

Abstract
Murine autoreactive anti-Smith (Sm) B cells are negatively regulated by anergy and developmental arrest, but are also positively selected into the marginal zone (MZ) and B-1 B-cell populations. Despite positive selection, anti-Sm production occurs only in autoimmune-prone mice. To investigate autoreactive B-cell activation, an anti-Sm transgene was combined with the lpr mutation, a mutation of the proapoptotic gene Fas (Faslpr), on both autoimmune (MRL) and nonautoimmune backgrounds. Faslpr induces a progressive and autoantigen-specific loss of anti-Sm MZ and B-1 B cells in young adult Faslpr and MRL/Faslpr mice that does not require that Faslpr be B-cell intrinsic. This loss is accompanied by a bypass of the early pre–plasma cell (PC) tolerance checkpoint. Although the MRL bkg does not lead to a progressive loss of anti-Sm MZ or B-1 B cells, it induces a robust bypass of the early pre-PC tolerance checkpoint. Faslpr mice have a high frequency of apoptotic lymphocytes in secondary lymphoid tissues and a macrophage defect in apoptotic cell phagocytosis. Since Sm is exposed on the surface of apoptotic cells, we propose that anti-Sm MZ and B-1 B-cell activation is the result of a Faslpr-induced defect in apoptotic cell clearance.
Introduction
A goal of B-cell tolerance studies is to understand how tolerance is lost in autoimmune diseases such as systemic lupus erythematosus (SLE). Mice of SLE-susceptible strains carry unique sets of susceptibility genes.1 MRL/Faslpr mice develop a disease that closely resembles human SLE including the production of anti-Sm, a marker SLE antibody.2 The MRL bkg is required for anti-Sm production,3 but the genes predisposing to autoimmunity have not yet been identified. One gene that can contribute to both murine and human autoimmunity is the proapoptosis gene Fas.4-6 The lpr mutation of Fas (Faslpr) causes a loss of function4 and is sufficient to induce SLE, but the severity is dependent on the bkg genes. It accelerates and exacerbates the disease induced by the MRL bkg,7 but induces only a mild disease in C57BL/6 (B6) mice.7 Despite producing a wide spectrum of autoantibodies in B6 mice, Faslpr does not induce anti-Sm production in these mice.8
To examine how autoreactive B cells are regulated, we developed 2-12H transgenic (Tg) mice expressing an anti-Sm H chain transgene.9 Both positively and negatively selected anti-Sm B cells coexist in 2-12H mice, yet they are not activated to secrete antibody. Anti-Sm B cells constitute approximately 30% of the follicular (FO) B-cell subset, a majority of MZ B cells, and approximately 30% of peritoneal B-1 cells in 2-12H mice. Regulation of FO anti-Sm B cells occurs by anergy and developmental arrest,9,10 but how the anti-Sm MZ and B-1 B cells are regulated is unknown. We have recently determined that anti-Sm B cells are activated and begin plasma cell (PC) differentiation, but arrest at a pre-PC stage before becoming antibody secreting cells (ASCs).11 However, in 2-12H MRL/Faslpr mice, anti-Sm pre-PCs overcome this block and become ASCs, producing elevated anti-Sm levels with complete penetrance by 2 months of age.12
MZ and B-1 cells are likely sources of autoantibodies in MRL/Faslpr mice, as the normal B-1 and MZ B-cell repertoires contain a high frequency of anti-self B cells including anti-DNA.13,14 MZ B-cell involvement in autoantibody production has been documented in an Ig Tg model where these cells produce anti-ssDNA upon estrogen treatment.15 Also, excess BAFF expression rescues self-reactive B cells from peripheral deletion and leads to autoimmunity,16,17 and interestingly induces an expansion of the MZ B-cell subset,18 implying a role for this population. Antierythrocyte B-1 cells are responsible for the production of the hemolytic autoantibodies in Faslpr mice,19 implicating a role for B-1 cells in autoantibody production. Also, the sle2 locus of NZW origin contributes to autoimmunity and is responsible for, among other effects, an expansion of the B-1a cell population.20,21
We have recently demonstrated anti-Sm MZ and B-1 B cells are activated in 2-12H Tg mice carrying a mutation of the receptor tyrosine kinase Mer (Merkd).22 Merkd macrophages are impaired in their ability to phagocytize apoptotic cells, and Merkd mice develop a mild lupuslike disease with the production of autoantibodies.22,23 The anti-Sm MZ population is expanded in 2-12H/Merkd mice, and there is a progressive loss of peritoneal anti-Sm B-1 cells in young adult mice, which is caused at least in part by B-1–cell activation and differentiation to ASCs of the mesenteric lymph nodes (MLNs) and lamina propria (LP). Apoptotic cells are a likely source of Sm antigen in vivo, as Sm is exposed on the surface of apoptotic cell blebs,22 similar to other nuclear antigens,24 and immunization of nonautoimmune mice results in a transient anti-Sm response.22 Thus, the increased availability of apoptotic cells caused by Merkd mice could contribute to the induction of an anti-Sm response in 2-12H/Merkd mice, or Merkd could alter the function of dendritic cells or macrophages that indirectly lead to defects in T-cell and B-cell regulation. Since Fas has an opposing function in apoptosis to Mer, we sought to understand how Faslpr differs from Merkd in the dysregulation of anti-Sm B cells. In this report, we show that similar to Merkd, Faslpr induces the activation of anti-Sm MZ and B-1 B cells by an antigen-specific mechanism. This unexpected similarity can be explained by the observations that Faslpr also induces an increase in apoptotic cell frequency in lymphoid tissues and a defect in macrophage phagocytosis of apoptotic cells.
