

Abstract
GPVI is a 62-kDa membrane glycoprotein expressed in noncovalent association with the Fc receptor γ chain on human and murine platelets and serves as the major activating receptor for collagen. GPVI-specific antibodies have the capacity to specifically deplete GPVI from mouse and human platelets in vivo, rendering them unresponsive to collagen and GPVI-specific agonists. Such antibodies do not remove GPVI from noncirculating platelets in vitro, however, making it difficult to evaluate their antithrombotic potential and mechanism of action, particularly in human platelets. We devised a model system in which human platelets are introduced into the retroorbital plexus of nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice, allowed to circulate, and evaluated for the effects of GPVI-specific murine monoclonal antibodies (mAbs) on platelet survival and function. GPVI-specific mAbs triggered depletion of GPVI from human, but not murine, platelets. Soluble truncated human GPVI appeared concomitantly in mouse plasma. GPVI-depleted human platelets had markedly diminished responses to GPVI-specific agonists and unexpectedly exhibited somewhat depressed responses to G-protein–coupled agonists. The ability to evaluate in living mice the in vivo function and survival of circulating human platelets may prove valuable for determining mechanisms of antibody-mediated platelet passivation and aid in the development of novel anti-platelet therapeutics.
Introduction
An array of agonist receptors reside on the surface of platelets that, on exposure to ligands, initiate the activation of a complex network of signaling pathways leading to platelet activation, adhesion, and thrombus formation. Collagen, a major component of the extracellular matrix, acts as an important primary indicator to the presence of vessel injury. GPVI, the principal platelet collagen receptor, is a 62-kDa platelet-membrane glycoprotein expressed on the surface of human and murine platelets in a noncovalent complex with the immunoreceptor tyrosine-based activation motif (ITAM)–containing subunit, the FcRγ chain.1,2 GPVI is a member of the immunoglobulin gene (Ig) superfamily that is composed of 2 extracellular Ig-homology domains, a transmembrane domain, and a 51–amino acid cytoplasmic domain.3-5 The GPVI/FcRγ chain complex serves as the major platelet-activating receptor for collagen and signals via the Syk/SLP-76/PLCγ2 pathway to activate the integrins α2β1 and αIIbβ3 (also known as GPIIb-IIIa), leading to platelet activation and thrombus formation.6,7
Studies by Nieswandt et al8 Massberg et al,9 and Schulte et al10 have shown the injection into mice of rat anti–mouse GPVI monoclonal antibodies (termed JAQ1, JAQ2, and JAQ3) results in specific, long-term immunodepletion of GPVI from the surface of circulating murine platelets. Platelets isolated from mice treated in this manner exhibit a corresponding loss of responsiveness to collagen and the GPVI-specific agonist, collagen-related peptide (CRP).8,9 Antibody-mediated depletion of GPVI appears to be operable in humans as well, because platelets from a patient with a circulating autoantibody specific for GPVI, like the platelets from JAQ1-treated mice, are devoid of cell-surface GPVI, fail to become activated in response to collagen or CRP while remaining responsive to other agonists, and form thrombi less efficiently when passed over immobilized collagen under conditions of arterial shear.11
The ability of anti-GPVI antibodies to render platelets unresponsive to collagen suggests that such reagents, especially if nonactivating, might have therapeutic benefit in reducing mural thrombosis in a variety of clinical settings. Attempts to examine the efficiency with which GPVI-specific mAbs are able to passivate human platelets have unfortunately been compromised by the observation that most are unable to effect depletion of GPVI ex vivo8,10 (B.B. and P.J.N., unpublished observations, July 2004). Delineating the molecular mechanisms underlying GPVI immunodepletion in circulating human platelets has been similarly confounded. Although early studies suggested that rat anti–mouse GPVI mAbs initiate internalization of antibody/murine GPVI complexes,8 a number of recent in vitro studies suggest that agents that cause mitochondrial damage12 or activate platelets after binding GPVI13,14 are able to activate one or more yet-to-be-identified matrix metalloproteinases (MMPs) that proteolytically cleave the extracellular domain of GPVI, in some cases releasing the extracellular domain of GPIbα as well.12,15
To examine the ability of anti-GPVI mAbs to reduce GPVI expression on human platelets in an activation-independent manner and to dampen GPVI-mediated platelet activation responses, we have developed an in vivo model system in which human platelets are injected into nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice and allowed to circulate for up to 2 days. We show that coinjection of monovalent Fab fragments specific for human GPVI, which do not activate the circulating human platelets, leads to immunodepletion of this receptor from the platelet surface. As a result, soluble human GPVI accumulates in mouse plasma, and the GPVI-depleted human platelets exhibit a corresponding loss of CRP-induced activation. The ability to model in living mice the in vivo function and survival of circulating human platelets may prove valuable for determining mechanisms of antibody-mediated platelet passivation, and may aid in the development of other novel antiplatelet therapeutics.
Materials and methods
Animal protocols were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee.
