

Abstract
Mutations in the BCR/ABL kinase domain play a major role in resistance to imatinib mesylate (IM). We report here that BCR/ABL kinase stimulates reactive oxygen species (ROS), which causes oxidative DNA damage, resulting in mutations in the kinase domain. The majority of mutations involved A/T→G/C and G/C→A/T transitions, a phenotype detected previously in patients, which encoded clinically relevant amino acid substitutions, causing IM resistance. This effect was reduced in cells expressing BCR/ABL(Y177F) mutant, which does not elevate ROS. Inhibition of ROS in leukemia cells by the antioxidants pyrrolidine dithiocarbamate (PDTC), N-acetylcysteine (NAC), and vitamin E (VE) decreased the mutagenesis rate and frequency of IM resistance. Simultaneous administration of IM and an antioxidant exerted better antimutagenic effect than an antioxidant alone. Therefore, inhibition of ROS should diminish mutagenesis and enhance the effectiveness of IM. (Blood. 2006;108:319-327)
Introduction
The BCR/ABL gene is derived from the relocation of a portion of c-ABL gene from chromosome 9 to the portion of BCR gene locus on chromosome 22 (t(9;22), Philadelphia chromosome [Ph]) and is present in most chronic myeloid leukemia (CML) and in a cohort of acute lymphocytic leukemia (ALL) patients.1 BCR/ABL oncogenic tyrosine kinase (a product of BCR/ABL chimeric gene) exhibits 2 complementary roles in cancer. The first and best-characterized is stimulation of signaling pathways that eventually induce growth factor independence and affect the adhesive and invasive capability of leukemia cells. The second is the modulation of responses to DNA damage, rendering cells resistant to genotoxic therapies and causing genomic instability.2 Clinical observations and experimental findings suggest that BCR/ABL-induced genomic instability may lead to mutations and chromosomal translocations frequently observed during the transition from a relatively benign CML chronic phase (CML-CP) to an aggressive blast crisis (CML-BC).3 In addition, genomic instability also is manifested by numerous mutations detected in the BCR/ABL gene encoding for resistance to imatinib mesylate (IM).4
IM, a selective inhibitor of ABL kinase activity, revolutionized the treatment of BCR/ABL-positive leukemias.5 Unfortunately, clinical and experimental observations reveal that resistance to IM is increasingly problematic.4 Although the rate of progression of newly diagnosed CML-CP patients on IM is about 4% per year, IM resistance obscures this otherwise successful oncogenetargeted therapy.6
BCR/ABL kinase mutations appear to be the most frequent cause of acquired resistance to IM; resistant cells also may exhibit genomic amplification of nonmutated BCR/ABL and BCR/ABL independence due to overexpression of LYN kinase.4,7 Mutations also were detected in CML-CP patients before IM treatment, thus arguing for genetic instability early in the disease. Therefore, the BCR/ABL gene appears to be a casualty of genomic instability promoted by its own product—the BCR/ABL kinase.
Mutations usually result from enhanced DNA damage and/or deregulated mechanisms responsible for DNA repair.8,9 Much endogenous DNA damage arises from intermediates of oxygen reduction. Oxygen is metabolized inside the cell by a series of one-electron reductions with the generation of reactive and potentially damaging intermediates called reactive oxygen species (ROS),10 primarily generated by the mitochondrial respiratory chain (MRC).11 ROS units usually are short-lived and strike only molecules that are close in space and time, such as free nucleotides, which are subsequently incorporated into DNA during replication by unfaithful polymerases.9 Examples of ROS derivatives include 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxoG), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy), thymidine glycol, and 5-hydroxycytosine.12
BCR/ABL-mediated generation of ROS by MRC13 combined with aberrant regulation of DNA repair pathways14 may contribute to the mutator phenotype displayed by BCR/ABL cells.14-17 Therefore, the probability of accumulating DNA errors appears to be high in BCR/ABL-positive cells, because of enhanced “spontaneous” DNA damage and unfaithful repair mechanisms leading to misrepair. Thus, we investigated whether or not ROS-mediated DNA damage generates mutations in the BCR/ABL gene, leading to IM resistance.
Materials and methods
Cells
The 32Dcl3 murine growth factor-dependent hematopoietic cell line was transfected with pSRα-neo retroviral construct containing p210BCR/ABL, p190BCR/ABL, p190BCR/ABL(Y177F) mutant cDNA, or empty vector. Clones expressing similar amounts of BCR/ABL were used for the experiments 2 or 10 weeks after transfection. Mononuclear fractions of human normal bone marrow cells (hBMC from 4 donors), 4 patients in CML-CP, and 4 patients in myeloid CML-BC were obtained after informed consent and collected after centrifugation on Histopaque-1077 (Sigma, St Louis, MO). Murine cell lines and primary cells were cultured in the presence of interleukin-3 (IL-3) and stem-cell factor (SCF) + granulocytemacrophage colony-stimulating factor (GM-CSF), respectively, in pretested minimal concentrations required to maintain the proliferation of normal cells.
ROS assay
Levels of intracellular ROS were analyzed using the redox-sensitive fluorochrome 2′,7′-dichlorofluorescein-diacetate (DCFDA) (Sigma).14 Briefly, 2 × 105 cells/mL of culture medium were incubated with 5 μM DCFDA (5 mM stock in dimethyl sulfoxide [DMSO], stored in -20°C) for 15 minutes in 37°C. Next, samples were washed, resuspended in phosphate-buffered saline (PBS), and analyzed using flow cytometer EPICS XL (Beckman-Coulter, Miami, FL). The oxidized form of DCFDA, carboxy-DCFDA (Molecular Probes, Eugene, OR), was used as a control for uptake, retention, and decay.