Materials and methods
Mice
2-12H, 2-12H MRL/Faslpr, 2-12H/Merkd, and 6-1 mice have been described.9,12,22,25,26 2-12H/MRL mice were generated by backcrossing 2-12H MRL/Faslpr mice with Fas intact MRL/Mp+/+ (MRL) mice (Jackson Laboratory, Bar Harbor, ME) through 2 generations to eliminate the Faslpr mutation. 2-12H and 6-1 mice were crossed with C57BL/6 Faslpr mice (Jackson Laboratory) to generate 2-12H/Faslpr and 6-1/Faslpr mice. Ig H chain transgenes of offspring were identified by polymerase chain reaction (PCR) as previously described.9,25 Fas and Faslpr were identified by analysis of tail genomic DNA using the forward primer CAA GCC GTG CCC TAG GAA ACA CAG, and reverse primers GCA GAG ATG CTA AGC AGC AGC CGG and GTG GAG CTC CAA TGC AGC GTT CCT. All animal experiments were carried out with institutional IACUC approval.
Flow cytometry
The antibodies specific for IgMa (DS-1), IgMb (AF6-78), B220 (RA3-6B2), CD11b (M1/70), CD21 (7G6), CD23 (B3B4), CD43 (S7), and CD5 (53-7.3) were obtained from PharMingen (San Diego, CA), and were labeled with FITC, PE, APC, or biotin. Biotinylated Sm (Immunovision, Scottsdale, AZ) was used to stain for Sm-specific B cells. Biotinylated reagents were revealed with streptavidin-PerCP. Cells were stained as described before.22,26 Liposomes encapsulating FITC and composed of membranes containing PtC were used as described to identify PtC-specific B cells.25,27 CaspACE FITC-VAD-FMK was purchased from Promega (Madison, WI), and cells were stained according to the manufacturer's instructions. Data were analyzed using WinMDI (Scripps Institute, La Jolla, CA). All data represent cells that fall within the lymphocyte gate determined by forward and 90-degree light scatter. Per sample, 1 to 5 × 105 cells were analyzed.
Cell sorting
For cell sorting of FO, MZ, and CD138+ B cells for enzyme-linked immunospot (ELISpot) analysis, spleen cells were stained for CD19, CD21, CD23, and CD138. The CD19+, CD21hi, CD23–/lo cells were sorted as MZ B cells, the CD19+, CD21low, CD23+, CD138– cells were sorted as FO B cells, and the CD19+, CD138+ cells were sorted as pre-PCs. For cell sorting of peritoneal B-1 and B-2 cells for transfer, peritoneal cells were stained for CD19 and CD11b and the CD19+ CD11b+ (B-1) and CD19+ CD11b– (B-2) cells sorted. The cells were sorted using a MoFlo high-speed sorter (DakoCytomation, Fort Collins, CO). Sorted populations were more than 90% pure as determined by reanalysis.
Mouse peritoneal cell transfer
Sorted peritoneal B-1 and B-2 cells (1 × 105) were injected intraperitoneally into non-Tg recipients. In other experiments, approximately 5 × 106 peritoneal cells isolated from 2-12H Tg or 2-12H/Faslpr mice were transferred intraperitoneally into non-Tg wild-type or non-Tg Faslpr mice. MLN and LP ELISpot assays were conducted at 2 weeks after transfer.
ELISAs and ELISpots
Quantification of anti-Sm antibodies and total IgM in mouse serum was performed by enzyme-linked immunosorbent assay (ELISA) as previously described.9,22,26 A capture ELISA was used to measure levels of serum IL-10 as described previously.28 Anti-Sm and IgM ELISpot assays were conducted as described previously using Sm- and anti-IgM–coated plates.22 Small ELISpots were between 1 × 10–4 and 3.2 × 10–3 mm2, and large ELISpots were more than 3.2 × 10–3 mm2.
TUNEL assay
Spleen and MLN tissues were fixed in 10% formalin, embedded in paraffin, and sectioned. Deoxynucleotidyl-transferase–mediated UTP nick-end labeling (TUNEL) assays were performed using ApopTag PLUS peroxidase In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA) according to the manufacturer's instructions. Methyl green was used for counterstaining. Images were obtained with a Leica DMRE microscope (Leica, Bannockburn, IL) with a 40×/0.7 NA PH2 objective. Images were captured using an RT Spot CCD camera (Diagnostic Instruments, Sterling Heights, MI) and Spot 4.6 software (Diagnostic Instruments). Image processing was done using Adobe Photoshop 6.0 software (Adobe Systems, San Jose, CA).
Phagocytosis assay
In vitro phagocytosis experiments were done as described by Scott et al.23
Statistical analysis
Statistical analysis was performed using one-tailed Student t test. A value of P below .05 was considered significant.
Results
Anti-Sm antibody production in autoimmune 2-12H mice
We previously demonstrated that serum anti-Sm levels were elevated in 2-12H MRL/Faslpr mice.12 To investigate the contribution of Faslpr and the MRL bkg to anti-Sm production, we examined mice of a mixed C57BL/6-BALB/c bkg (2-12H and 2-12H/Faslpr) and an autoimmune MRL bkg (2-12H MRL and 2-12H MRL/Faslpr). Compared with 2-12H mice, serum anti-Sm levels were significantly elevated in all 2-12H/Faslpr, 2-12H MRL, and 2-12H MRL/Faslpr mice examined, indicating that both the MRL bkg and Faslpr induced anti-Sm production (Figure 1A). The levels were not significantly different between 2-12H MRL and 2-12H MRL/Faslpr mice, suggesting that the MRL bkg and Faslpr were not additive. Faslpr did not induce anti-Sm production in non-Tg mice, as previously demonstrated.8 Anti-Sm ASCs of 2-12H/Faslpr and 2-12H MRL/Faslpr mice were demonstrated in the bone marrow (BM), spleen, MLN, and LP (Figure 1B).
Figure 1.