Antibodies and reagents
11A12 and 6B12 are well-characterized mouse anti–human GPVI-specific monoclonal antibodies (mAbs) that have been previously described.7,16 The mAbs PECAM-1.3 and 235.1, specific for the extracellular and cytoplasmic domains, respectively, of PECAM-1, have been described.17 AP2, which recognizes a complex-dependent integrin GPIIb-IIIa epitope,18 was kindly provided by Dr Robert R. Montgomery (Blood Research Institute, BloodCenter of Wisconsin, Milwaukee, WI). Monovalent Fab fragments of 6B12, PECAM-1.3, and 235.1 were prepared using the Immunopure preparation kit (Pierce Biotechnology, Rockford, IL). The anti-GPVI cytoplasmic tail antibody was raised by immunizing New Zealand White rabbits with a previously described maltose-binding protein (MBP)–GPVI cytoplasmic tail fusion protein.19 Serum derived from these rabbits was subjected to sequential affinity chromatography on columns of MBP-GPVI cytoplasmic tail fusion protein and MBP alone coupled to Affi-Gel 10/15 (Bio-Rad Laboratories, Hercules, CA). The fibrinogen-mimetic mAb, PAC-1,20 prelabeled with fluorescein isothiocyanate (FITC), was purchased from BD Biosciences (San Jose, CA). In some cases, mAbs were directly conjugated to Alexa Fluors 647 (11A12 and AP2) and 488 (AP2) using a kit purchased from Molecular Probes (Eugene, OR). CRP was synthesized and crosslinked as previously described.21,22
Circulation of human platelets in NOD/SCID mice
Human blood drawn into acid-citrate-dextrose and supplemented with prostaglandin E1 (PGE1) at 50 ng/mL was incubated at room temperature for 10 minutes then spun at 200g for 10 minutes. Platelet-rich plasma (PRP) was collected, and PGE1 was added to 50 ng/mL. Platelets were incubated at room temperature for 10 minutes and then spun at 750g for 10 minutes. The platelets were resuspended in human plasma at 2.0 × 109/mL, supplemented with PGE1 to 50 ng/mL and incubated at room temperature for 30 minutes. Platelet concentrates (200 μL) were injected into the retro-orbital plexus of age- and sex-matched NOD/SCID mice (Stock no. 001303; The Jackson Laboratory, Bar Harbor, ME). Thirty micrograms monovalent Fabs or 10 μg intact IgG, each resuspended in 200 μL sterile DPBS, was injected intraperitoneally 30 minutes after introducing the human platelets.
Preparation of washed platelets
Whole blood (20-50 μL) was obtained from the mouse tail vein and collected into 1.0 mL of 1:9 mixture of 3.8% sodium citrate/Tyrode-HEPES buffer containing PGE1 at 50 ng/mL. The blood mixture (1.0 mL) was layered onto 2 mL Fico/Lite Platelets (Atlanta Biologicals, Norcross, GA) and spun for 15 minutes at 350g. The platelet layer (1.0 mL) was added to 3.0 mL Tyrode-HEPES buffer supplemented with 67 ng/mL PGE1. Platelets were washed by centrifugation at 750g for 10 minutes and resuspended in Tyrode-HEPES buffer.
Platelet activation assays
Washed platelets obtained from mice 24 hours after injection of human platelet concentrates were stimulated with CRP, ADP, thrombin, or a cocktail of 10 U/mL thrombin + 10 μM adenosine diphosphate (ADP) + 100 μM epinephrine and incubated for 20 minutes at room temperature in FITC-conjugated PAC-1 at 2.5 μg/mL and 1 mM CaCl2. Alexa Fluor 647–conjugated AP2 was then added at 5 μg/mL; platelets were incubated for an additional 20 minutes at room temperature and directly analyzed on a BD LSR II (Franklin Lakes, NJ).
Platelet aggregation
Washed human platelets (400 μL) in Tyrode-HEPES buffer at 3.0 × 108 platelets/mL were added to siliconized glass cuvettes at 37°C with constant stirring at 1000 rpm. CaCl2 and fibrinogen were added to a final concentration of 1 mM and 300 μg/mL, respectively. Platelet aggregation was measured in a whole-blood lumi-ionized calcium aggregometer (Chrono-Log, Havertown, PA) for 2 minutes in the presence of 10 μg/mL isotype control mAb or mAb 6B12, and then finally activated with 7 ng/mL CRP.
Immunoprecipitation of GPVI from plasma
Human platelets were introduced into the circulation of NOD/SCID mice as described in “Circulation of human platelets in NOD/SCID mice.” Twenty-four hours after transfusion, mice were lethally anesthetized with tribromo-ethanol administered intraperitoneally. Whole blood was collected from the inferior vena cava into 0.1 volume of 3.8% sodium citrate, incubated at room temperature for 10 minutes in the presence of 50 ng/mL PGE1, and then centrifuged at 200g for 10 minutes to remove red blood cells. Following readdition of 50 ng/mL PGE1, the supernatant containing platelet-rich plasma was spun at 750g for 10 minutes to pellet platelets and remaining leukocytes, and the supernatant was subjected to a final spin at 100 000g for 60 minutes. Soluble GPVI (sGPVI) was immunoprecipitated by nutating 250 μL mouse plasma overnight with 50 μL streptavidin-Sepharose that had been precoated for 12 hours at 4°C with 0.5 mg/mL biotinylated 11A12, and washed 3 × in Tyrode-HEPES buffer. Immunocaptured proteins were eluted with 0.1 M glycine, pH 2.7, for 10 minutes at room temperature and neutralized in 1.5 M Tris, pH 8.8.
