

Abstract
CXCR4 receptor expression is required for the retention of granulocyte precursors and mature neutrophils within the bone marrow, and disruption of the SDF-1/CXCR4 axis in the bone marrow results in the mobilization of myeloid lineage cells to the peripheral circulation. We report that G-CSF down-regulates CXCR4 expression in bone marrow–derived murine and human myeloid lineage cells. When exposed to G-CSF, murine Gr1+ bone marrow myeloid cells display a time-dependent reduction of cell-surface CXCR4 and respond poorly to SDF-1 in attachment and migration assays. Bone marrow–derived cells of nonmyeloid lineage display no change in surface CXCR4 expression upon exposure to G-CSF. Compared with controls, mice treated with G-CSF for mobilization of hematopoietic progenitor cells display reduced levels of CXCR4 selectively in bone marrow Gr1+ myeloid cells. Since bone marrow myeloid cells express G-CSF receptors and G-CSF rapidly reduces CXCR4 expression in purified Gr1+ cells populations, these results provide evidence that G-CSF acts directly on myeloid lineage cells to reduce CXCR4 expression. By down-regulating CXCR4 expression in bone marrow myeloid cells and attenuating their responsiveness to SDF-1, G-CSF promotes their mobilization from the bone marrow to the peripheral blood.
Introduction
Neutrophil release from the bone marrow is a highly regulated and dynamic process that ensures the maintenance of homeostatic levels of peripheral neutrophils. Although the mechanisms underlying this process are incompletely defined, compelling evidence from gene-targeting studies and other observations indicates that the chemokine receptor CXCR4 and its unique ligand SDF-1 play a critical role in the retention of hematopoietic cells within the bone marrow and their mobilization to the peripheral circulation.1 When CXCR4-deficient hematopoietic cells were injected into lethally irradiated recipient mice, the reconstituted marrows revealed the presence of the more immature myeloid lineage cells, but the more mature myeloid elements were noticeably reduced, likely a consequence of defective retention and premature release of the more mature, CXCR-4–negative, myeloid cells into the bloodstream.2 Patients with WHIM (warts, hypogammaglobulinemia, recurrent bacterial infections, and “myelokathexis”) syndrome, a genetic disorder characterized by heterozygous C-terminal truncations of CXCR4, are neutropenic, in spite of having normal neutrophils in the bone marrow,3,4 likely a consequence of defective neutrophil mobilization linked to a “gain of function” of the mutant CXCR4, with increased CXCR4 responsiveness to bone marrow SDF-1.3-5 AMD3100, a drug that selectively blocks CXCR4 signaling, and methionine-SDF-1β, a mutant chemokine that binds CXCR4 and induces prolonged down-regulation of surface CXCR4 expression, promote the mobilization of neutrophils and hematopoietic progenitor cells to the peripheral blood.6,7
G-CSF, the principal cytokine regulating the proliferation and differentiation of myeloid progenitors, is a potent inducer of the release of neutrophils, immature myeloid lineage cells, and hematopoietic progenitor cells from the bone marrow into the peripheral blood.8-11 Recently, a number of studies have provided evidence that modulation of CXCR4/SDF-1–derived retention signals contributes to G-CSF–induced mobilization of bone marrow hematopoietic progenitor cells, but the underlying mechanisms are not clear.12-16 Much attention has focused on the role of enzymatic cleavage of SDF-1. Following G-CSF administration, the marrow microenvironment is rich in proteolytic enzymes released by neutrophils, including metalloproteinase-9 (MMP-9), neutrophil elastase, and cathepsin G.17 MMP-9, neutrophil elastase, and cathepsin G can cleave and functionally inactivate SDF-1.18-20 A study in MMP-9–deficient mice detected defective G-CSF–induced mobilization of hematopoietic progenitor cells.21 Treatment with an inhibitor of neutrophil elastase reduced, in part, the mobilization of hematopoietic progenitor cells induced by G-CSF.13 However, the mobilization of hematopoietic progenitor cells was normal in mice deficient in either MMP-9 or neutrophil elastase plus cathepsin G, suggesting either a contribution by different enzymes or the occurrence of alternative pathways for G-CSF–induced mobilization of hematopoietic progenitor cells.16 Recently, it was reported that G-CSF reduces SDF-1 synthesis in bone marrow and that reduced SDF-1 in the bone marrow correlates with stem cell mobilization.22
In this study, we have investigated G-CSF regulation of CXCR4 expression in bone marrow myeloid lineage cells. We report that G-CSF significantly reduces levels of CXCR4 in bone marrow–derived primary myeloid lineage and identify down-regulation of CXCR4 expression as an important mechanism for myeloid cell mobilization by G-CSF.
Materials and methods
Preparation of murine and human bone marrow cells
All animal experiments were performed according to NIH guidelines for the care and handling of mice. Bone marrow cells obtained by flushing femurs and tibias of mice (C57BL/6NCr, females, 6-10 weeks old; The Jackson Laboratory, Bar Harbor, ME) were cultured in DMEM culture medium with 10% fetal bovine serum (FBS; Difco Laboratories, Detroit, MI). Gr1+ cells were selected from unfractionated bone marrow cells by positive selection using anti-FITC microbeads following the manufacturer's instructions (Miltenyi Biotech, Auburn, CA). Purity of Gr1+ cells was evaluated by flow cytometry and was found to be more than 90%. Murine myeloid cells were also purified from unfractionated bone marrow cells by electronic sorting (FACSVantage SE; BD Biosciences, San Jose, CA). The sorted cell population consisted of more than 95% Gr1+ cells. Human bone marrow cells were derived by density centrifugation of heparinized bone marrow aspirates obtained with institutional approval or purchased from Cambrex Bio Sciences (Walkersville, MD). CD33+ cells were selected from unselected bone marrow cell populations by positive selection using CD33 microbeads following the manufacturer's instructions (Miltenyi Biotech). Cell purity was more than 85% as assessed by flow cytometry.
Flow cytometric analysis
Murine cells were double stained using allophycocyanin (APC)–conjugated mAb directed against Gr1 (rat anti–mouse Gr1; BD Biosciences) and FITC-conjugated mAb against CXCR4 (rat anti–mouse 2B11; BD Biosciences). Human cells were double stained with phycoerythrin (PE)–conjugated mAb directed against CD33 (mouse anti–human CD33; BD Biosciences) and APC-conjugated mAb against CXCR4 (mouse anti–human 12G5; BD Biosciences). Cell suspensions were analyzed with a FACSCalibur cytofluorometer (BD Biosciences). Data were collected from 10 × 103 viable cells and analyzed with CELLQuest software (BD Biosciences). APC-conjugated CD19 and PE-conjugated CD3e (both from BD Biosciences) mAbs were also used. Isotype-matched FITC- and APC-conjugated immunoglobulin (BD Biosciences) served as controls.