In vitro mutagenesis assay
The following compounds were added to tissue culture: 2 μM ABL-tyrosine kinase inhibitor IM (Novartis Pharma, Basel, Switzerland), 0.2 μM antioxidant pyrrolidine dithiocarbamate (PDTC), 200 μM vitamin E (VE), 200 μM N-acetylcysteine (NAC), and 100 μM oxidant-inducer buthionine sulfoximine (BSO) (all from Sigma). IM or ouabain-resistant cells were identified by clonogenic assay in semisolid methylcellulose medium (StemCell Technologies, Vancouver, BC, Canada). 106 cells were plated in methylcellulose in the absence of growth factor(s) and the presence or absence of 2 μMIMor1 μM ouabain (Sigma), and colonies were counted after 7 days. Percentage of resistant cells was calculated as follows: number of colonies in the presence of inhibitor × 100/number of colonies without an inhibitor. IM-resistant single colonies were selected, expanded, and total RNA was isolated and reverse transcribed using random hexamers. To assess the mutagenicity in BCR/ABL-transformed murine cell lines, the primers specific for human c-ABL kinase domain (gi: 28236) (5′ primer: 5′-CGCAACAAGCCCACTGTC-3′,3′ primer: 5′-TCCACTTCGTCTGAGATACTGGATT-3′) and murine α subunit of Na+/K+ ATPase (gi: 21450276) (first pair: 5′ primer: 5′-GGTTGGACGAGACAAGTATGAG-3′,3′ primer: 5′-GCACGTTGGCTACGATGATA-3′; second pair: 5′ primer: 5′-TGATCCTTGAGTACACCTGGC-3′,3′ primer: 5′-CCCTCCACAATGATGAGCTT-3′; third pair: 5′ primer 5′-TACCACACAGAGATCGTCTTTGC-3′,3′ primer: 5′-GCCGCCTGAGTAATGAGCTT-3′) were used for polymerase chain reaction (PCR). cDNA from CML patient cells was amplified using 5′ primer in BCR exon 2 (5′-CACAGCATTCCGCTGACCATCA-3′) and 3′ primer in human c-ABL. Then, nested PCR was performed using human c-ABL-specific 5′ primer and 3′ primer (5′-AAATGCCCAGACGTCGGACTTG-3′). Amplified fragments were cloned using TA cloning kit (Invitrogen, Carlsbad, CA) and sequenced. The mutation rate in IM-resistant cells was determined according to the Luria-Delbruck fluctuation test with modifications.18
In vivo mutagenesis assay
Outbred SCID mice (Taconic Farms, Germantown, NY) were fed with control chow or VE-enriched diet (Bioserv, Frenchtown, NJ)19 starting from 1 week before and continuously after intravenous injection of 5 × 104 32Dcl3-BCR/ABL cells obtained 2 weeks after transfection. Eight weeks after leukemia-cell inoculation, splenocytes (SPLs) and bone marrow cells (BMCs) were harvested, and mononuclear-cell (MNC) populations were obtained after centrifugation of Lympholyte-M gradient (Cedarlane, Hornby, ON, Canada). MNCs were used for ROS and mutagenesis assays as described in “In vitro mutagenesis assay.” The experiments conform to the regulatory standards and were approved by the Institutional Animal Care and Use Committee at Temple University (Philadelphia, PA).
Comet assay
The comet assay was performed under alkaline conditions as described.14 For enzymatic treatment, cells were drained in agarose and covered with an enzyme buffer (control) or the enzyme (1 μg/mL Endo III or Fpg) in buffer and incubated for 30 minutes at 37°C.
Immunofluorescence
Monoclonal anti-8-oxoG (Chemicon International, Temecula, CA) followed by the secondary rabbit anti-mouse Alexa594 (Molecular Probes) antibodies were applied to detect the nuclear localization of the 8-oxoG lesions by immunofluorescence combined with deconvolution technology, as described.17 DNA was conuterstained with the DNA fluorochrome 4′,6′-diamedino-2-phenylindole (DAPI). Specific staining was visualized using an inverted Olympus IX70 fluorescence microscope (Olympus America, Melville, NY) equipped with a 100 ×/1.35 numeric aperture UPlan Apo objective and Cooke Sensicam QE camera (Cooke, Auburn Hills, MI). A series of 3-dimensional images of each indivdual picture (cell) was stored in SlideBook software version 3.0.1 (Intelligent Imaging Innovations, Denver, CO). Deconvolution was applied to increase the resolution and contrast of the images. A collection of 3-dimensional images describing individual cells was converted to one 2-dimensional picture. At least 20 individual cells were analyzed per experimental group. Pictures were prepared using Adobe Photoshop (Adobe Systems, San Jose, CA).
LL-PCR assay
Genomic DNA was purified by DNeasy Tissue Kit (Qiagen, Valencia, CA). The protocol for linker-ligation PCR (LL-PCR) to detect broken-ended double-stranded DNA was performed as described with modifications.14 Briefly, 2 oligomers, BW-1 (5′-GCGGTGACCCGGGAGATCTGAATTC-3′) and BW-2 (5′-GAATTCAGATC-3′) were annealed to form a linker, then stored frozen. Purified DNA (2 μg) was incubated with Klenow polymerase and ligated to the linker. Ligated DNA (200 ng) was used in a 50-μL PCR assay containing 3 pmol of the linker reverse primer (BW-1), human c-ABL-specific 5′ primer: 5′-CGCAACAAGCCCACTGTC-3′, and 2 units of Taq polymerase. After 2 rounds of PCR (2 × 20 cycles), the products were detected by Southern analysis using α-dCTP-labeled probe specific for the ABL kinase (5′-TGGAAGAAATACAGCCTGACG-3′).
Results
ROS-mediated oxidative DNA damage generates mutations in the BCR/ABL kinase domain, causing IM resistance in murine leukemia cells in vitro
In vitro models were used before to characterize mutations in BCR/ABL emerging after IM treatment in the absence of growth factor(s).20,21 To investigate the biologic relevance of the oxidative DNA damage in Ph-positive leukemias, an experimental model was established where the mutagenic parameters were not affected by selective pressure of IM. Therefore, while previously published reports mimic the IM-resistant mutations emerging during IM treatment, our model is focused on mutagenesis in IM-naive leukemia cells. p210BCR/ABL-32Dcl3 clones (3 clones per group) and parental counterparts (transfected with empty vector) were cultured for 2 weeks (B/A-early) and 10 weeks (B/A-late) in the presence of pretested concentrations of IL-3 required to maintain proliferation of nontransformed cells.
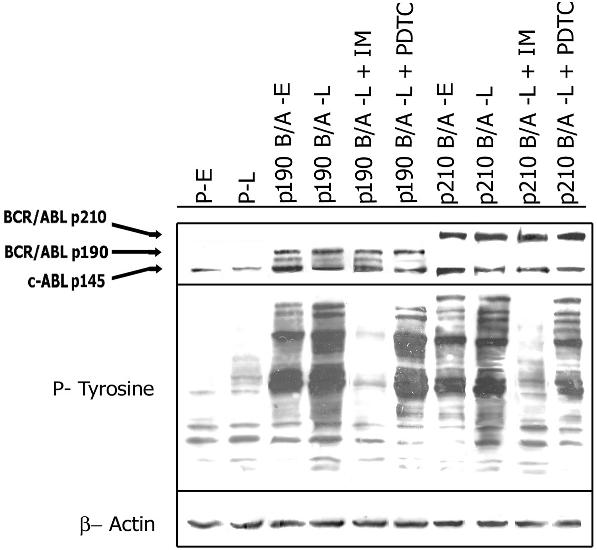
B/A-early cells did not display full transformation phenotype (eg, growth factor independence), whereas B/A-late cells were fully growth factor independent (Figure 1A). Cell-cycle analysis did not reveal major differences between parental and BCR/ABL-transformed counterparts in the presence of optimal concentrations of IL-3, although fully transformed B/A-late cells displayed a more abundant tyrosine phosphorylation pattern compared with B/A-early cells, as compared by Western analysis (Figure S1, available on the Blood website; see the Supplemental Figures link at the top of the online article). Our model mimics those described before and indicates that leukemogenesis driven by BCR/ABL kinase is a stepwise process reflecting multistage disease progression.22,23 In accordance, CML cells display disease stage-specific differences in growth factor-independent survival.24
Figure 1.