Results
Human platelets survive and circulate in NOD/SCID mice
Although GPVI is lost from the platelet surface in vitro following treatment with agents that activate platelets or damage mitochondria, attempts to deplete GPVI under in vitro conditions using mAbs that do not injure or activate platelets have been unsuccessful,8,10 perhaps because of a requirement for circulation and/or the presence of cells or factors that are present only in vivo. To effect antibody-mediated depletion of human GPVI in vivo, we developed a mouse model system in which approximately 200 μL concentrated human platelet-rich plasma is introduced, together with anti–human GPVI-specific mAbs, into the circulation of immune-deficient NOD/SCID mice via the retro-orbital plexus. As shown in Figure 1A, human platelets introduced in this way typically comprise approximately 10% to 15% of the total circulating platelet population and decrease to about 7% over a 24-hour time period, probably reflecting their clearance against a background of murine platelets that are continually being replenished. Thirty minutes after transfusing human platelets into the mice, 10 μg of a single mAb was injected intraperitoneally. Tail bleeds were performed at different time points, and flow cytometry was used to track human platelet survival over time. As shown in Figure 1B, mice injected with an intact mAb specific for human PECAM-1 showed rapid loss of human platelets, most likely resulting from Fc receptor-mediated clearance of antibody-coated platelets by splenic and hepatic macrophages and other elements of the reticuloendothelial system, which is fully intact in NOD/SCID mice. In contrast, injection of a nonbinding, isotype-matched control mAb (mAb 235.1, specific for the cytoplasmic domain of human PECAM-1) caused only a modest decrease in the number of circulating human platelets over time. Mice treated with a mAb specific for GPVI had an intermediate rate of human platelet clearance. To determine whether the human platelets in the anti-GPVI–treated mice were being removed from circulation because the kinetics of antibody-mediated immunodepletion of GPVI lagged behind that of FcR-mediated platelet destruction and to increase the number and survival time of human platelets in the mouse circulation, we repeated these experiments using monovalent Fab fragments, which lack the Fc region recognized by Fc receptors. As shown in Figure 1C, use of Fab fragments effectively eliminated the antibody-mediated component of human platelet clearance in mice injected with either anti-GPVI or anti–PECAM-1 mAbs; therefore, Fabs were used in all subsequent experiments.
Figure 1.

Survival of human platelets in NOD/SCID mice. Human PRP (200 μL) at 2 × 109 platelets/mL was injected into the retro-orbital plexus of NOD/SCID mice, and human platelet survival was measured by flow cytometry using AP2 (a monoclonal antibody specific for the human GPIIb-IIIa complex). (A) Flow cytometric data showing a typical time course of human platelet survival following intraperitoneal injection of an isotype-matched control mAb. Note that approximately 50% of the human platelets are lost from circulation over a period of 24 hours, most likely reflecting the kinetics of normal clearance resulting from platelet aging. (B) Comparison of human platelet survival over time in mice treated with an isotype-matched intact monoclonal IgG, the GPVI-specific mAb 11A12, or the anti–PECAM-1 mAb PECAM 1.3. (C) Human platelet survival in the mouse circulation following exposure to 30 μg Fab fragments of the GPVI-specific mAb 6B12 or PECAM 1.3. Note that the rate of platelet clearance (∼ 40% in 24 hours) is not significantly greater than observed in mice treated with nonbinding isotype-matched mAbs shown in Figure 1B. Data shown is representative of 5 (A,B) and 7 (C) separate experiments.
Immunodepletion of GPVI from circulating human platelets using monovalent Fab fragments of the murine GPVI-specific mAb, 6B12
Nonactivating, GPVI-specific mAbs that dampen platelet responses to collagen have potential therapeutic applications as antithrombotic agents; however, they have not to date been shown to be capable of removing GPVI from the surface of human platelets. The finding that human platelets can circulate in NOD/SCID mice for more than 24 hours (Figure 1) allowed us to investigate the consequences of antibody binding to human GPVI under in vivo conditions. Evidence that the non–function-blocking, GPVI-specific mAb, 6B12, does not activate platelets, even in divalent form, is provided in Figure 2A. When Fab fragments of 6B12 were introduced into NOD/SCID mice together with approximately 4 × 108 human platelets, approximately 50% of circulating human platelets lost GPVI expression within 24 hours (Figure 2B, lower right). In contrast, Fab fragments specific for PECAM-1 (Figure 2B, upper right) or an isotype control Fab (not shown) had no effect of GPVI surface expression. The ability of monovalent Fab fragments to remove GPVI from the surface of human platelets demonstrates that clustering of the GPVI/FcRγ chain complex is not required for antibody-mediated immunodepletion, a result in keeping with the observations of Nieswandt et al8 and Schulte et al,10 who found that rat anti–mouse GPVI Fabs could achieve the same effect in murine platelets in vivo.
Figure 2.