Adhesion and migrations assays
Cell adhesion was carried out essentially as described.23 Wells (96-well plates, Immulon 4HBX; ThermoLabsystems, Franklin, MA) were coated with 4 μg/mL SDF-1 (R&D Systems, Minneapolis, MN) or diluent (PBS with 0.1% BSA) overnight at 4°C, blocked with PBS containing 5% BSA for 1 hour at room temperature. After washing, 105 cells were added in 50 μL DMEM with 10% FBS and incubated at 37°C in 5% CO2 for 30 minutes. Wells were washed 3 times with DMEM, and adherent cells were detached (5 mM EDTA in PBS), counted, stained with APC-conjugated Gr1 mAb, and analyzed by flow cytometry. Adhesion was calculated as a percentage of input Gr1+ cells. Migration assays were carried out as described.24 Briefly, bone marrow cells were loaded onto the upper well layered with a 3-μM pore polycarbonate membrane (Transwell; Costar, Boston, MA). The lower chamber was supplemented with 0.6 mL DMEM with or without 100 ng/mL SDF-1 (R&D Systems). After 2-hour incubation, all cells in the lower chamber were counted, stained with APC-conjugated Gr1 mAb, and analyzed by flow cytometry. The percent Gr1+ cells migrated was calculated (number of migrated Gr1+ cells/total number of Gr1+ cells × 100).
Neutrophil elastase (NE) and cathepsin G (CG) enzymatic activity, and SDF-1 ELISA
The enzymatic assays were carried out as described.25 For standards, purified NE (Elastin Products, Owensville, MI) was diluted to 2 μg/mL in 0.1 M Tris-HCl (pH 7.5), 0.5M NaCl, 0.01% NaN3 (NE buffer); purified CG (Elastin Products) was diluted to 5 μg/mL in 0.1 M Tris-HCl (pH 8.3), 0.01% NaN3 (CG buffer). Test samples or standards (50 μL) were incubated with 50 μL NE substrate or CG substrate (0.4 mM) in the buffer used for standards dilution (Elastin Products) for 30 minutes at 25°C. Reactions were stopped with 0.2 mM PMSF, and absorbance of free pNA was read at 405 nm. As controls, aliquots of NE standards (50 μL) were incubated for 20 minutes at room temperature with or without the Elastase Inhibitor III (CalBiochem, San Diego, CA) before addition of substrate. At 500 nM, the inhibitor could inhibit 10 μg/mL NE. SDF-1 levels were measured by enzyme-linked immunosorbent assay (ELISA), as described.26
CXCR4 immunoblotting
Cells were washed with cold PBS supplemented with protease inhibitor cocktail (Complete; Roche, Indianapolis, IN), and lysed on ice for 30 minutes in 1% Triton X-100 TNE lysis buffer (25 mM Tris [pH 7.5], 150 mM NaCl, 5 mM EDTA) with protease inhibitor cocktail set III (CalBiochem). After centrifugation (10 minutes at 10 000g at 4°C), supernatants were incubated (30 minutes at 37°C). Samples were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE, NuPAGE 10% Bis-Tris Gels; Invitrogen, Grand Island, NY) and blotted onto polyvinylidene difluoride membranes (Fisher Scientific International, Two Rivers, WI). After blocking (5% milk), membranes were incubated overnight with anti–mouse CXCR4 mAb (rat anti–mouse 2B11; BD Biosciences). Bands were visualized using horseradish peroxidase–conjugated goat antirat antibody (Santa Cruz Biotechnology, Santa Cruz, CA), followed by enhanced chemiluminescence detection system (ECL; Amersham Biosciences, Little Chalfont, Buckinghamshire, United Kingdom). Blots were stripped and probed with goat antibodies to actin (Santa Cruz Biotechnology). Films were subjected to densitometric scanning with Image J 1.33u program (NIH, Bethesda, MD) and normalized for actin.
Quantitative reverse-transcriptase–polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from Gr1+ cells using TRIzol (Life Technologies, Gaithersburg, MD). Synthesis of cDNA was performed with 1 μg total RNA by using TaqMan reverse-transcription reagents (Applied Biosystems, Branchburgh, NJ). Real-time PCR amplification was carried out using the ABI Prism 7000 Sequence Detection system (Perkin-Elmer Applied Biosystems, Lincoln, CA), TaqMan Universal PCR Master Mix, and CXCR4-specific primer and FAM-labeled probe sets for quantitative gene expression. Primers for amplification of murine CXCR4 mRNA were (forward) TCAACCTCTACAGCAGCGTTCTC TT and (reverse) TGTTGGTGGCGTGGACAAT. After initial activation with uracyl-N-glycosylase at 50°C for 2 minutes, AmpliTaq Gold was activated at 95°C for 10 minutes. The subsequent PCR conditions consisted of 45 cycles of denaturation at 95°C for 15 seconds and annealing extension at 60°C for 1 minute per cycle. During PCR amplification, the amplified products were measured continuously by determination of fluorescence emission. Expression levels of the CXCR4 gene were normalized to internal GAPDH levels and are presented as relative expression.
Mobilization of hematopoietic cells in mice
C57BL/6NCr female mice, 6 to 10 weeks old (The Jackson Laboratory), were treated intraperitoneally with 5 μg human G-CSF (filgrastim; Amgen, Thousand Oaks, CA) daily for 5 days. Control mice were observed untreated or received a daily intraperitoneal injection of PBS containing 2% human albumin for 5 days. On day 6, mice were killed, and peripheral blood, spleens, femurs, and tibias were collected. Bone marrow unfractionated cells were obtained from femurs and tibias and Gr1+ cells purified as described under “Preparation of murine and human bone marrow cells.”
Statistics
Results are presented as mean ± SEM (standard error of the mean). Comparisons were carried out using Student t test or Mann-Whitney U test, with P values less than .05 considered significant.
Results
G-CSF reduces surface CXCR4 expression in bone marrow myeloid cells
Unfractionated bone marrow cells from mouse tibias and femurs (4- to 6-week-old C57 BL/6 mice) were incubated (1 × 106 cells/mL in DMEM medium supplemented with 10% FBS) at 37°C for 6 hours with or without G-CSF (0.1, 10, and 100 ng/mL). G-CSF dose-dependently reduced surface levels of CXCR4 in Gr1+ cells (Figure 1A), as measured by double staining for CXCR4 (2B11 antibodies27) and mouse Gr1. Myeloid cells express higher levels of surface Gr1 as they mature.28 We found that G-CSF (18-hour incubation) reduces surface CXCR4 in Gr1hi and Gr1lo myeloid cell populations, but the relative reduction in CXCR4 levels was somewhat greater in the Gr1hi compared with Gr1lo myeloid cells (MFI for Gr1hi cells was reduced by 38%, whereas the MFI for Gr1lo cells was reduced by 28.5%). Since during 18-hour culture with G-CSF the proportion of Gr1lo myeloid cells increased by only 6%, these results indicate that the CXCR4 reduction in myeloid cells is not simply due to a relative increase of Gr1lo. G-CSF minimally reduced surface CXCR4 expression in bone marrow T- and B-cell lineage cells (CD3e+ and CD19+ cells, respectively), as measured by double staining for CXCR4 (2B11) and CD3e or CXCR4 and CD19 (Figure 1B), whereas exposure to SDF-1 (100 ng/mL, 37°C, 2 hours) reduced surface CXCR4 on these cells (not shown). We measured SDF-1 levels in culture supernatants of unfractionated bone marrow cells incubated (1 × 106 cell/mL) for 18 hours with or without G-CSF (100 ng/mL) and found them to be undetectable (< 300 pg/mL) as measured by a specific ELISA (not shown).
Figure 1.