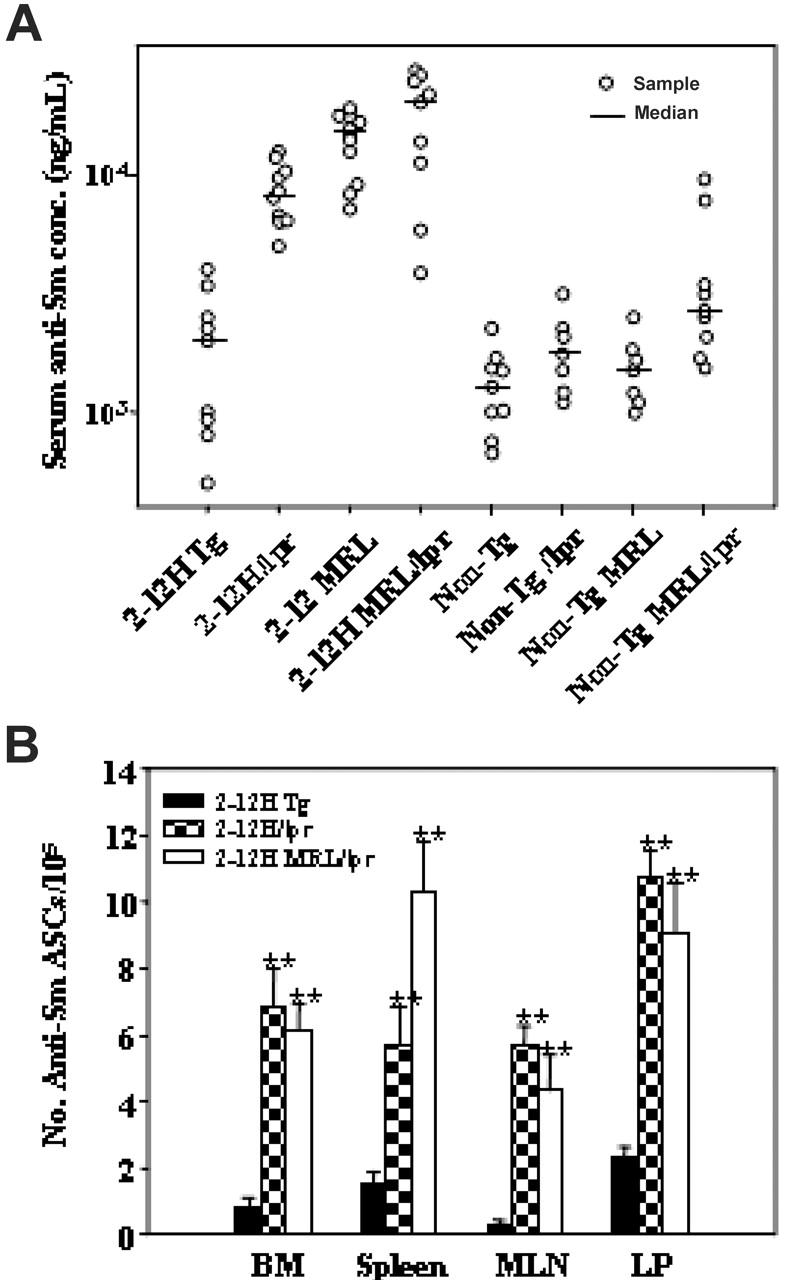
Anti-Sm antibody ASC production in autoimmune mice. (A) Serum anti-Sm levels are shown for mice 2 to 4 months of age. (B) The number ± SEM of anti-Sm ASCs in the bone marrow (BM), spleen, MLN, and LP as determined by ELISpot assay is shown (**P < .01 in comparison with 2-12H mice; n ≥ 3).
Faslpr preferentially affects anti-Sm MZ B cells in the spleen
To understand the effects of the MRL bkg and Faslpr on anti-Sm B cells, we examined their effects on B-cell differentiation. We observed a higher frequency and number of anti-Sm B cells in 2-12H MRL than 2-12H mice (41% ± 3% vs 30% ± 3%, respectively; P = .013) (Figure 2A-B). This increase was against the backdrop of an overall 2-fold increase in the total number of splenic B cells in 2-12H MRL and non-Tg MRL mice compared with their nonautoimmune counterparts (Figure 2B). Thus, the MRL bkg preferentially expanded the anti-Sm B-cell subset. This expansion was opposed by Faslpr, as both the frequency (16% ± 3% in 2-12H MRL/Faslpr vs 41% ± 3% in 2-12H MRL mice; P = .002) and number of anti-Sm B cells were reduced by half in 2-12H MRL/Faslpr mice (Figure 2A-B). This Faslpr-induced reduction of anti-Sm B cells in MRL mice did not occur in nonautoimmune 2-12H mice (30% ± 3% in 2-12H vs 36% ± 4% in 2-12H/Faslpr mice; P = .114; Figure 2A-B). Thus, Faslpr prevented the expansion of both the splenic B-cell and anti-Sm B-cell populations induced by the MRL bkg, but had no effect on the sizes of these populations in nonautoimmune mice.
Figure 2.

Anti-Sm B-cell development in spleen of 2-12H Tg, 2-12H MRL, 2-12H/Faslpr, and 2-12H MRL/Faslpr mice. (A) Anti-Sm B cells are present in the spleen of 2-12H Tg, 2-12H MRL, 2-12H/Faslpr, and 2-12H MRL/Faslpr mice. All histograms are gated on CD19+ B cells, and anti-Sm B cells are boxed. All mice are 2 to 4 months of age. (B) Numbers of splenic B cells and anti-Sm B cells are presented. Means ± SEM of 5 6-month-old mice of the indicated strain are plotted. Asterisks indicate a statistically significant difference (*P < .05 and **P < .01) from cell numbers of their nonautoimmune counterparts. Cell numbers at 3 months are not statistically different from those shown for 6 months (data not shown). (C) B-cell subset analysis of splenic anti-Sm B cells from 1-month-old and 3-month-old mice based on CD21 and CD23 expression. Anti-Sm B cells are gated as indicated in panel A. The CD21hi, CD23lo cells are MZ B cells, and the CD21int, CD23hi cells are FO B cells. The percent of total anti-Sm B cells for each subset is given. (D) Total number of anti-Sm MZ B cells from 1- to 6-month-old mice of each strain is given. Gating for anti-Sm B cells is as shown in panel A. Mean ± SE is plotted (n = 6). (E) Sm staining of MZ and FO B cells is shown. Gating on total MZ and FO B cells was based on CD21 and CD23 expression as indicated in panel B. The shaded regions are Sm staining of non-Tg B cells; the solid lines are Sm staining by B cells of their Tg counterparts.