BCR/ABL induces ROS-mediated DNA oxidative damage in hematopoietic cell lines. (A) Parental 32Dcl3 cells freshly transfected with empty plasmid (P) or p210BCR/ABL retroviral construct (B/A) were cultured continuously for 2 weeks (P-early [groups 1 and 2] and B/A-early [groups 3 and 4], respectively) or 10 weeks (P-late [groups 5 and 6] and B/A-late [groups 7 and 8], respectively) in the presence of IL-3. B/A-late cells were eventually pre-incubated for 48 hours with IM or PDTC (groups 9 and 10, or 11 and 12, respectively). ROS was measured by fluorescence in cells starved (-) or not (+) from IL-3 for 12 hours. Survival of cells was examined after 24 hours of incubation in the absence (starvation) or presence of IL-3 and represents the percentage of cells excluding trypan blue. P < .05 compared with other groups (*), corresponding group incubated without IL-3 (**), and groups 10 and 12 (***). (B) 8-oxoG staining is detected by immunofluorescence in the nuclei, whose borders are marked in blue. (C) Oxidative DNA lesions were detected after addition of either EndoIII or Fpg to the comet reaction. Bars represent the enzyme-dependent increase of DNA damage over that detected in the undigested samples. *P < .05 compared with groups 1, 3, 4, 5, 9, and 11. (D) A DSB (arrow) detected in the BCR/ABL kinase sequence by LL-PCR followed by Southern analysis; parental 32Dcl3-late cells served as negative control. G/C-rich stretch at the predicted DSB site is listed. Numbering is consistent in the panels.
ROS was not elevated by BCR/ABL at the early (2 weeks) stage of transformation (Figure 1A, compare bar 3 with 1, and 4 with 2), whereas it was induced at the late (10 weeks) stage (Figure 1A, compare bar 7 with 5, and 8 with 6). BCR/ABL exerted approximately 3 times stronger stimulatory effect on ROS levels at the late stage compared with the early stage (Figure 1A, compare bar 8 with 4). IL-3 and BCR/ABL seemed to display an additive effect on ROS stimulation (Figure 1A, compare bar 7 with 8 and 5). Thus, our experimental model reflects a stepwise transformation, including time-dependent growth-factor independence accompanied by an increase in ROS levels. IM (48 hours' incubation with 2 μM) down-regulated ROS in B/A-late cells in the presence and absence of IL-3 (compare bars 9 with 7, and 10 with 8, respectively), implicating the kinase-dependent phenomenon. Addition of ROS scavenger PDTC (48 hours' incubation with 0.2 μM) diminished ROS in B/A-late cells in the presence and absence of IL-3 (Figure 1A, compare bars 11 with 7, and 12 with 8, respectively). The differences in ROS levels presented in Figure 1A do not appear to be affected by DCFDA uptake, cellular esterase activity, and efflux because cells in various groups displayed similar fluorescence intensity when carboxy-DCFDA, a constitutively fluorescent form of the dye, was used (data not shown).
Enhanced 8-oxoG staining was detected in B/A-late cells in the presence and absence of IL-3, whereas reduced staining was observed in parental cells, B/A-early cells, and in B/A-late cells after inhibition of the kinase with IM and down-regulation of ROS by PDTC (Figure 1B). To directly detect oxidative damage of DNA, we employed 2 enzymes, endonuclease III (EndoIII) and formamidopyrimidine-DNA glycosylase (Fpg), which convert oxidative lesions into gaps detectable by the alkaline version of the comet assay.25,26 Addition of both enzymes detected the presence of more oxidative DNA lesions in fully transformed growth factorindependent B/A-late cells, compared with parental counterparts and freshly generated B/A-early cells (Figure 1C, compare group 7 with group 5 and 3, respectively). This effect is dependent upon the BCR/ABL kinase-induced ROS, because inhibition of the kinase by IM and ROS by PDTC reduces the number of oxidative lesions (Figure 1C, compare groups 9 and 11 with group 7, respectively). Removal of IL-3 caused modest, statistically nonsignificant reduction of the oxidative damage in B/A-late cells (Figure 1C, compare group 8 with 7). Results obtained using only buffer were subtracted from those obtained using enzymes (enzyme treatment usually increased the detection of DNA damage by approximately 2-fold).
Elevated oxygen stress is probably a major source of spontaneous DNA double-strand breaks (DSBs).27 Interestingly, LL-PCR followed by Southern analysis revealed “spontaneous” DSB in a G/C-rich stretch in the sequence encoding BCR/ABL kinase domain in B/A-late cells (Figure 1D, lanes 7 and 8). Incubation with IM or PDTC prevented this effect (Figure 1D, lanes 9 and 11, respectively), again implicating BCR/ABL kinase and ROS. Altogether, it appears that BCR/ABL kinase-dependent enhancement of ROS induced an excess of oxidative DNA damage, which eventually led to a DSB in the BCR/ABL kinase sequence.
32Dcl3 cells transformed with p190BCR/ABL displayed similar properties: elevated levels of ROS, enhanced nuclear staining for 8-oxoG lesions, and a “spontaneous” DSB in the p190BCR/ABL kinase (data not shown).
B/A-early cells neither contained detectable mutations in the sequence encoding for BCR/ABL kinase domain nor displayed IM resistance. However, about 0.001% (1 × 10-5) of B/A-late cells were resistant to 2 μM IM after 8 weeks of continuous culture in the presence of pretested optimal concentrations of IL-3 (Figure 2A, % of IM-resistant cells, B/A group). IM resistance was associated with mutations in the region encoding for the BCR/ABL kinase domain: approximately 65% of them involved G/C→A/T transitions (Figure 2A, Mutation rate and Mutation phenotype, respectively). Mutations caused amino acid substitutions (Figure 2A, Aa substitutions), including those proven to be clinically relevant for resistance to IM28-30 and those identified in BCR/ABL mutagenesis screening using DNA repair-deficient bacteria.31 Mutagenesis in the sequence encoding BCR/ABL kinase domain was dependent upon BCR/ABL kinase activity, because inhibition of the kinase by continuous presence of IM (Figure S1) abrogated the mutagenic effect (Figure 2A, left panel, compare B/A + IM with B/A).
Figure 2.

Antioxidants prevent BCR/ABL-induced ROS-mediated mutations in BCR/ABL kinase domain and mouse α1 subunit of Na+/K+ ATPase in vitro. 32Dcl3 parental (P-early) and 2-week-old 32Dcl3-p210BCR/ABL cells (B/A-early, no mutations detected in BCR/ABL kinase and mouse α1 Na+/K+ ATPase) were cultured for 8 weeks in medium supplemented with IL-3 (B/A) and IM (B/A + IM), PDTC (B/A + PDTC), or IM and BSO (B/A + IM + BSO). The percentage of IM-resistant cells (A, % of IM-resistant cells) and ouabain-resistant cells (B, % of ouabain-resistant cells) was determined by clonogenic assay performed in the absence of IL-3 and the presence or absence of IM or ouabain, respectively. Mutation rate and mutation phenotype in BCR/ABL kinase and mouse α1 subunit of Na+/K+ ATPase are shown in panels A and B, respectively. Mutations leading to previously described amino acid substitutions are listed (Aa substitutions in panels A and B, respectively). *P < .05 compared with B/A group; **P < .05 compared with other groups.