G-CSF selectively reduces levels of surface CXCR4 in myeloid lineage cells. (A) Flow cytometric analysis of surface CXCR4 expression in bone marrow Gr1+ myeloid cells. Unfractionated bone marrow cell populations were incubated (1 × 106/mL) at 37°C for 6 hours in medium alone or with G-CSF at concentrations of 0.1, 10, and 100 ng/mL. Cells were double stained for Gr1 (APC-labeled) and CXCR4 (FITC-labeled 2B11 antibody) or with APC- and FITC-labeled control antibodies. Gr1+ cells (R1 gate) represented 40.1% of total nucleated cells. (B) Analysis of CXCR4 expression in CD19+ B-lineage (top panel) and CD3e+ T-lineage (bottom panel) cells. Unfractionated bone marrow cell populations were incubated at 37°C for 6 hours with or without G-CSF (100 ng/mL). Cells were double stained for CD19 (APC-labeled) and CXCR4 (FITC-labeled) or CD3e (APC-labeled) and CXCR4 (FITC-labeled) and gated on CD19+ cells (44% of total nucleated cells) or CD3e+ (5.2% of total nucleated cells) cells. (C) Expression of CXCR4 in Gr1+ cells as a function of culture time (0-18 hours) in medium alone or with G-CSF (100 ng/mL). The results are expressed as mean fluorescence intensity (MFI) of CXCR4 detected on Gr1+ cells (mean ± SEM of 3 experiments). (D) Time-dependent reduction of surface CXCR4 in Gr1+ cells induced by G-CSF (100 ng/mL). Unfractionated bone marrow cell populations were incubated at 37°C for 18 hours with or without G-CSF (100 ng/mL). The results are expressed as the mean (± SEM of 5 experiments) percent reduction of surface CXCR4 induced by G-CSF compared with medium alone. *P < .05 medium versus G-CSF. (E) G-CSF reduces surface CXCR4 in precultured bone marrow cells. Unfractionated bone marrow cell populations were preincubated (37°C, 18 hours) in medium alone to achieve high-level surface CXCR4 expression, and further incubated for 18 hours with or without G-CSF (100 ng/mL). The results reflect levels of surface CXCR4 expression (expressed as MFI) in Gr1+ cells cultured with or without G-CSF after the preculture (mean ± SEM of 3 experiments). (F) Unfractionated bone marrow cell populations were incubated (37°C) for 18 hours with or without G-CSF (100 ng/mL), or for 40 minutes with or without SDF-1α (400 ng/mL) to achieve a reduction of surface CXCR4. After washing (PBS containing 50 mM glycine), the cells were incubated for 2 hours at 37°C in medium containing cycloheximide (100 μg/mL). The results reflect surface CXCR4 expression in Gr1+ myeloid cells (representative experiment of 3 performed).
Kinetic analysis showed that G-CSF (100 ng/mL, 18 hours) reduces surface CXCR4 expression in bone marrow Gr1+ cells after 2-hour incubation and continuing for 18 hours (Figure 1C). This effect was seen consistently (Figure 1D). Notably, surface CXCR4 levels are low in Gr1+ cells immediately after procurement from the bones, but progressively increase during 18-hour incubation at 37°C in medium alone (Figure 1C).
We tested whether G-CSF can reduce surface CXCR4 in Gr1+ cells that already express high-level surface CXCR4. Unfractionated bone marrow cells were preincubated (37°C, 18 hours) in medium only, at which time the Gr1+ cells expressed high-level surface CXCR4, and then incubated for additional 18 hours with or without G-CSF (100 ng/mL). G-CSF reduced surface CXCR4 expression in Gr1+ cells that expressed high-level surface CXCR4 (Figure 1E), but to a somewhat lower degree compared with freshly separated bone marrow cells. We compared the effect of removal of G-CSF or SDF-1 on CXCR4 surface levels in bone marrow Gr1+ cells. As expected, cell incubation for 40 minutes with SDF-1 (400 ng/mL) reduced surface CXCR4, and cell washing followed by further incubation (2 hours, 37°C in medium alone) was associated with considerable reconstitution of surface CXCR4, attributable to SDF-1–induced CXCR4 internalization and recycling to the cell surface (Figure 1F, lower panel). Cell incubation for 18 hours with G-CSF (30 ng/mL) reduced surface CXCR4, but in contrast to SDF-1, cell washing followed by further incubation (2 hours, 37°C in medium alone) was not associated with reconstitution of surface CXCR4 (Figure 1F, upper panel). We purified Gr1+ cells (> 95% purity) from mouse bone marrow cells by electronic sorting, and confirmed that G-CSF (100 ng/mL, 18-hour incubation) significantly reduces surface CXCR4 expression in this highly purified Gr1+ cell population (not shown).
In functional assays, we examined whether reduction of cell surface CXCR4 in Gr1+ cells was reflected by diminished responses to SDF-1, the CXCR4-specific ligand. After preincubation (37°C, 18 hours) with medium only or medium with G-CSF (100 ng/mL) and washing, unfractionated bone marrow cell populations were tested for specific adhesion to SDF-1–coated wells (4 μg/mL) or buffer only over 30 minutes at 37°C. The proportion of Gr1+ cells that specifically adhered to SDF-1–coated wells was significantly (P < .05) reduced after preincubation with G-CSF as opposed to medium alone (Figure 2A). Although cell attachment in this assay system does not simply reflect SDF-1/CXCR4 interactions but depends on contributions by other molecules induced by CXCR4 activation,23 these results provide evidence that G-CSF reduces functional CXCR4 levels in myeloid cells. Additionally, we found that the specific migration of Gr1+ cells in response to SDF-1 (60 and 300 ng/mL) was significantly reduced (P < .05) after preincubation (37°C, 18 hours) with G-CSF (100 ng/mL) as opposed to medium alone (Figure 2B). These experiments demonstrated that G-CSF reduces surface CXCR4 expression in bone marrow myeloid cells and specific responses to SDF-1.
Figure 2.

Effect of G-CSF on bone marrow myeloid cell adhesion and migration to SDF-1. Unfractionated bone marrow cell populations (1 × 106/mL) were preincubated at 37°C for 18 hours with or without G-CSF (100 ng/mL). (A) After washing, the cells were plated onto SDF-1–coated (4 μg/mL) or diluent only–coated (PBS with 0.1% BSA) microtiter wells and incubated at 37°C for 30 minutes. After removal of nonadherent cells, the remaining cells were detached with 5 mM EDTA, counted, and stained for surface Gr1. The results reflect the differential attachment of Gr1+ cells preincubated in medium only or with G-CSF and are expressed as the mean (± SE) percent Gr1+ cells attached of input Gr1+ cells (results from 5 experiments). (B) After preincubation with or without G-CSF and washing, cells were tested for trans-well migration to SDF-1 (2.4-300 ng/mL) or medium only. Cells recovered in the lower well after 2-hour incubation at 37°C were counted and stained for surface Gr1 expression. Results are expressed as the mean (± SE) percent of migrated Gr1+ cells (results reflect the means from 3 experiments). *P < .05 (cells preincubated with medium versus G-CSF).