The MRL bkg and Faslpr preferentially affected the anti-Sm MZ B-cell subset. MRL mice had an approximately 4-fold expansion of the MZ B-cell subset compared with nonautoimmune mice (Figure 2C-D bottom), as has been previously reported,29,30 but had an approximately 8-fold expansion of the anti-Sm MZ B-cell subset (Figure 2D top). These increases exceeded the approximately 2-fold expansion of total splenic B-cell numbers (Figure 2B), indicating a preferential effect on MZ B cells and in particular anti-Sm MZ B cells. Again, Faslpr opposed the expansion of the MZ B-cell subset, as 2-12H MRL/Faslpr mice had one third the number of MZ B cells and one seventh the number of anti-Sm MZ B cells as 2-12H MRL mice (Figure 2D). Faslpr affected only the MRL-induced expansion of MZ B cells, since non-Tg/Faslpr and non-Tg mice did not differ in the number of MZ B cells (Figure 2D). Thus, the MRL bkg preferentially expanded the anti-Sm MZ B-cell subset, and this expansion was opposed by Faslpr.
While Faslpr opposed an anti-Sm MZ expansion in autoimmune 2-12H MRL mice, this same mutation was responsible for the loss of anti-Sm MZ B cells in 2-12H/Faslpr mice. 2-12H and 2-12H/Faslpr mice have equal numbers of anti-Sm MZ B cells up to 2 months of age, but while 2-12H anti-Sm MZ numbers remained the same thereafter, 2-12H/Faslpr numbers declined to background levels by 3 months of age (Figure 2C-D). Because there was no decline in MZ B-cell numbers in non-Tg Faslpr mice (Figure 2D bottom) and the majority of MZ B cells in 2-12H but not non-Tg mice were anti-Sm (Figure 2E), we conclude that Faslpr induces an autoantigen-specific loss of MZ B cells.
Faslpr and the MRL bkg promoted bypass of the pre-PC tolerance checkpoint
We previously determined that anti-Sm B cells in 2-12H mice differentiate to an early pre-PC stage. Similar to PCs, these cells express intermediate levels of the PC marker CD138 (CD138int), have up-regulated CXCR4, and have become larger and more granular; yet, similar to B cells, they still express near-normal levels of CD19, B220, and IgM.11 However, their differentiation is arrested prior to becoming ASCs. In contrast, the anti-Sm B cells of autoimmune 2-12H MRL/Faslpr mice can differentiate beyond this checkpoint, become CD138hi, increase in size and granularity, and express low levels of CD19, B220, and IgM.11 Most significantly, some differentiate to ASCs.11 To assess the role of the MRL bkg and Faslpr on pre-PC differentiation, we examined anti-Sm pre-PCs in these autoimmune mice. We found that the frequency of CD138int anti-Sm B cells was similar in 2-12H and 2-12H/Faslpr mice (P = .48) (Figure 3A) and that there were no differences in size or expression of IgM, CXCR4, CD19, and CD80 (Figure 3B and data not shown). Moreover, Faslpr did not induce an increase in CD138hi pre-PCs (P = .38). In contrast, the MRL bkg induced a significant decrease in the frequency of CD138int anti-Sm B cells in 2-12H MRL mice (P = .008 versus 2-12H) and a significant increase in CD138hi anti-Sm B cells (P = .049 versus 2-12H). Compared with CD138int pre-PCs, the CD138hi pre-PCs were larger, more granular, and had higher levels of CXCR4 and lower levels of CD19, B220, and IgM (Figure 3B and data not shown). This phenotype was similar to that of the anti-Sm CD138hi pre-PCs of 2-12H MRL/Faslpr mice (Figure 3A-B and Culton et al11). Thus, only the MRL bkg exhibits evidence of significant differentiation beyond the early pre-PC tolerance checkpoint.
Figure 3.

ELISpot analysis of anti-Sm ASCs in 2-12H, 2-12H/Faslpr, and 2-12HMRL/Faslpr mice. (A) Spleen cells were stained for expression of CD19, CD138, and Sm. The CD138 expression of CD19+, Sm+ cells is shown. Boxes indicate the CD138int and CD138hi populations. The percent of each population is provided. Data are representative of 3 or more mice of each strain. The percentages of CD138int B cells are as follows: 17% ± 2.4% for 2-12H, 14% ± 3.1% for 2-12H/Faslpr, 7.0% ± 1.1% for 2-12H MRL, and 7.7% ± 0.50% for 2-12H MRL/Faslpr. The percentages of CD138hi B cells are as follows: 0.13% ± 0.07% for 2-12H, 0.09% ± 0.01% for 2-12H/Faslpr, 1.4% ± 0.46% for 2-12H MRL, and 1.9% ± 0.66% for 2-12H MRL/Faslpr. (B) Size, IgM expression, and CXCR4 expression are shown for anti-Sm CD138int and CD138hi B cells from mice of the indicated strains. The gates are illustrated in panel A. Data are representative of 3 or more mice. (C) Spleen cells were stained for CD19, CD138, CD21, and CD23. The gating scheme used to sort the FO, MZ, and CD138+ cells is indicated using 2-12H spleen cells. The top histogram shows the identification of CD19+, CD138+ cells sorted for ELISpot analysis. The CD19+ CD138– cells were further fractionated by CD21 and CD23 expression to identify FO and MZ B cells as shown. (D) The number of small and large anti-Sm ELISpots was determined for each of the indicated cell populations. The definition of large and small ELISpots is given in “Materials and methods.” The average size of small ELISpots characteristic of MZ and FO B cells is 2 × 10–3 ± 4 × 10–4 mm2, and the average size of the large ELISpots characteristic of CD138+ pre-PCs is 1.7 × 10–2 ± 4 × 10–4 mm2. Examples of the small FO/MZ-like spots and large CD138+ pre-PC ELISpots are shown. The number of FO anti-Sm ELISpots per 105 cells in 2-12H and 2-12H/Faslpr mice is 23.3 ± 2.36 and 3.51 ± 2.48, respectively (P < .001). The number of MZ anti-Sm ELISpots per 105 cells in 2-12H and 2-12H/Faslpr mice is 183 ± 19.2 and 22.5 ± 10.9, respectively (P = .002). The number of CD138+ pre-PC anti-Sm ELISpots per 105 cells from 2-12H, 2-12H/Faslpr, and 2-12H MRL/Faslpr mice is 26.0 ± 5.6, 1540 ± 135, and 7600 ± 265, respectively (P < .01 for all comparisons). ND indicates none detected.