To determine the role of ROS in mutagenesis of BCR/ABL kinase domain leading to IM resistance, B/A-early cells were cultured for 8 weeks in the presence of pretested optimal concentrations of IL-3 and ROS scavenger PDTC, or IM + ROS inducer BSO (to inhibit BCR/ABL kinase activity while keeping high levels of ROS). ROS were monitored periodically during the experiment to ensure the desirable effect of factors modulating ROS levels: IM inhibited the ABL kinase, resulting in downmodulation of ROS (Figure 1A, bar 9), PDTC quenched ROS (Figure 1A, bar 11), and BSO elevated ROS after inhibition of BCR/ABL kinase by IM (data not shown). Addition of PDTC to the culture medium reduced IM resistance frequency by approximately 5-fold (Figure 2A, left panel, compare B/A + PDTC with B/A). These results suggest that ROS plays a crucial role in BCR/ABL kinase self-mutagenesis leading to IM resistance. When BSO was added together with IM, the IM resistance frequency was increased only by approximately 2-fold (Figure 2A, % of IM-resistant cells, compare B/A + IM + BSO with B/A + IM), implicating that although ROS is essential for IM resistance, another BCR/ABL kinase-induced mechanism also may play a role. In concordance, mutation rate also was reduced greatly by the presence of PDTC and only modestly increased when BSO was added to tissue culture containing IM (Figure 2A, Mutation rate).
To determine whether or not ROS-induced mutagenesis is restricted to the BCR/ABL gene, the Na+/K+ ATPase mutagenesis model was employed where amino acid substitutions in the α1 subunit of the enzyme causes resistance to ouabain.32-34 Parental and B/A-early cells did not display detectable resistance to ouabain; however, resistant cells accumulated during cell culture in a time-dependent and accelerated fashion in the BCR/ABL-positive population in comparison to parental counterpart (Figure S2). The percentage of BCR/ABL cells resistant to ouabain was approximately 28 times higher than that of parental cells after 8 weeks of culture in the presence of IL-3 (Figure 2B, % of ouabain-resistant cells). Sequencing of the α1 subunit detected a mutation rate increased by approximately 400 times in BCR/ABL cells compared with parental counterparts (Figure 2B, Mutation rate). Again, mutation phenotype revealed the prevalence of G/C→A/T transitions (Figure 2B, Mutation phenotype); mutations produced some of the previously identified amino acid substitutions, such as C111A, D128S, and D128E (equivalents of sheep C104A, D121S, and D121E, which induced resistance to ouabain32-34). Na+/K+ ATPase mutagenesis in BCR/ABL cells was inhibited by incubation with IM or PDTC. In conclusion, BCR/ABL-induced ROS-dependent mutagenesis appears not to be restricted to the BCR/ABL gene.
BCR/ABL kinase mutagenesis and IM resistance also were examined in cells expressing BCR/ABL(Y177F) mutant, which does not elevate ROS.35 BCR/ABL[Y177F]-32Dcl3 cells displayed lower levels of ROS compared with BCR/ABL-32Dcl3 cells when cultured in the presence of pretested minimal concentrations of IL-3 necessary to support the proliferation of parental cells (Figure 3A). After 8 weeks of continuous culture, BCR/ABL[Y177F]-32Dcl3 cells contained approximately 3 times fewer clones resistant to IM; in addition, mutation rate was diminished by approximately 1.7-fold (Figure 3B). These results support the notion that ROS causes mutations in the BCR/ABL kinase domain and IM resistance.
Figure 3.

Cells expressing BCR/ABL(Y177F) mutant displayed reduced ROS and mutagenic activity. Freshly transformed p190BCR/ABL wild-type (wt) and p190BCR/ABL(Y177F) cells were cultured continuously for 8 weeks in the presence of IL-3. (A) ROS was measured by fluorescence. (B) The percentage of IM-resistant cells (% of IM-resistant cells) was determined by clonogenic assay performed in the absence of IL-3 and the presence or absence of IM. Mutation rate in BCR/ABL kinase is shown. *P = .002 compared with wild-type group.
Next, we determined if simultaneous scavenging of ROS by an antioxidant and inhibition of BCR/ABL kinase by IM exert better antimutagenic effects than each agent used individually. For this reason, B/A-early cells were incubated in the presence of IL-3 for 8 weeks with IM and/or various antioxidants such as PDTC, NAC, or VE. As expected, each compound inhibited ROS and reduced the number of IM-resistant clones (Figure 4). VE and NAC appear to be more effective compared with IM (Figure 4, compare B/A + NAC and B/A + VE with B/A + IM), probably due to the fact that IM inhibits only BCR/ABL-driven generation of ROS, while antioxidants scavenge ROS generated by BCR/ABL and IL-3-dependent mechanisms. PDTC did not exert better effect than IM, probably because of dose limitations (higher concentrations of PDTC exerted a detrimental effect on cell proliferation). Addition of IM did not further diminish ROS levels in leukemia cells treated with antioxidants, but it significantly reduced the percentage of cells resistant to IM (Figure 4, compare B/A + IM + PDTC/NAC/VE with B/A + IM). Similar mutations to those described in Figure 2A were detected here in IM-resistant colonies (data not shown). These results suggest that although ROS appears to be necessary to trigger mutagenesis, additional BCR/ABL kinase-dependent mechanism(s) may work in concert with ROS to cause mutations in the kinase.
Figure 4.

IM enhanced the effect of antioxidants in prevention of IM resistance. B/A-early cells were cultured for 8 weeks in medium supplemented with IL-3 (B/A) and PDTC, NAC, VE, and IM administered individually or in combinations of an antioxidant with IM. ROS levels were examined by fluorescence (Relative fluorescence). Cells were plated in methylcellulose in the presence or absence of 2 μM IM and absence of growth factors, and colonies were counted after 7 days. Results represent percent of IM-resistant cells (mean ± SD); P < .05 compared with other groups (*), B/A + IM group (**), and corresponding group treated with antioxidant only (***).
ROS-mediated oxidative DNA damage generates mutations in the BCR/ABL kinase domain, causing IM resistance in CML primary cells in vitro
A significant increase of ROS was detected in CML-BC cells, whereas CML-CP cells displayed rather modest elevation of ROS compared with cells from healthy donors (Figure 5A). However, the latter population sometimes consisted of 2 subpopulations displaying low and higher levels of ROS (Figure S3). CML-BC myeloid bone marrow cells contained, on average, 3.8 times more ROS than normal counterparts (Figure 5A), whereas BCR/ABL-transformed 32Dcl3 cell line displayed approximately only 2-fold increase compared with parental cells (Figure 1A). This discrepancy is probably due to the heterogeneity of primary-cell populations and/or slower proliferation rate of cells from healthy donors compared with those from CML-BC patients; 32Dcl3 cells and BCR/ABL-transformed counterparts represent more homogenous populations with a similar proliferation rate. Cells obtained from CML-BC patients in lymphoid blast crisis also contained high levels of ROS (2 patients analyzed, data not shown).
Figure 5.

Primary CML cells contain elevated levels of ROS and oxidative DNA lesions. (A) ROS levels in mononuclear bone marrow cells from healthy volunteers (N), CML-CP, and CML-BC patients were analyzed by fluorescence; bars show mean ± SD; *P = .02, **P = .001 compared with N group. (B) 8-oxoG nuclear staining detected by immunofluorescence. The nuclei borders are marked in blue.