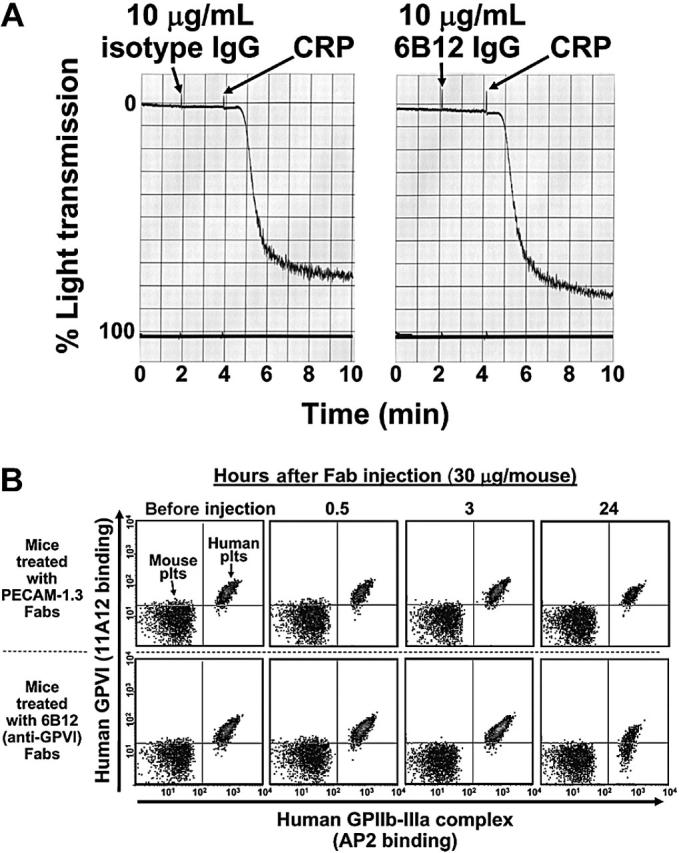
Activation-independent antibody-induced depletion of GPVI from human platelets circulating in NOD/SCID mice. (A) Washed human platelets resuspended in Tyrode-HEPES buffer at 3.0 × 108 platelets/mL were incubated for 2 minutes in the presence of 10 μg/mL isotype control mAb (left) or the mAb 6B12 (right). Note that 6B12 IgG neither induces nor blocks platelet aggregation. Platelets were fully reactive, as shown by later addition of CRP. (B) Concentrated human PRP (200 μL) containing 2 × 109 platelets/mL was injected into the retro-orbital plexus of a NOD/SCID mouse, followed by an intraperitoneal injection of 30 μg indicated Fab. Platelets in whole blood drawn from the tail vein at 0, 0.5, 3, and 24 hours after injection were assessed for surface expression of human GPIIb-IIIa and GPVI. Note that GPVI surface expression is not noticeably affected on platelets from mice treated with anti–PECAM-1 Fabs (top 4 panels) but decreases significantly in mice treated with the GPVI-specific Fab, 6B12.
6B12 treatment results in loss of GPVI-specific agonist responses as well as diminished responsiveness to agonists for G-protein–coupled receptors
To determine whether in vivo introduction of mAb 6B12 could effectively dampen the response of circulating human platelets to GPVI-mediated activation signals, we examined their ability to bind the fibrinogen-mimetic mAb, PAC-1, following exposure to the GPVI-specific agonist, CRP. As shown in Figure 3A, human platelets taken from mice treated with 6B12 Fabs for 24 hours lost almost half of their reactivity to high-dose CRP, as reported by PAC-1 binding, compared with human platelets derived from control mice that had been treated with PECAM 1.3 Fabs for 24 hours. Somewhat unexpectedly, human platelets taken from 6B12-treated mice also consistently exhibited slightly deceased (∼ 20%) PAC-1 binding on activation with a cocktail of agonists composed of 10 U/mL thrombin, 10 μM ADP, and 100 μM epinephrine, all of which stimulate G-protein–coupled receptors on the platelet surface. To further characterize the nature of this activation defect, more detailed dose-response studies were performed for ADP, thrombin, and CRP. As shown in Figure 3D, PAC-1 binding to 6B12-treated human platelets was markedly reduced at all concentrations of CRP examined, as expected, more or less in proportion to the degree of GPVI depletion effected by the antibody (Figure 2B, bottom right). In contrast, ADP- and thrombin-induced PAC-1 binding to 6B12-treated human platelets was nearly normal at low-dose agonist concentrations, but it was reduced by 38% and 43%, respectively, when high concentrations of ADP or thrombin were used. Thus, it appears that immunodepletion of the GPVI/FcRγ chain complex exerts somewhat more widespread effects on platelet function than had previously been thought.
Figure 3.

Attenuation of agonist-induced platelet activation. Human platelets were allowed to circulate in NOD/SCID mice in the presence of monovalent Fab fragments specific for either PECAM-1 (black bars/•) or GPVI (gray bars/▿) for 24 hours and examined for PAC-1 binding induced by (A) 2.5 ng/mL CRP or a cocktail made up of 10 U/mL thrombin + 10 μM ADP + 100 μM epinephrine, or by increasing doses of (B) ADP, (C) thrombin, or (D) CRP. Percentages indicate reduction of PAC-1 binding to human platelets that circulated in the presence of anti-GPVI Fab compared with those that circulated with anti–PECAM-1 Fab. Human platelets were distinguished from murine platelets by their ability to bind the human GPIIb-IIIa complex-specific mAb, AP2. Note that treatment with the anti-GPVI Fab fragment, 6B12, results in generalized loss of agonist-induced platelet activation, an effect that is most pronounced in response to the GPVI-specific agonist CRP.
