. 2003;14: 1297-1305. [DOI] [PubMed] [Google Scholar]
- 2.Ye P, Thompson AR, Sarkar R, et al. Naked DNA transfer of factor VIII induced transgene-specific, species-independent immune response in hemophilia A mice. Mol Ther. 2004;10: 117-126. [DOI] [PubMed] [Google Scholar]
- 3.Mariani G, Siragusa S, Kroner BL. Immune tolerance induction in hemophilia A: a review. Semin Thromb Hemost. 2003;29: 69-76. [DOI] [PubMed] [Google Scholar]
- 4.Saint-Remy JM. Hemophilia factor VIII therapy: B- and T-cell tolerance, from basic concepts to clinical practice. Haematologica. 2000;85: 93-96. [PubMed] [Google Scholar]
- 5.Ewenstein BM, Hoots WK, Lusher JM, et al. Inhibition of CD40 ligand (CD154) in the treatment of factor VIII inhibitors. Haematologica. 2000;85: 35-39. [PubMed] [Google Scholar]
- 6.Brox AG, Laryea H, Pelletier M. Successful treatment of acquired factor VIII inhibitors with cyclosporin. Am J Hematol. 1998;57: 87-88. [DOI] [PubMed] [Google Scholar]
- 7.Wiestner A, Cho HJ, Asch AS, et al. Rituximab in the treatment of acquired factor VIII inhibitors. Blood. 2002;100: 3426-3428. [DOI] [PubMed] [Google Scholar]
- 8.Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood. 2004;103: 4424-4428. [DOI] [PubMed] [Google Scholar]
- 9.Berntorp E, Astermark J, Carlborg E. Immune tolerance induction and the treatment of hemophilia: Malmo protocol update. Haematologica. 2000;85: 48-50. [PubMed] [Google Scholar]
- 10.Carlborg E, Astermark J, Lethagen S, Ljung R, Berntorp E. The Malmo model for immune tolerance induction: impact of previous treatment on outcome. Haemophilia. 2000;6: 639-642. [DOI] [PubMed] [Google Scholar]
- 11.Qian J, Collins M, Sharpe AH, Hoyer LW. Prevention and treatment of factor VIII inhibitors in murine hemophilia A. Blood. 2000;95: 1324-1329. [PubMed] [Google Scholar]
- 12.Rossi G, Sarkar J, Scandella D. Long-term induction of immune tolerance after blockade of CD40-CD40L interaction in a mouse model of hemophilia A. Blood. 2001;97: 2750-2757. [DOI] [PubMed] [Google Scholar]
- 13.Reipert BM, Sasgary M, Ahmad RU, Auer W, Turecek PL, Schwarz HP. Blockade of CD40/CD40 ligand interactions prevents induction of factor VIII inhibitors in hemophilic mice but does not induce lasting immune tolerance. Thromb Haemost. 2001;86: 1345-1352. [PubMed] [Google Scholar]
- 14.Hausl C, Ahmad RU, Sasgary M, et al. High-dose factor VIII inhibits factor VIII-specific memory B cells in hemophilia A with factor VIII inhibitors. Blood. 2005;106: 3415-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rawle FEM, Labelle AD, Davie E, Pratt K, Lillicrap DP. Prevention of anti-FVIII inhibitor formation post protein infusion in hemophilic mice by prior feeding of the FVIII-C2 domain [abstract]. Blood. 2004;104: 15. [Google Scholar]
- 16.Lei TC, Su Y, Scott DW. Tolerance induction via a B-cell delivered gene therapy-based protocol: optimization and role of the Ig scaffold. Cell Immunol. 2005;235: 12-20. [DOI] [PubMed] [Google Scholar]
- 17.Moayeri M, Hawley TS, Hawley RG. Correction of murine hemophilia A by hematopoietic stem cell gene therapy. Mol Ther. 2005;12: 1034-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toole JJ, Pittman DD, Orr EC, Murtha P, Wasley LC, Kaufman RJ. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci U S A. 1986;83: 5939-5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bi L, Sarkar R, Naas T, et al. Further characterization of factor VIII-deficient mice created by gene targeting: RNA and protein studies. Blood. 1996;88: 3446-3450. [PubMed] [Google Scholar]