Analysis of the contribution of enzymatic cleavage to surface CXCR4 reduction by G-CSF
G-CSF treatment promotes the accumulation of neutrophil-derived proteolytic enzymes, including neutrophil elastase, cathepsin-G, and MMP-9 in the bone marrow.17,25 Purified neutrophil elastase and cathepsin G can cleave CXCR4 at the N-terminus.12,20 We tested supernatants of bone marrow cells cultured with G-CSF (100 ng/mL; 20-hour incubation) for their ability to reduce cell-surface CXCR4 levels in freshly isolated Gr1+ cells. Supernatants from G-CSF–treated cultures reduced significantly the levels of surface CXCR4 in Gr1+ cells compared with supernatants from control cultures incubated (6 hours at 37°C) without G-CSF (Figure 3A, left). To distinguish between the 2 possibilities that this CXCR4 reduction was attributable to G-CSF in these supernatants (either human added to the cultures or murine from endogenous secretion) or to enzymes released in the culture supernatants as a consequence of G-CSF stimulation, we used G-CSF neutralizing antibodies. In the presence of G-CSF neutralizing antibodies to both human (5 μg/mL; Peprotech, Rocky Hill, NJ) and mouse (3 μg/mL; R&D Systems) G-CSF (Figure 3A), supernatants from G-CSF–activated bone marrow cultures did not reduce levels of surface CXCR4 in Gr1+ cells compared with control supernatants. We verified the effectiveness of G-CSF neutralization by measuring G-CSF bioactivity in the culture supernatants. Using the G-CSF–responsive NFS-60 cells, supernatants from G-CSF–activated bone marrow cultures induced significant proliferation compared with control supernatants, indicative of the presence of G-CSF. However, addition of anti–human plus anti–mouse G-CSF neutralizing antibodies markedly reduced stimulation of NFS-60 cells by G-CSF–induced supernatants (Figure 3B). These experiments provided evidence for a role of G-CSF as opposed to soluble products induced by G-CSF in the reduction of surface levels of CXCR4 in Gr1+ cells.
Figure 3.

Analysis of the relative contribution of G-CSF and soluble mediators induced by G-CSF to the reduction of surface CXCR4 on bone marrow myeloid cells. Unfractionated bone marrow cell populations were preincubated (1 × 106/mL) at 37°C for 18 hours in medium alone or with G-CSF (100 ng/mL). (A) Cell-free supernatants from these cultures were preincubated (1 hour, 37°C) with rabbit neutralizing antibody against human (final concentration, 5 μg/mL) and rat neutralizing antibodies against mouse (final concentration, 3 μg/mL) G-CSF or with PBS only, and then added undiluted (1 mL) to freshly isolated bone marrow cell populations (1 × 106 cells); the mixture was further incubated (6 hours, 37°C). Levels of CXCR4 expression in Gr1+ cells were measured by flow cytometry. The results are expressed as MFI (± SE) of triplicate determinations. *P < .05 (medium versus G-CSF). (B) Detection of G-CSF bioactivity in culture supernatants from bone marrow cell populations incubated with or without neutralizing antibodies against mouse and human G-CSF (as described under “Preparation of murine and human bone marrow cells”). G-CSF bioactivity was measured by 3H thymidine incorporation in murine NFS-60 cells, and the results are expressed as mean (± SE) of triplicate cultures. *P < .05. (C-D) Neutrophil elastase reduces levels of surface CXCR4 in bone marrow myeloid (C) and lymphoid (D) cells. Unfractionated bone marrow cell populations were incubated (1 × 106/mL in DMEM medium with 0.2% BSA for 4 hours at 37°C) with medium alone, leukocyte elastase (enzyme, 40 μg/mL), and with leukocyte elastase plus leukocyte elastase inhibitor III (10 μM). Levels of surface CXCR4 were measured by flow cytometry on Gr1+ cells and lymphoid cells. (E) Dose dependency of leukocyte elastase–induced reduction of surface CXCR4 on Gr1+ cells and lymphoid cells. Unfractionated bone marrow cell populations were incubated with leukocyte elastase (5-100 μg/mL) for 2 hours at 37°C. The results are expressed as percent CXCR4 expression compared with untreated cells (MFI of elastase-treated cells/MFI of untreated cells × 100). (F) Surface CXCR4 expression in the presence of specific enzyme inhibitors. Unfractionated bone marrow cell populations were preincubated (2 hours, 37°C) in medium only (DMEM with 0.2% BSA); or with leukocyte elastase inhibitor III (LEI 10 μM); cathepsin-G (CGI, 10 μM); LEI plus CGI (10 μM each); the CD26/dipeptdyl peptidase inhibitor AB192 (50 μM); and the metalloproteinase inhibitor BB94 (10 μM). Subsequently, cells were incubated with or without G-CSF (100 ng/mL, 4 hours, 37°C). Surface CXCR4 was measured on Gr1+ cells by flow cytometry. The results are expressed as percent MFI compared with cells incubated in medium alone without inhibitors.
To examine further the potential contribution of neutrophil elastase and cathepsin G to the reduction of surface CXCR4 induced by G-CSF, we measured these enzymes in culture supernatants of bone marrow cells cultured in medium only or with G-CSF (100 ng/mL) for 6 to 18 hours. Mean neutrophil elastase levels in control supernatants (medium only) were 0.85 ± 0.5 μg/mL (mean ± SD; n = 21) and in G-CSF–supplemented culture supernatants were 0.76 ± 0.23 μg/mL (n = 22) (P = .576 vs control). Mean cathepsin G levels in control supernatants were 0.25 ± 0.32 μg/mL (n = 21) and in G-CSF–supplemented culture supernatants were 0.17 ± 0.21 μg/mL (n = 22) (P = .351 vs control). In the absence of serum (DMEM with 0.2% bovine serum albumin), purified neutrophil elastase (40 μg/mL, 4 hours at 37°C) reduced levels of surface CXCR4 in bone marrow Gr1+ cells (Figure 3C) and, unlike G-GSF, in lymphocytes (Figure 3D) as well, albeit to a lower degree. The leukocyte elastase inhibitor (elastase inhibitor III, leukocyte elastase inhibitor [LEI] 10 μM, added 10 minutes prior to addition of the enzyme) prevented this reduction (Figure 3C-D). Under these experimental conditions, surface CXCR4 reduction in myeloid and lymphoid cells was dependent upon the dose of neutrophil elastase (Figure 3E). However, unlike G-CSF, the enzyme was ineffective at reducing CXCR4 in the presence of 10% serum (not shown), likely a consequence of enzyme inhibitors present in serum.
The LEI (10 μM; CalBiochem), cathepsin G inhibitor I (CGI, 10 μM, CalBiochem), CD26/dipeptidylpeptidase IV inhibitor AB192 (50 μM, a gift from Dr A. M. Lambeir, University of Antwerp, Belgium), and the metalloproteinase inhibitor BB94 (10 μM; British Biotech, Oxford, United Kingdom) were added to cultures of bone marrow cells for 20 minutes prior to the addition of medium only or with G-CSF (100 ng/mL), and incubation continued for 4 hours. Culture supernatants with or without inhibitors were tested for their ability to reduce surface CXCR4 in freshly isolated bone marrow cells after 6-hour incubation. As shown (Figure 3F), individual inhibitors and the combination of NEI plus CGI did not prevent the occurrence of G-CSF–induced reduction of surface CXCR4 in Gr1+ cells. These results, together, provide evidence that soluble products induced by G-CSF, including the enzymes neutrophil elastase, cathepsin G, CD26/dipeptidylpeptidase IV, and metalloproteinases, are not responsible for reducing surface CXCR4 levels in the current system, and rather point to a more direct role of G-CSF.