Soluble, truncated form of human GPVI is shed into mouse plasma as a result of 6B12-induced immunodepletion
Although a number of recent in vitro studies suggest that agents that either cause mitochondrial damage12 or activate platelets via GPVI13,14 can lead to platelet-associated metalloproteinase-mediated shedding of GPVI into the extracellular milieu, the mechanism by which nonactivating, GPVI-specific mAbs are able to effect GPVI depletion, particularly from the surface of human platelets, still is not known. Because the latter class of reagents is likely to be more clinically useful, and to investigate whether 6B12 treatment caused GPVI shedding in vivo, human platelets were injected into mice in the presence of anti-GPVI or anti–PECAM-1 Fabs and allowed to circulate for 24 hours. Plasma samples were then collected and subjected to immunoprecipitation using streptavidin-Sepharose beads that had been saturated with biotinylated 11A12-a murine anti-GPVI mAb that binds an epitope on human GPVI distinct from that for 6B12 and therefore does not compete for binding. Immunocaptured proteins were acid-eluted from the streptavidin-11A12 beads, subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE), and Western blotted with 6B12. As shown in Figure 4, plasma obtained from 6B12-treated (lanes 3), but not PECAM-1.3–treated (lanes 2), mice contained a prominent 50-kDa soluble form of human GPVI that reacted with an antibody to the extracellular, but not cytoplasmic, domain of GPVI, indicating that this fragment is composed of a truncated, cleaved form of GPVI. GPVI derived from human platelet lysate (HPL) reacted with both antibodies, as expected. These data are consistent with proteolytic cleavage being, at least in part, responsible for the mechanism by which 6B12 depletes GPVI from the platelet surface.
Figure 4.

Soluble, truncated form of GPVI accumulates in the plasma of mice when human platelets circulate in the presence of anti-GPVI Fab fragments. NOD/SCID mice injected with human platelet concentrates along with Fab fragments directed against human GPVI (6B12) or PECAM-1 (1.3) were examined for the presence of soluble GPVI accumulation in the plasma. Streptavidin-Sepharose beads coated with a biotinylated form of the anti–human GPVI mAb, 11A12, were used to immunoprecipitate GPVI from human platelet lysates (HPL) or mouse plasma obtained from mice injected with human platelets and 6B12 or 1.3 fragments. On acid elution, samples were separated by SDS PAGE, transferred to a PVDF membrane, and probed for human GPVI using the mAb 6B12, which recognizes an epitope on the extracellular portion of human GPVI (A) or a rabbit polyclonal antibody directed against the cytoplasmic domain of GPVI (B). These data illustrate that a GPVI fragment corresponding to the extracellular domain accumulates in mouse plasma when human platelets circulate in the presence of an anti-GPVI Fab fragment of mAb 6B12.
Discussion
The major findings of the present investigation are that (1) monovalent murine anti–human GPVI Fabs can effectively deplete GPVI from the surface of human platelets in vivo, (2) they appear to act largely by inducing ectodomain shedding, and (3) the NOD/SCID mouse model that we describe herein may be useful for future investigations in which the function and/or survival of circulating human platelets following treatment with various antiplatelet agents needs to be examined.
Immunodepletion of GPVI from the platelet surface was first observed in a murine model that involved injecting mice with rat anti–mouse GPVI mAbs, JAQ1, JAQ2, or JAQ3.8-10 JAQ mAb-treated mice exhibited mild, transient thrombocytopenia, long-term immunodepletion of GPVI, and diminished platelet responsiveness to collagen. More recent studies suggest that antibody-mediated loss of GPVI can also take place on human platelets, as evidenced by a recently described patient with autoimmune thrombocytopenia caused by an autoantibody specific for GPVI, concomitant with a loss of platelet responsiveness to GPVI-specific agonists and a mild bleeding diathesis.11 That antibody-induced GPVI removal has now been demonstrated in both humans and mice raised the intriguing possibility that antibodies to GPVI might be used as adjunct therapy to temporarily pacify platelets during a thrombotic episode (ie, myocardial infarction or stroke, during unstable angina, or prior to clinical procedures such as percutaneous transluminal coronary angioplasty that are associated with increased incidence of mural thrombus formation). Several obstacles to making further progress in this area, however, exist. First, studies performed in mice using rat anti–mouse GPVI reagents of the JAQ series, which by themselves do not activate platelets, have demonstrated that these antibodies immunodeplete GPVI only when administered in vivo (ie, addition of anti-GPVI mAbs to platelets in vitro has no effect on GPVI surface expression).8 Second, there currently exists no suitable animal model to evaluate the effectiveness of anti–human GPVI reagents or the consequences of GPVI elimination in human platelets. To circumvent these problems, we undertook the development of a novel NOD/SCID mouse model of GPVI-immunodepletion that uses circulating human platelets in the presence of a mouse anti-GPVI mAb and used it to begin to address the mechanism of antibody-mediated elimination of GPVI from the human platelet surface.