- 20.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6: 1258-1266. [DOI] [PubMed] [Google Scholar]
- 21.Zhang G, Budkar V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Human Gene Ther. 1999;10: 1735-1737. [DOI] [PubMed] [Google Scholar]
- 22.Miao CH, Ohashi K, Patijn GA, et al. Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro. Mol Ther. 2000;1: 522-532. [DOI] [PubMed] [Google Scholar]
- 23.Kasper CK, Aledort L, Aronson D, et al. Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh. 1975;34: 612. [PubMed] [Google Scholar]
- 24.Hamilton BL, Ochs HD. Immune dysfunction associated with graft-versus-host reaction in mice transplanted across minor histocompatibility barriers. Transplantation. 1989;47: 1061-1067. [DOI] [PubMed] [Google Scholar]
- 25.Connelly S, Andrews JL, Gallo AM, et al. Sustained phenotypic correction of murine hemophilia A by in vivo gene therapy. Blood. 1998;91: 3273-3281. [PubMed] [Google Scholar]
- 26.VandenDriessche T, Vanslembrouck V, Goovaerts I, et al. Long-term expression of human coagulation factor VIII and correction of hemophilia A after in vivo retroviral gene transfer in factor VIII-deficient mice. Proc Natl Acad Sci U S A. 1999;96: 10379-10384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans GL, Morgan RA. Genetic induction of immune tolerance to human clotting factor VIII in a mouse model for hemophilia A. Proc Natl Acad Sci U S A. 1998;95: 5734-5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu H, Reding M, Qian J, et al. Mechanism of the immune response to human factor VIII in murine hemophilia A. Thromb Haemost. 2001;85: 125-133. [PubMed] [Google Scholar]
- 29.Andrews RG, Winkler A, Potter J, et al. Normal immunologic response to a neoantigen, bacteriophage phiX-174, in baboons with long-term lymphohematopoietic reconstitution from highly purified CD34+ Linallogeneic marrow cells. Blood. 1997;90: 1701-1708. [PubMed] [Google Scholar]
- 30.Reding MT, Wu H, Krampf M, et al. CD4+ T cells specific for factor VIII as a target for specific suppression of inhibitor production. Adv Exp Med Biol. 2001;489: 119-134. [DOI] [PubMed] [Google Scholar]
- 31.Scott D. The Nature of Immunologic Tolerance. Austin, TX: RG Landes Co; 1994.
- 32.Smith R. Immunologic tolerance in non-living antigens. Adv Immunol. 1961;1: 67. [Google Scholar]
- 33.Dall'Era M, Davis J. CTLA4Ig: a novel inhibitor of costimulation. Lupus. 2004;13: 372-376. [DOI] [PubMed] [Google Scholar]
- 34.Blaha P, Bigenzahn S, Koporc Z, Sykes M, Muehlbacher F, Wekerle T. Short-term immunosuppression facilitates induction of mixed chimerism and tolerance after bone marrow transplantation without cytoreductive conditioning. Transplantation. 2005;80: 237-243. [DOI] [PubMed] [Google Scholar]
- 35.Foy T, Shepherd D, Durle Fea. In vivo CD40-gp39 interactions are essential for thymus dependent humoral immunity, II: prolonged suppression of the humoral immune response by an antibody to the ligand for CD40, gp39. J Exp Med. 1993;178: 1567-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kay MA, Meuse L, Gown AM, et al. Transient immunomodulation with anti-CD40 ligand antibody and CTLA4Ig enhances persistence and secondary adenovirus-mediated gene transfer into mouse liver. Proc Natl Acad Sci U S A. 1997;94: 4686-4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schowalter DB, Meuse L, Wilson CB, Linsley PS, Kay MA. Constitutive expression of murine CTLA4Ig from a recombinant adenovirus vector results in prolonged transgene expression. Gene Ther. 1997;4: 853-860. [DOI] [PubMed] [Google Scholar]