8-oxoG, a frequent DNA lesion resulting from ROS, may be detected by immunofluorescence as nuclear diffuse and focal staining; the latter may be associated with extensive oxidative DNA damage.36 8-oxoG staining was readily detectable in CML-BC cells displaying high levels of ROS; conversely, reduced staining was observed in normal hematopoietic counterparts displaying low levels of ROS (Figure 5B). Please note that CML-CP cells displaying weak and elevated 8-oxoG staining were detected, perhaps reflecting 2 subpopulations of CML-CP cells containing low and higher levels of ROS. Thus, enhanced ROS levels in CML cells appear to be associated with elevated oxidative DNA damage.
CML-BC patient cells (3 patients in myeloid blast crisis), previously not treated with IM, were used to test the role of ROS in generating BCR/ABL kinase mutants resistant to IM in primary cells. These cells did not display detectable IM resistance at the beginning of the experiment and were cultured for 6 weeks in the presence of pretested optimal concentrations of SCF + GM-CSF with the addition, or not, of IM or PDTC. IM and PDTC decreased ROS levels during the experiment (Figure 6, Relative fluorescence, compare CML + IM with CML and CML + PDTC with CML), resulting in reduction of the frequency of IM resistance by 8- to 11-fold (Figure 6, compare % of IM-resistant cells from CML + IM with CML and CML + PDTC with CML). IM resistance was associated with mutations in the BCR/ABL kinase domain, often involving G/C→A/T transitions (30%), A/T→G/C transitions (25%), and A/T→C/G transversions (25%). Mutations caused amino acid substitutions, including Y253H found in patients28-30 and S265T and I360F identified in bacterial screen.31 In addition, the following amino acid substitutions were found in the previously identified locations: M237V, M244L, M351L, E373A, and L387W. Moreover, an additional 24 amino acid substitutions were found in positions not described before. However, the role of these aberrations in IM resistance is unknown, especially because they often were accompanied by previously characterized mutations. A few IM-resistant colonies grew from IM- or PDTC-treated cells, but we did not obtain enough material to examine the cause of resistance.
Figure 6.

An antioxidant prevents mutations in BCR/ABL kinase domain and IM resistance in CML cells. Cells from CML-BC patients previously untreated with IM (no resistance detected prior to the experiment) were cultured for 6 weeks in medium supplemented with SCF + GM-CSF in the absence (CML) or presence of IM (CML + IM) or PDTC (CML + PDTC) (similar to that described in Figure 2). ROS levels were examined by fluorescence (Relative fluorescence). Cells were plated in methylcellulose without growth factors in the presence or absence of 2 μM IM, and colonies were counted after 7 days. Results represent percent of IM-resistant cells (mean ± SD). Mutation rate and mutation phenotype in BCR/ABL kinase is shown. Mutations leading to previously described amino acid substitutions are listed (Aa substitutions). *P < .05 and **P < .001 compared with CML group.
An antioxidant reduces ROS-dependent mutations in BCR/ABL kinase and IM resistance in vivo
BCR/ABL leukemia-bearing SCID mice were employed to determine whether or not antioxidants could prevent mutagenesis and IM resistance in vivo. Animals were injected with BCR/ABL-early cells and fed a VE-rich diet as described by Factor et al.19 VE diet reduced ROS levels in SPLs and BMCs in leukemic mice (Figure S4). The percentage of IM-resistant BCR/ABL leukemia cells was reduced approximately 4-fold in BMCs harvested from the mice fed with VE diet than with control chow; this effect was associated with approximately 2-fold reduction in the mutation frequency (Figure 7, % of IM-resistant cells and Mutation frequency, respectively). In splenocytes, VE diet caused an approximately 3-time reduction of the percentage of IM-resistant leukemia cells, but not of mutation frequency. Mutation frequency was calculated by dividing the number of mutations in the experimental group by the total number of sequenced bases. The Luria-Delbruck fluctuation test could not be applied here because of the unknown number of cell divisions. Therefore, mutation frequency presented here is higher than the mutation rate presented in Figure 2A, because these 2 values were calculated using different tests. Mutation phenotype also was analyzed and showed prevalence of G/C→A/T transitions (Figure 7, Mutation phenotype). Similar to the in vitro experiment, mutations caused amino acid substitutions previously characterized as responsible for IM resistance (Figure 7, Aa substitutions). This shows that VE diet inhibited BCR/ABL-induced ROS-dependent mutagenesis in vivo.
Figure 7.

An antioxidant inhibits ROS-induced mutations in BCR/ABL kinase domain in vivo. Leukemia-bearing SCID mice were fed with control chow (▪) or vitamin E-enriched diet (VE diet,  ). Then, mononuclear cells of SPL and BMC were plated in methylcellulose in the absence of IL-3 (only leukemia cells grow) and the presence or absence of IM, and colonies were counted after 7 days. BCR/ABL kinase domain in IM-resistant cells was amplified by reverse transcriptase-polymerase chain reaction and sequenced. Mutation frequency and the percentages of base substitutions (Mutation phenotype) in the sequence encoding BCR/ABL kinase are shown. Mutations leading to previously described amino acid substitutions in BCR/ABL kinase are listed (Aa substitutions). *P < .01 compared with control chow group.
). Then, mononuclear cells of SPL and BMC were plated in methylcellulose in the absence of IL-3 (only leukemia cells grow) and the presence or absence of IM, and colonies were counted after 7 days. BCR/ABL kinase domain in IM-resistant cells was amplified by reverse transcriptase-polymerase chain reaction and sequenced. Mutation frequency and the percentages of base substitutions (Mutation phenotype) in the sequence encoding BCR/ABL kinase are shown. Mutations leading to previously described amino acid substitutions in BCR/ABL kinase are listed (Aa substitutions). *P < .01 compared with control chow group.
Discussion
Mutations encoding for IM resistance represent a major cause of disease relapse.4 They likely arise as a result of genomic instability induced by BCR/ABL kinase.2 Since genomic instability, like other events, may depend on BCR/ABL expression levels,37 it is likely that mutations causing IM resistance will occur more often in less-differentiated leukemia stem cells such as the CD34+Lin- population, which expresses high levels of BCR/ABL.38 In agreement with this speculation, cell lines expressing BCR/ABL at the levels found in CML-BC were more likely to develop IM-resistant BCR/ABL mutants than those expressing BCR/ABL at the levels detectable in CML-CP.39 Mutations detected in CD34+ CML-cell population are likely to be passed on to successive generations of leukemia cells.40-42 IM-resistant mutations also were detected in Ph-positive ALL cells.28
Here we report that BCR/ABL oncogenic tyrosine kinases induce self-mutagenesis in murine leukemia cells and CML primary cells over time. This observation is supported by the clinical results showing no mutations in 40 patients in early CML-CP diagnosis (defined as < 12 months from diagnosis); conversely, 14 mutations were identified in 64 patients in late CML-CP (> 12 months from diagnosis) and in 13 patients in accelerated phase.29 Mutagenesis is not restricted to the kinase sequence, because leukemia cells acquire mutations in other genes such as Na+/K+ ATPase (this article), c-kit,43 hypoxanthineguanine phosphoribosyl transferase,17,44 green fluorescent protein,14 and LacI.45 Mutations in Na+/K+ ATPase arose faster in leukemia cells than in normal counterparts, indicating that mutagenesis in the former cells is an active process. In accordance with previous reports,44,45 IM applied in nonselective conditions (in the presence of growth factors) reduced mutagenesis in BCR/ABL-transfected cell lines and in CML primary cells, indicating that BCR/ABL kinase activity is essential for acquiring mutations.