Human platelets are able to circulate in NOD/SCID mice for more than 1 day (Figures 1, 2), most likely because the immune deficiency of these mice23 precludes production of “heterophile” antibodies that would normally result in immediate clearance of blood cells from another species. As expected, injection of PECAM-1.3, a mouse mAb specific for the extracellular domain of PECAM-1, bound to the circulating human platelets and resulted in rapid, marked thrombocytopenia reminiscent of human ITP24 (Figure 1B). In contrast, there was noticeably less reduction in platelet number induced by mouse anti-GPVI IgG (Figure 1B), especially when Fab fragments were used (Figure 1C). Equally important, Fab fragments of mAb 6B12 triggered rapid, selective loss of GPVI from the surface of human platelets (Figure 2B), rendering them substantially less responsive to the GPVI agonist, CRP (Figure 3D). For reasons that are unclear, platelets from 6B12-treated mice also exhibit somewhat dampened reactivity to ADP and thrombin, agonists that exclusively stimulate G-protein–coupled receptor signaling pathways (Figure 3B-C). Although crosstalk between the thrombin and ADP receptors and the GPVI/FcRγ chain has not been previously reported, the GPVI/FcRγ chain signaling complex has recently been shown to mediate platelet activation responses not only to collagen but also to laminin,25 suggesting that ITAM-mediated signaling may be a more widespread amplifier of platelet activation than has previously been thought. It is possible, therefore, that antibody-mediated shedding of the GPVI ectodomain inadvertently cripples FcRγ-chain–mediated amplification events that occur downstream of a wide range of agonist receptors. Studies are in progress in our laboratory to examine in more detail the effects of FcRγ chain depletion on platelet function.
The degree of GPVI elimination achieved in the circulating human platelet-NOD/SCID model is less than that observed by Nieswandt et al,8 Massberg et al,9 and Schulte et al10 for murine platelets using JAQ mAbs. This may due to the fact that GPVI expression on human platelets circulating in NOD/SCID mice has to be determined 18 to 36 hours following their injection (ie, before they age and became cleared). In contrast, JAQ-induced depletion of murine platelet surface GPVI has normally been evaluated 3 or more days following JAQ treatment and thus may account for the more efficient GPVI immunodepletion observed. The kinetics of human platelet clearance in NOD/SCID mice, therefore, may represent a limitation of this model for examining the effects of antiplatelet agents whose action requires 1 or more days of systemic exposure. Nonetheless, the finding that human platelets can circulate and survive in NOD/SCID mice, albeit for a limited period of time, may prove useful for evaluating the in vivo efficacy of other antithrombotic agents and additionally provide important mechanistic insights into their mode of action. It may also be of use in evaluating the efficacy of enzymatic, biochemical, and physical manipulations that attempt to correct the so-called platelet storage lesion.26,27
The precise mechanism(s) by which anti-GPVI antibodies cause specific depletion of the GPVI/FcRγ-chain complex from the surface of murine and human platelets remains an important, and unresolved, matter. Several possibilities come to mind, including (1) antibody-induced receptor clustering leading to internalization of the antibody/antigen complex, (2) Fc receptor-mediated removal of the antibody/receptor complex by tissue macrophages present within the reticuloendothelial (RE) system, and (3) involvement of endothelial cell– and/or circulating blood cell–derived proteases acting on cryptic protease cleavage sites that become exposed as a consequence of antibody binding. Using a variety of approaches, we have to date been unable to find convincing evidence for internalization of 6B12 Fab fragments in human platelets circulating in NOD/SCID mice (B.B. and P.J.N., unpublished observations, June 2005); however, the fact that monovalent Fab fragments induce efficient GPVI clearance from the platelet surface (Figure 2) would appear to rule out both a requirement for receptor clustering, as well as an Fc-dependent mechanism of RE-mediated clearance as prerequisites for GPVI immunodepletion.
In support of a proteolytic mechanism for GPVI immunodepletion, we found (Figure 4) that a readily detectable, truncated, soluble 50-kDa fragment containing the GPVI extracellular, but not cytoplasmic, domain became liberated from human platelets into mouse plasma following in vivo exposure to monovalent Fab fragments of 6B12. To our knowledge, our observation that nonactivating anti-GPVI reagents can induce shedding of the ectodomain of GPVI from the human platelet surface has not been previously demonstrated. Several recent studies, however, have reported GPVI shedding as a consequence of (1) platelet activation following stimulation with a GPVI-specific agonist,13,14 (2) exposure to an activating GPVI-specific mAb,14 (3) inhibition of an interaction between calmodulin and the GPVI cytoplasmic domain,13 or (4) exposure to mitochondrial-damaging agents.12,15 In each of these studies, immunodepletion of GPVI could be attributed to the action of MMPs, a class of enzymes originally characterized by their ability to degrade various elements of the extracellular matrix (for a review, see Mott and Werb28), because GPVI shedding could be inhibited by the broad range MMP inhibitor, GM6001.12-14 Because MMPs are secreted from α-granules following platelet activation,29-32 it is tempting to speculate that calcium mobilization caused by platelet activation induces not only secretion of granule-associated MMPs but also concurrent dissociation of calmodulin from the GPVI cytoplasmic tail, resulting in a conformational change in the extracellular domain that exposes a cryptic protease cleavage site and GPVI shedding. In contrast, rat anti–mouse GPVI antibodies,8,10 patient anti-GPVI autoantibodies,11 and the mouse anti–human GPVI mAbs used in this study all involve nonactivating antibodies that, because they do not cause granule release, may effect GPVI removal via a related protease whose action is facilitated by shear force, or by exposure to other cells that are encountered during circulation. Thus far, we have been unable to determine whether activation-independent GPVI shedding in vivo is mediated by an extracellularly derived MMP-mediated event because most readily available MMP inhibitors become rapidly inactivated after injection. We have also attempted to examine whether activation-independent, antibody-induced GPVI shedding might be mediated by the same MMP that cleaves GPIb. If this were true, then GPIb might also be inadvertently clipped when GPVI antibodies are added. Flow cytometric analysis to measure the surface expression of both GPIb and GPV on human platelets taken out of mice treated with mAb 6B12, however, has revealed that GPIb levels are actually increased, not decreased, in the very same platelets in which GPVI has been depleted (B.B. and P.J.N., unpublished observations, November 2005). These data suggest, therefore, either (1) that nonactivating anti-GPVI mAbs do not activate the same protease as that responsible for activation-induced GPIb shedding, as recently suggested by Bergmeier et al,12 or (2) that the protease is the same but that the 6B12/GPVI/protease complex is not close enough to GPIb to enable it to be shed. Studies are in progress to identify the source and identity of the responsible GPVI-cleaving protease.