- 38.Jiang ZL, Reay D, Kreppel F, et al. Local highcapacity adenovirus-mediated mCTLA4Ig and mCD40Ig expression prolongs recombinant gene expression in skeletal muscle. Mol Ther. 2001;3: 892-900. [DOI] [PubMed] [Google Scholar]
- 39.Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11: 191-212. [DOI] [PubMed] [Google Scholar]
- 40.Lin H, Bolling SF, Linsley PS, et al. Long-term acceptance of major histocompatibility complex mismatched cardiac allografts induced by CTLA4Ig plus donor-specific transfusion. J Exp Med. 1993;178: 1801-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenschow DJ, Zeng Y, Thistlethwaite JR, et al. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science. 1992;257: 789-792. [DOI] [PubMed] [Google Scholar]
- 42.Aruffo A, Farrington M, Hollenbaugh D, et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell. 1993;72: 291-300. [DOI] [PubMed] [Google Scholar]
- 43.Noelle RJ. CD40 and its ligand in host defense. Immunity. 1996;4: 415-419. [DOI] [PubMed] [Google Scholar]
- 44.Abrams JR, Lebwohl MG, Guzzo CA, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999;103: 1243-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boumpas DT, Furie R, Manzi S, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003;48: 719-727. [DOI] [PubMed] [Google Scholar]
- 46.IDEC PWs. Available at: http://wwwidecpharm-com/site/science/idec131htm. Accessed January 20, 2003.
- 47.Davis JC Jr, Totoritis MC, Rosenberg J, Sklenar TA, Wofsy D. Phase I clinical trial of a monoclonal antibody against CD40-ligand (IDEC-131) in patients with systemic lupus erythematosus. J Rheumatol. 2001;28: 95-101. [PubMed] [Google Scholar]
- 48.Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6: 114. [DOI] [PubMed] [Google Scholar]
- 49.Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47: 85-118. [DOI] [PubMed] [Google Scholar]
- 50.Qian J, Burkly LC, Smith EP, et al. Role of CD154 in the secondary immune response: the reduction of pre-existing splenic germinal centers and antifactor VIII inhibitor titer. Eur J Immunol. 2000;30: 2548-2554. [DOI] [PubMed] [Google Scholar]
- 51.Hirsch R, Bluestone JA, DeNenno L, Gress RE. Anti-CD3 F(ab′)2 fragments are immunosuppressive in vivo without evoking either the strong humoral response or morbidity associated with whole mAb. Transplantation. 1990;49: 1117-1123. [DOI] [PubMed] [Google Scholar]
- 52.Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A. 1994;91: 123-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346: 1692-1698. [DOI] [PubMed] [Google Scholar]
- 54.Saxena R, Mishra DK, Kashyap R, Choudhry VP, Mahapatra M, Bhargava M. Acquired haemophilia—a study of ten cases. Haemophilia. 2000;6: 78-83. [DOI] [PubMed] [Google Scholar]
- 55.Hoyer LW, Qian J. Characterization of the immune response to factor VIII using hemophilia A* mice. Haematologica. 2000;85: 100-102. [PubMed] [Google Scholar]
- 56.Neuhaus P, Klupp J, Langrehr JM. mTOR inhibitors: an overview. Liver Transpl. 2001;7: 473-484. [DOI] [PubMed] [Google Scholar]
- 57.Shapiro AM, Suarez-Pinzon WL, Power R, Rabinovitch A. Combination therapy with low dose sirolimus and tacrolimus is synergistic in preventing spontaneous and recurrent autoimmune diabetes in non-obese diabetic mice. Diabetologia. 2002;45: 224-230. [DOI] [PubMed] [Google Scholar]