Since 2-12H/Faslpr mice generate anti-Sm ASCs (Figure 1B), we sought to determine whether bypass of the early pre-PC checkpoint occurs inefficiently, so as not to alter the frequency of CD138int and CD138hi pre-PCs, yet still generate ASCs. Thus, we determined the frequency of anti-Sm ASCs among CD138+ pre-PCs from both nonautoimmune and autoimmune 2-12H mice by ELISpot analysis. Since anti-Sm MZ B cells in nonautoimmune mice secrete small amounts of antibody,22 we also examined the frequency of ASCs among FO and MZ B cells. The sorting criteria were as illustrated in Figure 3C. PCs were excluded from the sorted CD138int B-cell population since the sorted population excluded all CD19– cells (Figure 3B). The FO and MZ B cells of 2-12H and 2-12H/Faslpr mice formed only small anti-Sm ELISpots, with the highest frequency among MZ B cells (Figure 3D). Of interest, 2-12H/Faslpr mice had a significantly lower frequency of anti-Sm ELISpots among MZ B cells than 2-12H mice, and anti-Sm ELISpots were undetectable among MZ and FO B cells of 2-12H MRL/Faslpr mice. In contrast, large anti-Sm ELISpots were detectable only among the CD138+ pre-PCs from 2-12H/Faslpr and 2-12H MRL/Faslpr mice, with the highest frequency among the latter (Figure 3D). Thus, although Faslpr does not induce an increase in CD138hi pre-PCs, it induces some pre-PCs to become ASCs, indicating bypass of the early pre-PC tolerance checkpoint. Moreover, the inverse relationship between the numbers of small anti-Sm ELISpots of FO and MZ B cells and large anti-Sm ELISpots of the CD138+ pre-PCs suggests that MZ and FO B cells are induced to differentiate to CD138int pre-PCs in autoimmune mice.
Faslpr opposed the MRL-induced expansion of the B-1–cell population and induced anti-Sm B-1–cell activation
Faslpr and the MRL bkg also affected the number of anti-Sm B-1 cells. B-1 cells were identified as CD5+, CD43+, CD23–, and CD11b+ (Figure 4A and data not shown). As with the MZ B-cell population, the MRL bkg genes induced a 1.5- to 2-fold expansion of both the anti-Sm B-1–cell population and the total B-1–cell population (Figure 4B), and Faslpr opposed this increase (Figure 4B). However, it had no effect on the number of B-1 cells in nonautoimmune 2-12H or non-Tg mice (Figure 4B). As with anti-Sm MZ B cells, the anti-Sm B-1–cell population underwent limited expansion in perinatal 2-12H/Faslpr and 2-12H MRL/Faslpr mice and declined to background numbers by 3 months of age, whereas the anti-Sm B-1–cell numbers were constant in 2-12H and 2-12H MRL mice. Thus, Faslpr induces the loss of anti-Sm B-1 cells, but not the total number of B-1 cells, suggesting that like the loss of anti-Sm MZ B cells, anti-Sm B-1–cell loss is autoantigen specific.
Figure 4.