Mutagenic force producing IM-resistant clones in SCID mice (∼2-4.5 × 10-5, reported here) is similar to that (∼5-12 × 10-5) described in BCR/ABL transgenic mice using the Big Blue mutation detection system.16 Several lines of evidence associated ROS, which appear to play a major role in genomic instability,9 with BCR/ABL self-mutagenesis. BCR/ABL kinase can induce ROS in cell lines13 and CML patient cells as suggested by previous reports46,47 and confirmed here. Furthermore, higher levels of ROS in CML-BC cells compared with CML-CP cells implicate the role in accelerated mutagenesis and genomic instability, leading to malignant progression of the disease.48
We found that cells expressing BCR/ABL[Y177F] mutant, which exerts the kinase activity but does not elevate mitochondrialderived ROS,35 displayed reduced frequency of IM resistance and mutagenic rate. In addition, antioxidants such as PDTC, NAC, and VE caused a reduction of mutagenesis and IM resistance. Moreover, PDTC reduced the frequency of mutations in the gene encoding Na+/K+ ATPase, indicating that ROS may play a more general role in mutagenesis in Ph-positive leukemias. Treatment with the antioxidants exerted a stronger antimutagenic effect than Y177F amino acid substitution in BCR/ABL. This effect may depend on more general anti-ROS effect of the scavengers compared with the mutation, which can ablate ROS generated in mitochondria.
Results obtained in vitro suggest that inhibition of ROS may prevent BCR/ABL mutations encoding for IM resistance. This is further supported by the experimental data showing that inhibition of ROS by VE diet in mice harboring BCR/ABL leukemia cells reduced the generation of IM-resistant cells in bone marrow and spleen compared with mice fed with control diet. Furthermore, VE diet reduced the mutation frequency in the BCR/ABL kinase in IM-resistant clones harvested from bone marrow, but not spleencell populations. Although the reason for this effect is unknown, it appears rather unlikely that it results from the capability of cells carrying some IM-resistant BCR/ABL mutants to outgrow those containing wild-type BCR/ABL,49 because of the detection of a variety of mutants in bone marrow and spleen-cell populations. Therefore, it seems that the VE diet displayed slightly better antimutagenic effect in bone marrow cells than in spleen cells, suggesting that localization of leukemia cells may have an impact on the efficiency of antioxidative treatment.
It has been shown that CML patients display significant reduction of VE level in serum,50 possibly contributing to elevation of ROS in leukemia cells. Therefore, enhanced VE supply may exert an antimutagenic effect in Ph-positive patients. This hypothesis is supported by others showing that antioxidants, including VE, reduce the mutagenic effects of ROS in humans.51
The majority of mutations detected in this work involve A/T→G/C and G/C→A/T transitions, in accordance with the reports from IM-resistant patients.28,29,52 Similar transition phenotypes often were detected in the kinase gene family and in p53 in breast cancer.53 Such mutator phenotype might emerge from ROS-mediated oxidative DNA damage.9,54 ROS-dependent mutagenesis usually results from inefficient and/or unfaithful repair of oxidized bases and DNA DSBs. The base excision repair (BER) likely produces G/C→A/T transitions.54 In addition, DSBs generated by ROS may occur in BCR/ABL-transformed hematopoietic cells in the regions containing G/C stretches.14,27 Interestingly, a G/C-rich “hot spot” generating ROS-induced DSBs in the sequence encoding BCR/ABL kinase is identified here, and A/T→G/C transitions are frequently introduced during homologous recombination repair (HRR) in BCR/ABL-positive leukemia cells.55 Therefore, it appears that unfaithful repair of oxidative DNA damage by BER and HRR may contribute to the appearance of point mutations in BCR/ABL kinase causing IM resistance. However, quiescent Ph-positive cells also accumulate mutations.40 Since ROS-mediated DSBs and HRR depend on DNA replication, unfaithful/inefficient BER may play a primary role in quiescent leukemia cells.
Although ROS appears to play a major role in elevation of oxidative DNA damage and BCR/ABL kinase mutagenesis, other factors may be important, too. This hypothesis is supported by the observation that increased ROS levels are necessary but may not be sufficient to enhance oxidative DNA damage. For example, we showed that antioxidant PDTC inhibited ROS and reduced oxidative DNA damage in BCR/ABL-late cells in the presence and absence of IL-3. However, while BCR/ABL-late cells in the absence of IL-3 displayed similar levels of ROS as parental cells and IM-treated BCR/ABL-late cells in the presence of IL-3, accumulation of oxidized bases was detected only in the former cells. Thus, we speculate that not only BCR/ABL kinase-induced elevation of ROS, but also stimulation of incorporation of oxidized nucleotides into DNA, and/or inhibition of their excision, may be responsible for accumulation of oxidative DNA damage in BCR/ABL-late cells.
In addition, mutagenesis in the BCR/ABL gene was only modestly induced if ROS was elevated by BSO in the absence of BCR/ABL kinase activity. Moreover, although inhibition of BCR/ABL kinase by IM did not cause significantly stronger inhibition of ROS when used with the antioxidants in concentrations applied here, it further reduced the frequency of IM-resistant clones. These observations implicate additional BCR/ABL kinase-dependent mechanisms in mutagenesis. For example, DNA polymerase β is overexpressed in BCR/ABL cells,15 which may diminish the fidelity of BER and HRR.15,56 Moreover, we showed that point mutations might be introduced during HRR in BCR/ABL-positive cells.48 Therefore, we hypothesize that although ROS may be essential to initiate mutagenesis, inefficient and/or unfaithful DNA repair mechanisms introduce mutations into the sequence encoding BCR/ABL kinase during attempts of repair of numerous oxidative DNA lesions in leukemia cells.
IM resistance associated with BCR/ABL mutations plague an otherwise effective antileukemia therapy.4,5,7 In addition, resistance to FLT3 and epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors was associated with mutations in the kinase domains.57,58 Novel inhibitors are being generated to overcome IM resistance,59-61 but mutations causing resistance against new drugs are likely to emerge.62,63 Therefore, a rational strategy also should target the mechanisms leading to mutagenesis in order to prevent generation of new mutations during inhibitor(s) therapy. Here we demonstrated that antioxidant treatment may reduce/eliminate IM resistance driven by mutations in the BCR/ABL kinase. Interestingly, cells transformed by BCR/ABL-related fusion tyrosine kinases (FTKs), such as TEL/ABL and TEL/PDGFβR, and those expressing EGFR, HER1-2 kinase, also display elevated levels of ROS64 (data not shown). Thus, it is conceivable that tumor cells harboring oncogenic tyrosine kinases will be prone to develop resistance to kinase inhibitors. Although inhibition of an oncogenic kinase reduces the frequency of mutations in vitro, antimutagenic effect of the inhibitors may be limited in vivo. For example, IM does not penetrate to the central nervous system and also may not achieve effective concentrations in portions of other tissues/organs.65 In addition, nonproliferating CML CD34+ progenitors probably containing leukemia stem cells are resistant to IM.66 Thus, identifying approaches that reduce mutations in the oncogenic kinases may have a great impact on the therapeutic efficiency of IM and other small molecule inhibitors.