Finally, although GPIIb-IIIa receptor antagonists have been an important addition to the regimen of antithrombotics used to reduce the incidence of complications following coronary angioplasty, an undesirable side effect of this class of therapeutics is their propensity, for poorly understood reasons, to cause in approximately 1% of recipients acute, antibody-mediated platelet destruction and/or bleeding.33,34 Although still a long way from preclinical testing, GPVI-directed therapeutics have the potential to avoid these untoward complications. First, anti-GPVI mAbs, be they rat,8-10 human,11 or mouse (this study) in origin, are rapidly shed from the platelet surface on binding their target receptor, thereby sparing the platelet from clearance, thus avoiding thrombocytopenia. Second, although platelet reactivity to mural collagen,9 and perhaps mural laminin,25 exposed at sites of vascular injury is largely lost following targeted depletion of GPVI, platelets treated with anti-GPVI antibodies retain much of their ability to respond to soluble agonists (Figure 3). Taken together with the findings of the present study, nonactivating mAbs directed against GPVI may represent a novel class of compounds having antithrombotic properties without the increased incidence of unexpected bleeding. Further in vivo studies will be required to establish the efficacy of these antibodies in controlling thrombosis in primates and humans.
Acknowledgments
We thank Dr Debra K. Newman for her constructive thoughts and suggestions and for carefully reviewing this manuscript prior to its submission.
Prepublished online as Blood First Edition Paper, March 28, 2006; DOI 10.1182/blood-2005-07-2937.
Supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (grant HL-44612) (P.J.N.) and (grant HL-067311) (M.L.K.) and by the National Health and Medical Research Council of Australia (M.C.B.).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Gibbins JM, Okuma M, Farndale R, Barnes M, Watson SP. Glycoprotein VI is the collagen receptor in platelets which underlies tyrosine phosphorylation of the Fc receptor γ-chain. FEBS Lett. 1997;413: 255-259. [DOI] [PubMed] [Google Scholar]
- 2.Tsuji M, Ezumi Y, Arai M, Takayama H. A novel association of Fc receptor gamma-chain with glycoprotein VI and their co-expression as a collagen receptor in human platelets. J Biol Chem. 1997;272: 23528-23531. [DOI] [PubMed] [Google Scholar]
- 3.Clemetson JM, Polgar J, Magnenat E, Wells TN, Clemetson KJ. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcαR and the natural killer receptors. J Biol Chem. 1999;274: 29019-29024. [DOI] [PubMed] [Google Scholar]
- 4.Jandrot-Perrus M, Busfield S, Lagrue AH, et al. Cloning, characterization, and functional studies of human and mouse glycoprotein VI: a platelet-specific collagen receptor from the immunoglobulin superfamily. Blood. 2000;96: 1798-1807. [PubMed] [Google Scholar]
- 5.Ezumi Y, Uchiyama T, Takayama H. Molecular cloning, genomic structure, chromosomal localization, and alternative splice forms of the platelet collagen receptor glycoprotein VI. Biochem Biophys Res Commun. 2000;277: 27-36. [DOI] [PubMed] [Google Scholar]
- 6.Nieswandt B, Watson SP. Platelet collagen interaction: is GPVI the central receptor? Blood. 2003;102: 449-461. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Kahn ML. Reciprocal signaling by integrin and nonintegrin receptors during collagen activation of platelets. Mol Cell Biol. 2003;23: 4764-4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nieswandt B, Schulte V, Bergmeier W, et al. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. 2001;193: 459-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massberg S, Gawaz M, Gruner S, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197: 41-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulte V, Rabie T, Prostredna M, et al. Targeting of the collagen-binding site on glycoprotein VI is not essential for in vivo depletion of the receptor. Blood. 2003;101: 3948-3952. [DOI] [PubMed] [Google Scholar]
- 11.Boylan B, Chen H, Rathore V, et al. Anti-GPVI-associated ITP: an acquired platelet disorder caused by autoantibody-mediated clearance of the GPVI/FcRγ-chain complex from the human platelet surface. Blood. 2004;104: 1350-1355. [DOI] [PubMed] [Google Scholar]
- 12.Bergmeier W, Rabie T, Strehl A, et al. GPVI down-regulation in murine platelets through metalloproteinase-dependent shedding. Thromb Haemost. 2004;91: 951-958. [DOI] [PubMed] [Google Scholar]