G-CSF reduces CXCR4 protein levels in myeloid cells
Since CXCR4 is largely an intracellular protein,27 we examined the effect of G-CSF on the total CXCR4 content in purified Gr1+ bone marrow cells. By Western blotting with anti-CXCR4–specific antibodies (2B11), we typically detected 2 major CXCR4-related bands with approximate relative molecular weights of 47 and 42 kDa in cell lysates of purified Gr1+ bone marrow cells (Figure 4A). In repeated experiments, the relative intensity of these 2 bands was somewhat variable (not shown). After treatment with PNGase F (10 U/40 μg protein; New England Biolabs), an enzyme that hydrolyzes most types of N-glycan chains from glycopeptides, an intense and mostly unique CXCR4-related band is visualized at approximately 39 kDa (Figure 4B), providing evidence that both the 42- and 47-kDa CXCR4 bands visualized in Gr1+ bone marrow cells are glysosylated forms of the protein. CXCR4 has a predicted 39.7 kDa MW and contains 2 glycosylation sites.29,30
Figure 4.

Effects of G-CSF on CXCR4 protein levels in murine bone marrow myeloid cells. (A) Typical representation of CXCR4 protein in cell lysates of freshly isolated Gr1+ cells purified from bone marrow cell populations detected by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with specific antibodies to CXCR4 (2B11 antibody). (B) Effect of the enzyme PNGase F (10 U/40 mg protein) on CXCR4 protein detected by immunoblotting (2B11 antibody). (C) Time-dependent reduction of Gr1+ cell–associated CXCR4 induced by G-CSF. Cells lysates were prepared from purified Gr1+ bone marrow cells either immediately after purification or after culture in medium alone (M) or with G-CSF (G, 100 ng/mL). Samples were immunoblotted with anti-CXCR4 antibodies (2B11), stripped, and reprobed with antibodies to beta-actin (representative experiment of 5 performed). (D) Appearance of CXCR4-related bands after 18-hour incubation of electronically sorted Gr1+ cells (> 95% purity) in medium only (M) or with G-CSF (G, 100 ng/mL). Bottom panel reflects actin-related bands upon reprobing with anti–beta actin antibodies. (E) Levels of CXCR4 protein in Gr1+ cells immediately after purification and after culture (1-18 hours) with medium only or with 100 ng/mL G-CSF measured by band intensities relative to actin. The results are expressed as percent CXCR4 measured in Gr1+ cells immediately after purification (mean ± SD of 3-6 separate experiments; 1-hour time point: mean of 6 experiments; all other time points: mean of 3 experiments). *P < .05; **P < .1 (medium vs G-CSF).
When cultured with G-CSF, purified bone marrow Gr1+ cells reproducibly displayed a time-dependent reduction of the 42- and 47-kDa CXCR4-related bands compared with cells cultured in medium alone (representative image Figure 4C). This reduction was not attributable to unequal loading as reflected by membrane reprobing with actin antibodies (Figure 4C). Since there were no higher or lower molecular weight CXCR4-related bands specifically induced by G-CSF treatment during incubation, even after 18-hour incubation (Figure 4D), these results provided evidence that G-CSF can reduce the CXCR4 content in Gr1+ cells. Analysis of relative band intensities from 3 to 5 experiments revealed that CXCR4 levels are lower in the presence of G-CSF compared with medium alone (P < .05 at time points of 1 hour and 6 hours; P < .1 at 3 and 18 hours) (Figure 4E).
G-CSF reduces CXCR4 synthesis in murine myeloid cells
We examined whether G-CSF reduces CXCR4 synthesis in this system. When bone marrow Gr1+ cells were cultured with cycloheximide (CHX; 5 μg/mL), which inhibits protein synthesis, the appearance and size of the CXCR4-related bands were similar in cells cultured with or without G-CSF for 1 or 2 hours (Figure 5A). Of note, in the presence of CHX with or without G-CSF, the 42-kDa CXCR4 band was no longer visible (Figure 5A), suggesting that this band may reflect newly synthesized CXCR4 protein. Moreover, the spontaneous increase of surface and cell-associated CXCR4 during incubation was blocked by CHX. The observation that G-CSF no longer reduced the CXCR4 content in Gr1+ cells in the presence of CHX is consistent with G-CSF affecting CXCR4 synthesis.
Figure 5.
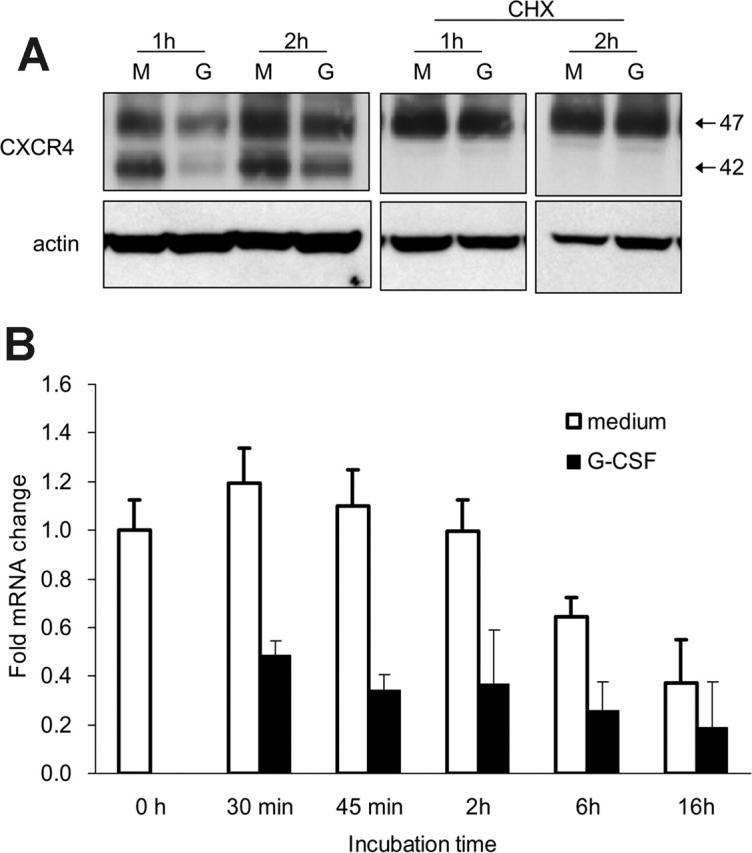
G-CSF down-regulates CXCR4 mRNA expression in bone marrow myeloid cells. (A) Effects of cycloheximide (CHX) on CXCR4 protein levels in Gr1+ cells detected by immunoblotting with specific antibodies to CXCR4 and actin. Purified bone marrow Gr1+ cells were cultured (1 × 106/mL) for 1 and 2 hours in medium alone (M) or with G-CSF (G, 100 ng/mL), with or without CHX (5 μg/mL). Total cell lysates were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and immunoblotted (representative experiment of 3 performed). (B) Levels of CXCR4 mRNA in purified bone marrow Gr1+ cells immediately after purification and after culture with G-CSF (100 ng/mL, 30 minutes to 16 hours) measured by real-time RT-PCR analysis. The results are expressed as mean fold (± SE) mRNA change relative to mRNA levels detected before culture (results from 3 independent experiments each tested in triplicate).