Supplementary Material
Acknowledgments
We thank Lori Rink and Elisabeth Bolton from Temple University for critical reading of the manuscript.
Prepublished online as Blood First Edition Paper, March 9, 2006; DOI 10.1182/blood-2005-07-2815.
Supported by grants from the National Cancer Institute (CA89052) and the Department of Defense (W81XWH-04-1-0805 and W81XWH-05-1-0214); a scholarship from the Leukemia and Lymphoma Society (T.S.); and a grant from Komitet Badan Naukowych (KBN) 3P04A 03225 (J.B.).
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Kurzrock R, Kantarjian HM, Druker BJ, Talpaz M. Philadelphia chromosome-positive leukemias: from basic mechanisms to molecular therapeutics. Ann Intern Med. 2003;138: 819-830. [DOI] [PubMed] [Google Scholar]
- 2.Skorski T. BCR/ABL regulates response to DNA damage: the role in resistance to genotoxic treatment and in genomic instability. Oncogene. 2002;21: 8591-8604. [DOI] [PubMed] [Google Scholar]
- 3.Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103: 4010-4022. [DOI] [PubMed] [Google Scholar]
- 4.Shah NP, Sawyers CL. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene. 2003;22: 7389-7395. [DOI] [PubMed] [Google Scholar]
- 5.Druker BJ. Imatinib and chronic myeloid leukemia: validating the promise of molecularly targeted therapy. Eur J Cancer. 2002;38: S70-S76. [DOI] [PubMed] [Google Scholar]
- 6.Branford S, Rudzki Z, Harper A, et al. Imatinib produces significantly superior molecular responses compared to interferon alfa plus cytarabine in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Leukemia. 2003;17: 2401-2409. [DOI] [PubMed] [Google Scholar]
- 7.Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003;4: 75-85. [DOI] [PubMed] [Google Scholar]
- 8.Schar P. Spontaneous DNA damage, genome instability, and cancer—when DNA replication escapes control. Cell. 2001;104: 329-332. [DOI] [PubMed] [Google Scholar]
- 9.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477: 7-21. [DOI] [PubMed] [Google Scholar]
- 10.Gutteridge JM, Halliwell B. Free radicals and antioxidants in the year 2000: a historical look to the future. Ann N Y Acad Sci. 2000;899: 136-147. [DOI] [PubMed] [Google Scholar]
- 11.Fleury C, Mignotte B, Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84: 131-141. [DOI] [PubMed] [Google Scholar]
- 12.Lu AL, Li X, Gu Y, Wright PM, Chang DY. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem Biophys. 2001;35: 141-170. [DOI] [PubMed] [Google Scholar]
- 13.Sattler M, Verma S, Shrikhande G, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275: 24273-24278. [DOI] [PubMed] [Google Scholar]
- 14.Nowicki MO, Falinski R, Koptyra M, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104: 3746-3753. [DOI] [PubMed] [Google Scholar]
- 15.Canitrot Y, Lautier D, Laurent G, et al. Mutator phenotype of BCR-ABL transfected Ba/F3 cell lines and its association with enhanced expression of DNA polymerase beta. Oncogene. 1999;18: 2676-2680. [DOI] [PubMed] [Google Scholar]
- 16.Salloukh HF, Laneuville P. Increase in mutant frequencies in mice expressing the BCR-ABL activated tyrosine kinase. Leukemia. 2000;14: 1401-1404. [DOI] [PubMed] [Google Scholar]
- 17.Canitrot Y, Falinski R, Louat T, et al. p210 BCR/ABL kinase regulates nucleotide excision repair (NER) and resistance to UV radiation. Blood. 2003;102: 2632-2637. [DOI] [PubMed] [Google Scholar]
- 18.Capizzi RL, Jameson JW. A table for the estimation of the spontaneous mutation rate of cells in culture. Mutat Res. 1973;17: 147-148. [DOI] [PubMed] [Google Scholar]
- 19.Factor VM, Laskowska D, Jensen MR, Woitach JT, Popescu NC, Thorgeirsson SS. Vitamin E reduces chromosomal damage and inhibits hepatic tumor formation in a transgenic mouse model. Proc Natl Acad Sci U S A. 2000;97: 2196-2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Bubnoff N, Barwisch S, Speicher MR, Peschel C, Duyster J. A cell-based screening strategy that predicts mutations in oncogenic tyrosine kinases: implications for clinical resistance in targeted cancer treatment. Cell Cycle. 2005;4: 400-406. [DOI] [PubMed] [Google Scholar]
- 21.Flamant S, Turhan AG. Occurrence of de novo ABL kinase domain mutations in primary bone marrow cells after BCR-ABL gene transfer and imatinib mesylate selection. Leukemia. 2005;19: 1265-1267. [DOI] [PubMed] [Google Scholar]
- 22.Gishizky ML, Witte ON. Initiation of deregulated growth of multipotent progenitor cells by bcr-abl in vitro. Science. 1992;256: 836-839. [DOI] [PubMed] [Google Scholar]
- 23.Laneuville P, Sun G, Timm M, Vekemans M. Clonal evolution in a myeloid cell line transformed to interleukin-3 independent growth by retroviral transduction and expression of p210bcr/abl. Blood. 1992;80: 1788-1797. [PubMed] [Google Scholar]
- 24.Gisslinger H, Kurzrock R, Wetzler M, et al. Apoptosis in chronic myelogenous leukemia: studies of stage-specific differences. Leuk Lymphoma. 1997;25: 121-133. [DOI] [PubMed] [Google Scholar]
- 25.Collins AR, Duthie SJ, Dobson VL. Direct enzymic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinogenesis. 1993;14: 1733-1735. [DOI] [PubMed] [Google Scholar]
- 26.Boiteux S, Gajewski E, Laval J, Dizdaroglu M. Substrate specificity of the Escherichia coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of purine lesions in DNA produced by ionizing radiation or photosensitization. Biochemistry. 1992;31: 106-110. [DOI] [PubMed] [Google Scholar]
- 27.Karanjawala ZE, Murphy N, Hinton DR, Hsieh CL, Lieber MR. Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr Biol. 2002;12: 397-402. [DOI] [PubMed] [Google Scholar]
- 28.Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99: 3472-3475. [DOI] [PubMed] [Google Scholar]
- 29.Branford S, Rudzki Z, Walsh S, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102: 276-283. [DOI] [PubMed] [Google Scholar]
- 30.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293: 876-880. [DOI] [PubMed] [Google Scholar]
- 31.Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112: 831-843. [DOI] [PubMed] [Google Scholar]
- 32.Lingrel JB, Croyle ML, Woo AL, Arguello JM. Ligand binding sites of Na, K-ATPase. Acta Physiol Scand Suppl. 1998;643: 69-77. [PubMed] [Google Scholar]
- 33.Price EM, Rice DA, Lingrel JB. Site-directed mutagenesis of a conserved, extracellular aspartic acid residue affects the ouabain sensitivity of sheep Na, K-ATPase. J Biol Chem. 1989;264: 21902-21906. [PubMed] [Google Scholar]