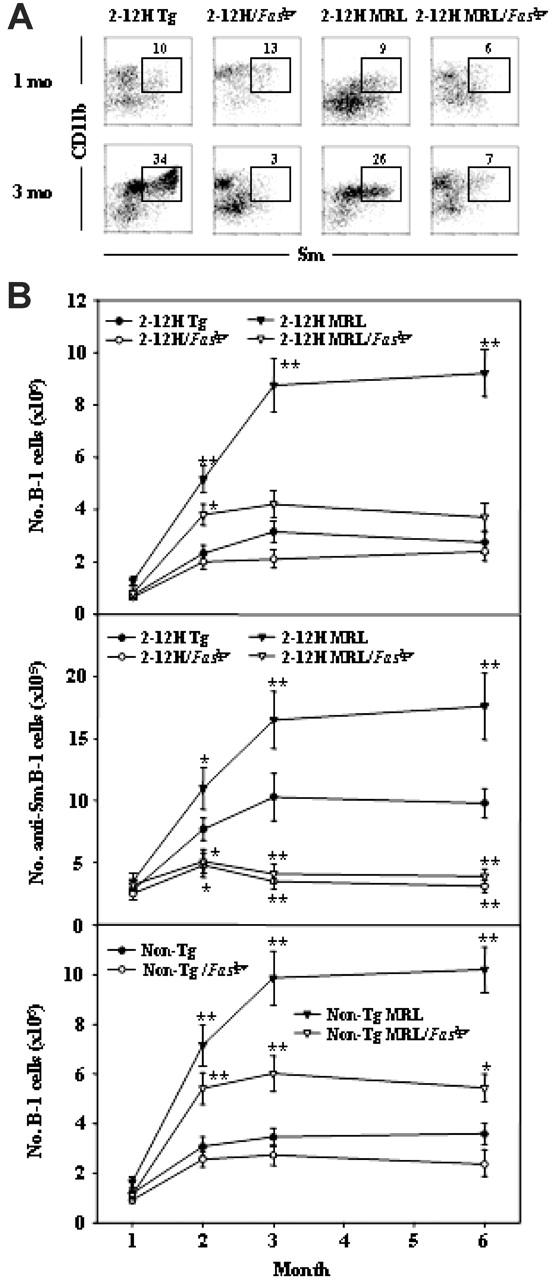
Peritoneal anti-Sm B-1 cells in 2-12H Tg, 2-12H MRL, 2-12H/Faslpr, and 2-12H MRL/Faslpr mice. (A) Phenotypic analysis of peritoneal B cells from 2-12H Tg, 2-12H MRL, 2-12H/Faslpr, and 2-12H MRL/Faslpr mice. Peritoneal cells from 1-month-old and 3-month-old mice were stained with CD19, IgM, CD11b, and Sm. Anti-Sm B-1 cells are boxed. All histograms are gated on CD19+ B cells. (B) Analysis of total B-1–cell numbers ± SEM (top and bottom graphs) and anti-Sm B-cell numbers ± SEM (middle) for mice between 1 and 6 months of age is shown.
We previously showed that peritoneal anti-Sm B-1–cell numbers declined in 2-12H/Merkd mice after approximately 2 months of age and that this was due at least in part to differentiation to ASCs of the MLNs and LP.22 To determine whether a similar explanation accounted for the loss of peritoneal anti-Sm B-1 cells in 2-12H/Faslpr mice, sorted B-1 and B-2 cells from 2-12H/Faslpr mice were transferred intraperitoneally to Faslpr recipient mice. As shown in Figure 5A, only B-1–cell transfers generated anti-Sm ASCs in the MLN and LP. Thus, Faslpr induced anti-Sm B-1 cells, but not B-2 cells, to differentiate to MLN and LP ASCs. Since these data indicate that only B-1 cells are activated after transfer, all subsequent experiments used unsorted peritoneal cells to maximize the number of B-1 cells transferred.
Figure 5.

Anti-Sm B-1–cell activation in Faslpr mice after peritoneal cell transfer. (A) 2-12H/Faslpr peritoneal cells were stained for CD19 and CD11b, and the CD19+ CD11b+ B-1 cells and CD19+ CD11b– B-2 cells were sorted and transferred to Faslpr recipient mice. Two weeks after transfer, anti-Sm ASCs in the bone marrow, spleen, MLN, and LP were quantified (± SEM) by ELISpot. ND indicates none detected. (B) Unsorted peritoneal cells from 2-12H/Faslpr mice were transferred to wild-type (wt) or Faslpr recipient mice. ELISA was used to measure (± SEM) serum IgMa+ anti-Sm (inset), and ELISpot was used to quantify (± SEM) anti-Sm ASCs from BM, spleen, MLN, and LP. (C) The same as panel B except that 2-12H peritoneal cells were transferred to wt or Faslpr recipients. Asterisks indicate the differences (P < .05) between wt and Faslpr recipients.
To determine whether the Faslpr environment is required for activation, 2-12H/Faslpr peritoneal cells were transferred to wt and Faslpr recipient mice. Two weeks after transfer, Faslpr recipients, but not wt recipients, had an elevated level of serum anti-Sm (Figure 5B inset) and an elevated number of anti-Sm ASCs of donor origin in the MLN and LP of Faslpr recipients (Figure 5B). Anti-Sm ASCs were not detected in the spleen or BM of either Faslpr or wt recipients. B-1–cell activation did not require that they be Fas-deficient, since transfer of 2-12H peritoneal cells to Faslpr mice, but not wt recipient mice, generated serum anti-Sm and anti-Sm ASCs of donor origin (Figure 5C). Thus, anti-Sm B-1 cells are activated only in Faslpr recipients and they need not be Fas-deficient.
B-1–cell activation by Faslpr was autoantigen specific
That Faslpr had no effect on total B-1–cell numbers in nonautoimmune mice (Figure 4B lower panel) suggested that the loss of anti-Sm B-1 cells caused by Faslpr is antigen specific. To further test this possibility, we examined B-1 cells specific for phosphatidyl choline (PtC), a common membrane phospholipid to which as many as 7% of B-1 cells are specific.27 For these experiments, we used mice with an H chain transgene that encodes anti-PtC antibodies (6-1). 6-1 Tg mice have a high frequency of splenic and peritoneal anti-PtC B-1 cells. These cells were identified as CD43+, CD5+ B cells that also stained with PtC-containing liposomes. Faslpr had no effect on the percentage of peritoneal anti-PtC B-1 cells (Figure 6A) or on the expansion of the anti-PtC B-1–cell population in neonatal and perinatal mice (Figure 6B), and it did not induce a loss of PtC-specific B-1 cells (Figure 6B).
Figure 6.

The effect of Faslpr on PtC-specific B cells. (A) Comparison of 6-1 and 6-1/Faslpr peritoneal B cells for liposome binding and CD43 and CD5 expression. Histograms are gated on CD19+ B cells. PtC-specific B-1 cells (CD43+ and CD5+) are boxed, and the percent of total CD19+ B cells is indicated. (B) The number, ± SEM, of PtC-specific B-1 cells present in the peritoneal cavity of 6-1 and 6-1/Faslpr between 1 and 6 months is graphed. PtC-specific B-1 cells were gated as shown in panel A. (C) Number, ± SEM, of IgMa ASCs in wt and Faslpr recipient mice 2 weeks after transfer of 6-1 peritoneal cells.