We used quantitative RT-PCR to test whether G-CSF reduces CXCR4 mRNA levels in Gr1+ cells. Bone marrow Gr1+ cells were incubated (37°C, 30 minutes to 18 hours) with or without G-CSF (100 ng/mL), and total RNA was extracted. As shown (Figure 5B reflecting results from 3 experiments each performed in triplicate), the CXCR4 mRNA content was significantly (P < .05, all time points) reduced in Gr1+ cells cultured with G-CSF as opposed to medium alone. These results demonstrate that G-CSF profoundly down-regulates CXCR4 synthesis in bone marrow Gr1+ cells.
Reduced CXCR4 expression in murine myeloid cells following G-CSF treatment in vivo
To test for the in vivo effects of G-CSF treatment on levels of CXCR4 in myeloid lineage cells, groups of 25 mice (C57/BL6, 6 weeks old) were treated systemically with G-CSF (5 μg/mouse, intraperitoneally) for 5 days. Consistent with previous observations,31 we documented that this treatment consistently induces mobilization of hematopoietic cells from the bone marrow to the peripheral blood (not shown). In 3 separate experiments, we purified Gr1+ cells from the bone marrow of mice treated for 5 days with G-CSF and from controls. By immunoblotting of total cell lysates, we found that levels of cell-associated CXCR4 were consistently lower in Gr1+ cells from G-CSF–treated mice compared with the untreated controls (Figure 6A). By contrast, levels of CXCR4 were similar in the Gr1– bone marrow cell populations from G-CSF–treated and control mice (Figure 6A). This reduction in CXCR4 content exhibited by bone marrow Gr1+ cells from mice treated with G-CSF compared with controls was confirmed in 2 other independent experiments (Figure 6B). These results provide evidence that G-CSF mobilization of hematopoietic cells in mice is associated with a reduction of CXCR4 levels in bone marrow Gr1+ cells.
Figure 6.

G-CSF–induced hematopoietic cell mobilization is associated with a reduction of CXCR4 in myeloid cells. Bone marrow Gr1+ cells were purified from the bone marrow of groups of mice (C57/BL6) either untreated or treated for 5 days with G-CSF (5 μg/mouse, daily intraperitoneally for 5 days). (A) Gr1+ cells and Gr1– cells were obtained from groups of mice treated for 5 days with G-CSF or buffer only. CXCR4 content in Gr1+ cells and Gr1– cell lysates evaluated by immunoblotting with specific CXCR4 antibodies, and reprobing with anti–beta actin antibodies. (B) Western blot analysis of CXCR4 and actin content in cell lysates from 2 groups of mice treated with G-CSF and a control group of untreated mice.
G-CSF reduces CXCR4 expression in human bone marrow myeloid cells
Unfractionated cells from normal human bone marrows were incubated (1 × 106 cells/mL in DMEM culture medium supplemented with 10% FBS) at 37°C for 18 hours with or without G-CSF (1 and 100 ng/mL). G-CSF dose-dependently reduced surface CXCR4 levels in human CD33+ cells after 18-hour incubation (Figure 7A), as measured by double staining for CXCR4 (12G5 antibodies27) and human CD33. By contrast, G-CSF did not substantially change surface CXCR4 levels in human CD33– bone marrow cell populations after 18-hour incubation (Figure 7B). Kinetic analysis showed that levels of surface CXCR4 spontaneously increase in CD33+ cells during incubation in medium alone (Figure 7C). When present in these cultures, G-CSF (100 ng/mL) reduced surface CXCR4 levels in CD33+ cells (Figure 7C), and this reduction was significant (P < .05) beginning at the 4-hour time point (Figure 7D).
Figure 7.

Effects of G-CSF on CXCR4 expression in human bone marrow myeloid cells. (A-B) Unfractionated bone marrow cell populations were incubated (1 × 106/mL) at 37°C for 18 hours in medium alone or with G-CSF (1 and 100 ng/mL). Cells were double stained for CD33 (PE-labeled) and CXCR4 (APC-labeled 12G5 antibody) or with PE- and APC-labeled control antibodies (isotype). (A) Surface CXCR4 expression in human bone marrow CD33+ myeloid cells cultured for 18 hours in medium alone (none) or with G-CSF (1 and 100 ng/mL). (B) Surface CXCR4 expression in human bone marrow CD33– cell populations cultured for 18 hours in medium alone (none) or with G-CSF (1 and 100 ng/mL). (C) Expression of surface CXCR4 in human CD33+ bone marrow cells as a function of culture time (0-18 hours) in medium alone or with G-CSF (100 ng/mL). The results are expressed as mean fluorescence intensity (MFI) of CXCR4 detected on CD33+ cells (mean ± SEM of 3 experiments). (D) Time-dependent reduction of surface CXCR4 in CD33+ cells induced by G-CSF (100 ng/mL). Unfractionated bone marrow cell populations were incubated at 37°C for 18 hours with or without G-CSF (100 ng/mL). The results are expressed as the mean (± SEM of 3 experiments) percent reduction of surface CXCR4 induced by G-CSF compared with medium alone. *P < .05 medium versus G-CSF. (E) CXCR4 detected in cell lysates of purified bone marrow CD33+ cells (> 85% purity) immediately after separation (0h), and after 1.5-hour and 18-hour incubation in medium only (M) or with G-CSF (G, 100 ng/mL). Bottom panel reflects the results upon membrane reprobing with anti–beta actin antibodies.
We tested the effects of G-CSF on the CXCR4 content of human bone marrow CD33+ cells. To this end, we affinity-purified human bone marrow CD33+ cells (> 85% pure). After 1.5- and 18-hour incubations at 37°C with G-CSF (100 ng/mL), the purified CD33+ cells displayed a specific reduction in the intensities of CXCR4-related bands compared with cells cultured in medium alone (Figure 7E). These results provide evidence that G-CSF can reduce CXCR4 levels in human bone marrow myeloid cells.
Discussion
There is substantial evidence for a critical role of the SDF-1/CXCR4 axis in the retention of myeloid lineage cells to the bone marrow.2 There is also evidence that disruption of the SDF-1/CXCR4 axis plays a role in the mobilization of myeloid lineage cells from the bone marrow to the peripheral circulation induced by G-CSF,1 but the mechanisms underlying this disruption are incompletely known.