- 34.Schultheis PJ, Lingrel JB. Substitution of transmembrane residues with hydrogen-bonding potential in the alpha subunit of Na, K-ATPase reveals alterations in ouabain sensitivity. Biochemistry. 1993;32: 544-550. [DOI] [PubMed] [Google Scholar]
- 35.Kim JH, Chu SC, Gramlich JL, et al. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005;105: 1717-1723. [DOI] [PubMed] [Google Scholar]
- 36.Conlon KA, Zharkov DO, Berrios M. Immunofluorescent localization of the murine 8-oxoguanine DNA glycosylase (mOGG1) in cells growing under normal and nutrient deprivation conditions. DNA Repair (Amst). 2003;2: 1337-1352. [DOI] [PubMed] [Google Scholar]
- 37.Cambier N, Chopra R, Strasser A, Metcalf D, Elefanty AG. BCR-ABL activates pathways mediating cytokine independence and protection against apoptosis in murine hematopoietic cells in a dose-dependent manner. Oncogene. 1998;16: 335-348. [DOI] [PubMed] [Google Scholar]
- 38.Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351: 657-667. [DOI] [PubMed] [Google Scholar]
- 39.Barnes DJ, Palaiologou D, Panousopoulou E, et al. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65: 8912-8919. [DOI] [PubMed] [Google Scholar]
- 40.Graham SM, Jorgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99: 319-325. [DOI] [PubMed] [Google Scholar]
- 41.Sorel N, Bonnet ML, Guillier M, Guilhot F, Brizard A, Turhan AG. Evidence of ABL-kinase domain mutations in highly purified primitive stem cell populations of patients with chronic myelogenous leukemia. Biochem Biophys Res Commun. 2004;323: 728-730. [DOI] [PubMed] [Google Scholar]
- 42.Chu S, Xu H, Shah NP, et al. Detection of BCR-ABL kinase mutations in CD34+ cells from chronic myelogenous leukemia patients in complete cytogenetic remission on imatinib mesylate treatment. Blood. 2005;105: 2093-2098. [DOI] [PubMed] [Google Scholar]
- 43.Inokuchi K, Yamaguchi H, Tarusawa M, et al. Abnormality of c-kit oncoprotein in certain patients with chronic myelogenous leukemia—potential clinical significance. Leukemia. 2002;16: 170-177. [DOI] [PubMed] [Google Scholar]
- 44.van der Kuip H, Moehring A, Wohlbold L, Miething C, Duyster J, Aulitzky WE. Imatinib mesylate (STI571) prevents the mutator phenotype of Bcr-Abl in hematopoietic cell lines. Leuk Res. 2004;28: 405-408. [DOI] [PubMed] [Google Scholar]
- 45.Brain J, Saksena A, Laneuville P. The kinase inhibitor STI571 reverses the Bcr-Abl induced point mutation frequencies observed in pre-leukemic P190(Bcr-Abl) transgenic mice. Leuk Res. 2002;26: 1011-1016. [DOI] [PubMed] [Google Scholar]
- 46.Mellqvist UH, Hansson M, Brune M, Dahlgren C, Hermodsson S, Hellstrand K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: role of reactive oxygen species and regulation by histamine. Blood. 2000;96: 1961-1968. [PubMed] [Google Scholar]
- 47.Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407: 390-395. [DOI] [PubMed] [Google Scholar]
- 48.Nowicki MO, Falinski R, Koptyra M, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104: 3746-3753. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto M, Kurosu T, Kakihana K, Mizuchi D, Miura O. The two major imatinib resistance mutations E255K and T315I enhance the activity of BCR/ABL fusion kinase. Biochem Biophys Res Commun. 2004;319: 1272-1275. [DOI] [PubMed] [Google Scholar]
- 50.Singh V, Kharb S, Ghalaut PS, Gupta S. Serum vitamin E in chronic myeloid leukaemia. J Assoc Physicians India. 2000;48: 201-203. [PubMed] [Google Scholar]
- 51.Claycombe KJ, Meydani SN. Vitamin E and genome stability. Mutat Res. 2001;475: 37-44. [DOI] [PubMed] [Google Scholar]
- 52.Al-Ali HK, Heinrich MC, Lange T, et al. High incidence of BCR-ABL kinase domain mutations and absence of mutations of the PDGFR and KIT activation loops in CML patients with secondary resistance to imatinib. Hematol J. 2004;5: 55-60. [DOI] [PubMed] [Google Scholar]
- 53.Stephens P, Edkins S, Davies H, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005;37: 590-592. [DOI] [PubMed] [Google Scholar]
- 54.Wang D, Kreutzer DA, Essigmann JM. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat Res. 1998;400: 99-115. [DOI] [PubMed] [Google Scholar]
- 55.Slupianek A, Nowicki MO, Koptyra M, Skorski T. BCR/ABL modifies the kinetics and fidelity of DNA double-strand breaks repair in hematopoietic cells. DNA Repair. 2006;5: 243-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsuda T, Vande Berg BJ, Bebenek K, Osheroff WP, Wilson SH, Kunkel TA. The base substitution fidelity of DNA polymerase beta-dependent single nucleotide base excision repair. J Biol Chem. 2003;278: 25947-25951. [DOI] [PubMed] [Google Scholar]
- 57.Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102: 7665-7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cools J, Mentens N, Furet P, et al. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004;64: 6385-6389. [DOI] [PubMed] [Google Scholar]
- 59.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305: 399-401. [DOI] [PubMed] [Google Scholar]
- 60.O'Hare T, Pollock R, Stoffregen EP, et al. Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potent ATP-based oncogenic protein kinase inhibitor: implications for CML. Blood. 2004;104: 2532-2539. [DOI] [PubMed] [Google Scholar]
- 61.Reddy EP. Payoff of molecular oncology research. Cancer Biol Ther. 2003;2: 115-118. [DOI] [PubMed] [Google Scholar]
- 62.Burgess MR, Skaggs BJ, Shah NP, Lee FY, Sawyers CL. Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance. Proc Natl Acad Sci U S A. 2005;102: 3395-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.von Bubnoff N, Veach DR, van der Kuip H, et al. A cell-based screen for resistance of Bcr-Abl-positive leukemia identifies the mutation pattern for PD166326, an alternative Abl kinase inhibitor. Blood. 2005;105: 1652-1659. [DOI] [PubMed] [Google Scholar]
- 64.Benhar M, Dalyot I, Engelberg D, Levitzki A. Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Mol Cell Biol. 2001;21: 6913-6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wolff NC, Richardson JA, Egorin M, Ilaria RL Jr. The CNS is a sanctuary for leukemic cells in mice receiving imatinib mesylate for Bcr/Abl-induced leukemia. Blood. 2003;101: 5010-5013. [DOI] [PubMed] [Google Scholar]
- 66.Holtz MS, Forman SJ, Bhatia R. Nonproliferating CML CD34+ progenitors are resistant to apoptosis induced by a wide range of proapoptotic stimuli. Leukemia. 2005;19: 1034-1041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}