To determine whether Faslpr increased the activation of anti-PtC B-1 cells to ASCs, we compared the activation of anti-PtC B-1 cells in 6-1 and 6-1/Faslpr mice by cell transfer. 6-1 peritoneal cells were transferred to wt and Faslpr recipient mice. Anti-PtC B-1 cells are normally activated in nonautoimmune mice,27 and, thus, 2 weeks after transfer ASCs of donor origin were present in wt recipients (Figure 6C). However, the numbers of ASCs of donor origin in Faslpr mice were not significantly different from those in wt recipients (Figure 6C). Thus, the activation and depletion of B-1 cells in Faslpr mice is limited to cells of certain specificities, consistent with antigen dependence.
Faslpr mice have a high frequency of apoptotic cells in the spleen and MLNs
The activation and loss of anti-Sm B-1 cells of 2-12H/Faslpr mice were remarkably similar to our findings with anti-Sm B-1 cells of 2-12H/Merkd mice.22 We attributed this loss in Merkd mice to an increase in availability of apoptotic cell antigens such as Sm and a defect in macrophage phagocytosis of apoptotic cells. Since Fas has nonapoptotic functions,31,32 we sought to determine whether Faslpr mice also increased the availability of apoptotic cell antigens. The frequency of apoptotic cells in wt, Faslpr, and MRL/Faslpr mice was measured by flow cytometry using the fluorescence-labeled caspase inhibitor VAD-FMK. Both 2- and 3-month-old mice were analyzed to span the time frame for anti-Sm MZ and B-1 B-cell loss (Figures 2,4). No difference in the frequency of splenic or MLN apoptotic cells was evident at 2 months, whereas a higher frequency of apoptotic cells was noted in Faslpr spleens and MLNs at 3 months (Figure 7A). This was corroborated by the detection of cells with fragmented DNA by TUNEL assay of frozen tissue sections of 3-month-old Faslpr mice (Figure 7B). In the spleen, TUNEL+ cells were located predominantly in the extrafollicular space, whereas in the MLN they were located predominantly in B-cell–rich follicles and in germinal centers.
Figure 7.

Apoptotic cells in wt and Faslpr mice. (A) Flow cytometry analysis for the presence of apoptotic cells in the spleens and MLNs of wt and Faslpr mice by FITC-VAD-FMK staining is shown. Representative histograms for wt and Faslpr spleen cells are shown (top). Each histogram is gated on lymphocytes and the percent VAD-FMK+ is provided. The graph below shows the average ± SEM of percent apoptotic lymphocytes in the spleen and MLN of mice at 2 months and 3 months of age (n = 3). **A significant difference with wt (P < .01). (B) TUNEL-positive cell distributions in spleen and MLN of both wt and Faslpr mice. Both low (× 10) and high (× 40) magnifications are shown. Boxes indicate the areas of higher magnification. WP indicates white pulp; RP, red pulp; GC, germinal center; and F, follicle. (C) Macrophages from wt, Faslpr, MRL/Faslpr, and Merkd mice were incubated with apoptotic thymocytes for 60 minutes, and the percent of phagocytized thymocytes was determined by fluorescent microscopy. Controls are the percentage of live thymocytes (nonapoptotic) phagocytized. The graph is representative of 4 experiments and shows the average ± SEM of 3 mice per strain at both 2 months and 3 months of age. *P < .05, compared with wt.
Defective phagocytosis of apoptotic cells by Faslpr macrophages
The increase in apoptotic cell frequency seen in Faslpr mice could be due to a defect in their clearance by phagocytic cells. Since macrophages play a key role in apoptotic cell clearance,23 we compared the ability of macrophages from 2- and 3-month-old Faslpr and wt mice to phagocytize apoptotic cells in an in vitro assay. As shown in Figure 7C, both 2- and 3-month-old Faslpr macrophages were significantly less able to phagocytize apoptotic cells than their wt counterparts, although their defect at both ages was less severe than that of Merkd macrophages. Nevertheless, these data implicate Faslpr macrophages in the high frequency of apoptotic lymphocytes in Faslpr mice and in the activation of autoreactive B cells.
Discussion
We show here that MRL, MRL/Faslpr, and Faslpr mice with the 2-12H transgene develop a spontaneous anti-Sm response with complete penetrance. Our findings point to a central involvement of MZ and B-1 B cells in this response and suggest a link to defective apoptotic cell clearance in their activation. Our comparison of anti-Sm B cells from 2-12H mice and B cells of diverse specificities from non-Tg mice indicates bias at 2 levels: B-cell subset and antigen specificity. Faslpr induces a loss of anti-Sm MZ and B-1 B cells in young adult 2-12H/Faslpr and 2-12H MRL/Faslpr mice between 2 and 3 months of age. Since Faslpr has no effect on non-Tg MZ and B-1 B cells (Figures 2D,4) or on anti-PtC B-1 cells, we conclude that the loss of anti-Sm B cells is antigen specific (Figure 6) and not due to a general effect on the generation or maintenance of the MZ or B-1 B-cell populations.
The most likely explanation for the Faslpr-induced loss of anti-Sm MZ and B-1 B cells is that they are activated to differentiate to ASCs. Autoantigen activation would account for the antigen-specific depletion of anti-Sm MZ and B-1 B cells and the presence of ASCs in multiple lymphoid tissues (Figure 1). It would also account for the inverse relationship between FO and MZ ASCs and CD138int ASCs (Figure 3B) since activated FO and MZ B cells would presumably pass through the CD138int pre-PC stage before differentiating to PCs. Additionally, the activation of anti-Sm MZ and B-1 B cells would explain the appearance of serum anti-Sm in 2-12H MRL/Faslpr mice between 1 and 2 months of age.12 Anti-Sm B-1–cell activation is confirmed by the transfer experiments showing that they generate MLN and LP ASCs and produce serum antibody in Faslpr recipients, but not wt recipients (Figure 5A). Antigen-independent mechanisms may have a role in this activation, but the number of anti-PtC B-1 cells is the same in 6-1 and 6-1/Faslpr mice, indicating that any such effect is likely to be small. Thus, we conclude that loss of anti-Sm B-1 cells in Faslpr mice is due to autoantigen-specific activation and differentiation to ASCs. The parallel loss of anti-Sm MZ B cells by Faslpr mice suggests that anti-Sm MZ B cells are also activated to become ASCs, although this remains to be confirmed.