Here we show that G-CSF significantly and consistently down-regulates CXCR4 mRNA expression in bone marrow Gr1-expressing myeloid cells, and significantly reduces CXCR4 protein levels within and on the surface of murine and human bone marrow myeloid cells. When mice were mobilized with G-CSF, the Gr1+ myeloid cells from the bone marrow displayed reduced levels of CXCR4. These results support an important contribution of reduced CXCR4 expression to G-CSF–induced mobilization of myeloid lineage cells. Consistent with these results, AMD3100, a selective inhibitor of CXCR4 signaling and a mutant SDF-1β molecule, which binds CXCR4 and produces extended receptor desensitization, can promote the release of mature and immature hematopoietic progenitor cells from the bone marrow into the circulation.7,32 Recently, neutrophil mobilization in response to G-CSF was reported impaired in mice deficient in GRK6 (G protein–coupled receptor kinase-6), which participates in regulation of CXCR4 signaling.33
Prior to the current studies, a direct role of CXCR4 in mediating G-CSF mobilization was unclear. In one report, the amino terminus of CXCR4 was reported cleaved and the receptor inactive on human bone marrow and peripheral CD34+ hematopoietic progenitor cells following G-CSF mobilization.12 Neutrophil elastase and cathepsin G were implicated in the cleavage.12 However, the occurrence of CXCR4 cleavage was not confirmed in mouse bone marrow following G-CSF administration, and G-CSF mobilization proceeded normally in mice deficient in neutrophil elastase and cathepsin G.22 Here, we found no evidence for enzymatic cleavage of CXCR4 in murine Gr1+ cells. Other studies noted that levels of cell surface CXCR4 fluctuate in mouse bone marrow cells during G-CSF mobilization, with transient reductions after each G-CSF injection, attributed to local changes in SDF-1 levels.13
Unlike CXCR4, several studies have documented reductions of SDF-1 levels in the bone marrow following G-CSF treatment,12-14 and a correlation was found between levels of SDF-1 reduction in the bone marrow and degree of hematopoietic progenitor cell mobilization.22 It was proposed that G-CSF indirectly causes SDF-1 degradation and inactivation through the peptidase enzymes neutrophil elastase, cathepsin-G, or matrix MMP-9, which accumulate in the bone marrow during G-CSF treatment.12,13,17,21 However, mice genetically deficient in neutrophil elastase and cathepsin-G, matrix MMP-9, or dipetidylpeptidase I, an enzyme required for activation of several serine proteases, mobilized normally in response to G-CSF.16 Cell surface–associated CD26/dipeptidyl peptidase IV was also proposed to contribute to G-CSF mobilization through cleavage and inactivation of SDF-1,34 but there is no direct evidence of CD26 modulation during G-CSF mobilization, and CD26-null mice have no apparent defect in hematopoiesis.35 However, in one study, CD26-null mice mobilized poorly in response to G-CSF.34 The naturally occurring serine protease inhibitors serpina1 and serpina3 were reported down-regulated within the bone marrow during hematopoietic cell mobilization, raising the possibility that a shift in the balance between serine proteases and their natural inhibitors may be responsible for G-CSF–induced hematopoietic cell mobilization.36 Recently, G-CSF was reported to indirectly reduce SDF-1 mRNA and protein levels in bone marrow cells, supporting a contribution of protease-independent mechanisms.22
We observed that G-CSF reduces CXCR4 expression in bone marrow myeloid cells but not in lymphocytes. Since myeloid cells express G-CSF receptors37 and G-CSF rapidly reduces CXCR4 expression in highly purified Gr1+ cell populations, it is likely that in the current system G-CSF acts directly on the target myeloid cells. Similarly, G-CSF was reported to directly down-regulate ribosomal RNA synthesis in the myeloid 32D cells, a function that was mapped to the carboxyl-terminal domain of the receptor.38 The carboxyl-terminal domain of the G-CSF receptor is required for G-CSF–induced differentiation and mobilization, but not for the mitogenic function of G-CSF.39,40
Several mechanisms may contribute to G-CSF mobilization besides interference with retention signals by SDF-1/CXCR4.41 A requirement for an active role of CXCR4 was suggested by the reduced ability of G-CSF to promote mobilization in mice treated with neutralizing antibodies to CXCR4 or to SDF-1.13 These experiments suggested that circulating SDF-1 may contribute essential signals for transendothelial migration of bone marrow hematopoietic cells to the peripheral blood.13 Indeed, adenoviral-mediated expression of SDF-142 and administration of sulfated polysaccharides43 increased SDF-1 plasma levels and promoted the mobilization of hematopoietic progenitor cells. Recently, a CXCR4 agonist peptide, CTCE-0021, more stable than natural SDF-1, which is rapidly cleaved by serum enzymes,44,45 was reported to be an effective mobilizing agent in mice.46 SDF-1 associated with the bone marrow venous sinuses,47 the presumed point of exit of leukocytes from the bone marrow, may provide essential transmigration signals, as SDF-1 does for peripheral blood lymphocytes migrating to the extravascular space.48 Furthermore, the orderly movement of hematopoietic cells within the bone marrow may require local SDF-1 gradients, as it was recently shown for cells of B-cell lineage.49
G-CSF is the most commonly used agent for therapeutic mobilization of hematopoietic progenitor cells to the peripheral circulation. We now provide evidence supporting a critical role of reduced CXCR4 expression in mediating G-CSF mobilization of myeloid lineage cells. This new information provides a basis for development of new drugs for selective mobilization of hematopoietic progenitor cells.
Acknowledgments
The authors wish to thank Drs A. Friedman, Dennis Hickstein, P. McCormick, D. Fowler, J. Ford, P. Gasperini, T. Bauer, and O. Salvucci for their help in various aspects of this work. C.K.W. is a National Institutes of Health–University of Oxford Health Science Scholar.
Prepublished online as Blood First Edition Paper, March 14, 2006; DOI 10.1182/blood-2005-10-4162.
Supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and in part by a grant from the Cancer Research Institute, Seoul National University College of Medicine (CRI-05-04), Korea.
H.K.K., M.D.L.L.S., and A.V.G. performed experiments; C.K.K. analyzed results and made the figures; G.T. and H.K.K. designed the research and wrote the paper.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Link DC. Neutrophil homeostasis: a new role for stromal cell-derived factor-1. Immunol Res. 2005;32: 169-178. [DOI] [PubMed] [Google Scholar]
- 2.Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10: 463-471. [DOI] [PubMed] [Google Scholar]
- 3.Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk JR Jr. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet. 2000;91: 368-376. [PubMed] [Google Scholar]
- 4.Gulino AV, Moratto D, Sozzani S, et al. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood. 2004;104: 444-452. [DOI] [PubMed] [Google Scholar]
- 5.Balabanian K, Lagane B, Pablos JL, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105: 2449-2457. [DOI] [PubMed] [Google Scholar]
- 6.Hendrix CW, Flexner C, MacFarland RT, et al. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother. 2000;44: 1667-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen H, Cheng T, Olszak I, et al. CXCR-4 desensitization is associated with tissue localization of hemopoietic progenitor cells. J Immunol. 2001;166: 5027-5033. [DOI] [PubMed] [Google Scholar]
- 8.Nagata S, Tsuchiya M, Asano S, et al. Molecular cloning and expression of cDNA for human granulocyte colony-stimulating factor. Nature. 1986;319: 415-418. [DOI] [PubMed] [Google Scholar]