- 13.Gardiner EE, Arthur JF, Kahn ML, Berndt MC, Andrews RK. Regulation of platelet membrane levels of glycoprotein VI by a platelet-derived metalloproteinase. Blood. 2004;104: 3611-3617. [DOI] [PubMed] [Google Scholar]
- 14.Stephens G, Yan Y, Jandrot-Perrus M, et al. Platelet activation induces metalloproteinase-dependent GP VI cleavage to down-regulate platelet reactivity to collagen. Blood. 2005;105: 186-191. [DOI] [PubMed] [Google Scholar]
- 15.Bergmeier W, Burger PC, Piffath CL, et al. Metalloproteinase inhibitors improve the recovery and hemostatic function of in vitro-aged or -injured mouse platelets. Blood. 2003;102: 4229-4235. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Locke D, Liu Y, Liu C, Kahn ML. The platelet receptor GPVI mediates both adhesion and signaling responses to collagen in a receptor density-dependent fashion. J Biol Chem. 2002;277: 3011-3019. [DOI] [PubMed] [Google Scholar]
- 17.Jackson DE, Kupcho KR, Newman PJ. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of platelet/endothelial cell adhesion molecule-1 (PECAM-1) that are required for the cellular association and activation of the protein-tyrosine phosphatase, SHP-2. J Biol Chem. 1997;272: 24868-24875. [DOI] [PubMed] [Google Scholar]
- 18.Pidard D, Montgomery RR, Bennett JS, Kunicki TJ. Interaction of AP-2, a monoclonal antibody specific for the human platelet glycoprotein IIb-IIIa complex, with intact platelets. J Biol Chem. 1983;258: 12582-12586. [PubMed] [Google Scholar]
- 19.Suzuki-Inoue K, Tulasne D, Shen Y, et al. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling 3. J Biol Chem. 2002;277: 21561-21566. [DOI] [PubMed] [Google Scholar]
- 20.Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein IIb-IIIa complex during platelet activation. J Biol Chem. 1985;267: 11107-11114. [PubMed] [Google Scholar]
- 21.Rao GH, Fields CG, White JG, Fields GB. Promotion of human platelet adhesion and aggregation by a synthetic, triple-helical “mini-collagen”. J Biol Chem. 1994;269: 13899-13903. [PubMed] [Google Scholar]
- 22.Morton LF, Hargreaves PG, Farndale RW, Young RD, Barnes MJ. Integrin α2β1-independent activation of platelets by simple collagen-like peptides: collagen tertiary (triple helical) and quaternary (polymeric) structures are sufficient alone for α2β1-independent platelet reactivity. Biochem J. 1995;306: 337-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shultz LD, Schweitzer PA, Christianson SW, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154: 180-191. [PubMed] [Google Scholar]
- 24.McMillan R. Autoantibodies and autoantigens in chronic immune thrombocytopenic purpura. Semin Hematol. 2000;37: 239-248. [DOI] [PubMed] [Google Scholar]
- 25.Inoue O, Suzuki-Inoue K, McCarty OJ, et al. Laminin stimulates spreading of platelets through integrin α6β1-dependent activation of GPVI. Blood. 2006;107: 1405-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffmeister KM, Felbinger TW, Falet H, et al. The clearance mechanism of chilled blood platelets. Cell. 2003;112: 87-97. [DOI] [PubMed] [Google Scholar]
- 27.Hoffmeister KM, Josefsson EC, Isaac NA, et al. Glycosylation restores survival of chilled blood platelets. Science. 2003;301: 1531-1534. [DOI] [PubMed] [Google Scholar]
- 28.Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16: 558-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawicki G, Salas E, Murat J, Miszta-Lane H, Radomski MW. Release of gelatinase A during platelet activation mediates aggregation. Nature. 1997;386: 616-619. [DOI] [PubMed] [Google Scholar]
- 30.Sawicki G, Sanders EJ, Salas E, et al. Localization and translocation of MMP-2 during aggregation of human platelets. Thromb Haemost. 1998;80: 836-839. [PubMed] [Google Scholar]
- 31.Fernandez-Patron C, Martinez-Cuesta MA, Salas E, et al. Differential regulation of platelet aggregation by matrix metalloproteinases-9 and -2. Thromb Haemost. 1999;82: 1730-1735. [PubMed] [Google Scholar]
- 32.Galt SW, Lindemann S, Allen L, et al. Outside-in signals delivered by matrix metalloproteinase-1 regulate platelet function. Circ Res. 2002;90: 1093-1099. [DOI] [PubMed] [Google Scholar]
- 33.Bougie DW, Wilker PR, Wuitschick ED, et al. Acute thrombocytopenia after treatment with tirofiban or eptifibatide is associated with antibodies specific for ligand-occupied GPIIb/IIIa. Blood. 2002;100: 2071-2076. [PubMed] [Google Scholar]
- 34.Aster RH. Immune thrombocytopenia caused by glycoprotein IIb/IIIa inhibitors. Chest. 2005;127: 53S-59S. [DOI] [PubMed] [Google Scholar]