- 9.Nicola NA, Begley CG, Metcalf D. Identification of the human analogue of a regulator that induces differentiation in murine leukaemic cells. Nature. 1985;314: 625-628. [DOI] [PubMed] [Google Scholar]
- 10.Cohen AH, Mackler SA, Selzer ME. Functional regeneration following spinal transection demonstrated in the isolated spinal cord of the larval sea lamprey. Proc Natl Acad Sci U S A. 1986;83: 2763-2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelus LM, Horowitz D, Cooper SC, King AG. Peripheral blood stem cell mobilization: a role for CXC chemokines. Crit Rev Oncol Hematol. 2002;43: 257-275. [DOI] [PubMed] [Google Scholar]
- 12.Levesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111: 187-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3: 687-694. [DOI] [PubMed] [Google Scholar]
- 14.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002;17: 413-423. [DOI] [PubMed] [Google Scholar]
- 15.Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19: 583-593. [DOI] [PubMed] [Google Scholar]
- 16.Levesque JP, Liu F, Simmons PJ, et al. Characterization of hematopoietic progenitor mobilization in protease-deficient mice. Blood. 2004;104: 65-72. [DOI] [PubMed] [Google Scholar]
- 17.Levesque JP, Hendy J, Takamatsu Y, Williams B, Winkler IG, Simmons PJ. Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp Hematol. 2002;30: 440-449. [DOI] [PubMed] [Google Scholar]
- 18.Delgado MB, Clark-Lewis I, Loetscher P, et al. Rapid inactivation of stromal cell-derived factor-1 by cathepsin G associated with lymphocytes. Eur J Immunol. 2001;31: 699-707. [DOI] [PubMed] [Google Scholar]
- 19.McQuibban GA, Butler GS, Gong JH, et al. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276: 43503-43508. [DOI] [PubMed] [Google Scholar]
- 20.Valenzuela-Fernandez A, Planchenault T, Baleux F, et al. Leukocyte elastase negatively regulates stromal cell-derived factor-1 (SDF-1)/CXCR4 binding and functions by amino-terminal processing of SDF-1 and CXCR4. J Biol Chem. 2002;277: 15677-15689. [DOI] [PubMed] [Google Scholar]
- 21.Heissig B, Hattori K, Dias S, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kitligand. Cell. 2002;109: 625-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semerad CL, Christopher MJ, Liu F, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106: 3020-3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell JJ, Qin S, Bacon KB, Mackay CR, Butcher EC. Biology of chemokine and classical chemoattractant receptors: differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J Cell Biol. 1996;134: 255-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez-Ramos JC. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med. 1997;185: 111-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levesque JP, Takamatsu Y, Nilsson SK, Haylock DN, Simmons PJ. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood. 2001;98: 1289-1297. [DOI] [PubMed] [Google Scholar]
- 26.Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002;99: 2703-2711. [DOI] [PubMed] [Google Scholar]
- 27.Forster R, Kremmer E, Schubel A, et al. Intracellular and surface expression of the HIV-1 coreceptor CXCR4/fusin on various leukocyte subsets: rapid internalization and recycling upon activation. J Immunol. 1998;160: 1522-1531. [PubMed] [Google Scholar]
- 28.Hestdal K, Ruscetti FW, Ihle JN, et al. Characterization and regulation of RB6–8C5 antigen expression on murine bone marrow cells. J Immunol. 1991;147: 22-28. [PubMed] [Google Scholar]
- 29.Federsppiel B, Melhado IG, Duncan AM, et al. Molecular cloning of the cDNA and chromosomal localization of the gene for a putative seven-transmembrane segment (7-TMS) receptor isolated from human spleen. Genomics. 1993;16: 707-712. [DOI] [PubMed] [Google Scholar]
- 30.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272: 872-877. [DOI] [PubMed] [Google Scholar]
- 31.de Haan G, Dontje B, Engel C, Loeffler M, Nijhof W. The kinetics of murine hematopoietic stem cells in vivo in response to prolonged increased mature blood cell production induced by granulocyte colony-stimulating factor. Blood. 1995;86: 2986-2992. [PubMed] [Google Scholar]
- 32.Liles WC, Broxmeyer HE, Rodger E, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102: 2728-2730. [DOI] [PubMed] [Google Scholar]
- 33.Vroon A, Heijnen CJ, Raatgever R, et al. GRK6 deficiency is associated with enhanced CXCR4-mediated neutrophil chemotaxis in vitro and impaired responsiveness to G-CSF in vivo. J Leukoc Biol. 2004;75: 698-704. [DOI] [PubMed] [Google Scholar]
- 34.Christopherson KW II, Hangoc G, Broxmeyer HE. Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1 alpha-mediated chemotaxis of human cord blood CD34+ progenitor cells. J Immunol. 2002;169: 7000-7008. [DOI] [PubMed] [Google Scholar]
- 35.Conarello SL, Li Z, Ronan J, et al. Mice lacking dipeptidyl peptidase IV are protected against obesity and insulin resistance. Proc Natl Acad Sci U S A. 2003;100: 6825-6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winkler IG, Hendy J, Coughlin P, Horvath A, Levesque JP. Serine protease inhibitors serpina1 and serpina3 are down-regulated in bone marrow during hematopoietic progenitor mobilization. J Exp Med. 2005;201: 1077-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukunaga R, Ishizaka-Ikeda E, Nagata S. Purification and characterization of the receptor for murine granulocyte colony-stimulating factor. J Biol Chem. 1990;265: 14008-14015. [PubMed] [Google Scholar]
- 38.Kroll SL, Barth-Baus D, Hensold JO. The carboxyl-terminal domain of the granulocyte colony-stimulating factor receptor uncouples ribosomal biogenesis from cell cycle progression in differentiating 32D myeloid cells. J Biol Chem. 2001;276: 49410-49418. [DOI] [PubMed] [Google Scholar]
- 39.Ward AC, Smith L, de Koning JP, van Aesch Y, Touw IP. Multiple signals mediate proliferation, differentiation, and survival from the granulocyte colony-stimulating factor receptor in myeloid 32D cells. J Biol Chem. 1999;274: 14956-14962. [DOI] [PubMed] [Google Scholar]
- 40.Semerad CL, Poursine-Laurent J, Liu F, Link DC. A role for G-CSF receptor signaling in the regulation of hematopoietic cell function but not lineage commitment or differentiation. Immunity. 1999;11: 153-161. [DOI] [PubMed] [Google Scholar]
- 41.Juarez J, Bendall L. SDF-1 and CXCR4 in normal and malignant hematopoiesis. Histol Histopathol. 2004;19: 299-309. [DOI] [PubMed] [Google Scholar]
- 42.Hattori K, Heissig B, Tashiro K, et al. Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood. 2001;97: 3354-3360. [DOI] [PubMed] [Google Scholar]
- 43.Sweeney EA, Lortat-Jacob H, Priestley GV, Nakamoto B, Papayannopoulou T. Sulfated polysaccharides increase plasma levels of SDF-1 in monkeys and mice: involvement in mobilization of stem/progenitor cells. Blood. 2002;99: 44-51. [DOI] [PubMed] [Google Scholar]
- 44.De La Luz Sierra M, Yang F, Narazaki M, et al. Differential processing of stromal-derived factor-1alpha and stromal-derived factor1beta explains functional diversity. Blood. 2004;103: 2452-2459. [DOI] [PubMed] [Google Scholar]
- 45.Davis DA, Singer KE, De La Luz Sierra M, et al. Identification of carboxypeptidase N as an enzyme responsible for C-terminal cleavage of stromal cell-derived factor-1alpha in the circulation. Blood. 2005;105: 4561-4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelus LM, Bian H, Fukuda S, Wong D, Merzouk A, Salari H. The CXCR4 agonist peptide, CTCE-0021, rapidly mobilizes polymorphonuclear neutrophils and hematopoietic progenitor cells into peripheral blood and synergizes with granulocyte colony-stimulating factor. Exp Hematol. 2005;33: 295-307. [DOI] [PubMed] [Google Scholar]
- 47.Ponomaryov T, Peled A, Petit I, et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function [in process citation]. J Clin Invest. 2000;106: 1331-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol. 2001;2: 515-522. [DOI] [PubMed] [Google Scholar]
- 49.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20: 707-718. [DOI] [PubMed] [Google Scholar]