The inefficient bypass of the early pre-PC tolerance checkpoint induced by Faslpr (Figure 3) indicates that Fas-FasL–induced cell death is not a major mechanism of regulation at this checkpoint. Whether Fas-FasL interactions have a regulatory role at an earlier checkpoint cannot be ruled out. The interaction of Fas on autoreactive B cells with FasL on helper T cells can induce cell death,33,34 but this is not always the case.35 Although helper T-cell regulation of these B cells may occur through Fas-FasL interactions, the finding that Fas+ B-1 cells are activated in Faslpr hosts (Figure 5) suggests that the primary defect leading to anti-Sm MZ and B-1 B-cell activation in Faslpr mice lies elsewhere.
A possible explanation for anti-Sm and B-1–cell activation in Faslpr mice is the defect in macrophage phagocytosis of apoptotic cells and the increase in the presence of apoptotic lymphocytes in lymphoid tissues (Figure 7). Apoptotic cells expose Sm and can induce an anti-Sm response in nonautoimmune mice.22 A Faslpr-induced increase in apoptotic cells has been previously noted in the brains of MRL/Faslpr mice.36 This would account for the antigen-specific effects of Faslpr and for the observation that both Fas-sufficient and -deficient B-1 cells are activated (Figure 5). Additionally, since anti-PtC antibodies do not bind apoptotic cells (Y.Q. and S.H.C., unpublished data, March 2001), it would explain why Faslpr has no effect on anti-PtC B-1 cells (Figure 6). These findings parallel our previous observation that anti-Sm MZ and B-1 B cells are activated in 2-12H/Merkd mice, which also have a defect in apoptotic cell clearance.22 In addition, the appearance of increased numbers of apoptotic cells in lymphoid tissues corresponds with when anti-Sm production begins and when anti-Sm MZ and B-1 cells are lost, further implicating apoptotic cells as the driving force in the activation of Faslpr anti-Sm MZ and B-1 cells. Of interest, Faslpr mice that lack p53, a proapoptotic gene,37 generate lower levels of serum autoantibodies compared with Faslpr mice.38 Thus, the absence of p53 may result in a lower frequency of apoptotic cells in Faslpr mice, accounting for the lower autoantibody levels. Defective clearance of apoptotic cells is a recurring feature in autoimmunity, and targeted impairment of apoptotic cell clearance invariably results in autoimmunity.23,39-41 Defective phagocytosis of apoptotic cells is also a feature of human lupus.42,43 MRL macrophages are defective in apoptotic cell clearance,44 and thus the combination of the MRL bkg- and the Faslpr-induced impairment of apoptotic cell clearance may contribute to the more severe autoimmunity of MRL/Faslpr mice.42,43 Of interest, while the defect in macrophage phagocytosis of apoptotic cells is evident in Faslpr and MRL/Faslpr mice at 2 months (Figure 7A), there is no apparent effect on the frequency of apoptotic lymphocytes relative to wt mice at 2 months (Figure 7C). Thus, either this macrophage defect is not responsible for the increase in apoptotic lymphocytes or that there are compensatory mechanisms for phagocytosis in younger mice.
How Faslpr induces this macrophage defect remains to be investigated. Serum from lupus patients significantly inhibits efficient phagocytosis by normal human neutrophils,45 and thus there may be a secreted factor in Faslpr mice that diminishes macrophage phagocytosis of apoptotic cells. Although incompletely characterized, Fas has nonapoptotic functions, such as cytokine production, induction of DC maturation and survival, and chemokine production46,47 involving multiple cell types.31,32,48,49 Thus, Fas-FasL interactions could be necessary for the induction of efficient macrophage phagocytosis of apoptotic cells. IL-10 enhances macrophage phagocytosis of apoptotic cells,50 although there is disagreement,51 but no difference in systemic IL-10 levels was noted (22.3 ± 0.81 pg/mL for wt mice vs 22.9 ± 2.37 pg/mL for Faslpr mice, P = .73).
Our finding that B-1 cells are involved in the production of anti-Sm appears to contradict an earlier report.52 This earlier study relied on reconstitution of irradiated recipient mice, and we find that anti-Sm B-1 cells undergo apoptosis upon encounter with apoptotic cells and poorly reconstitute irradiated mice after bone marrow transfer (Y.Q. and S.H.C., unpublished observation, September 2000). Also, B-1 cells may contribute minimally to serum autoantibody since many anti-Sm ASCs of B-1–cell origin are located in the LP and are likely to secrete into the intestinal lumen. Thus, previous studies could reasonably lead to different conclusions.
The activation of anti-Sm MZ and B-1 B cells in MRL and Fas-deficient mice adds to a growing body of evidence that MZ and B-1 B cells are involved in autoimmunity.13,20,22,23,30,53-56 Both MZ and B-1 B cells are antigen-experienced cells that can rapidly differentiate to ASCs in response to antigen and provide protection against blood-borne particulate antigens and enteric organisms.57 These features, as well as their location, may make them uniquely positioned to encounter and respond to apoptotic cells in the blood and lymph. MZ B cells are potent activators of CD4+ T cells,58 and thus activation of MZ B cells may also contribute to disease through the activation of autoreactive T cells. Understanding how autoreactive MZ and B-1 B cells are activated and the nature of the link to increased apoptotic cell frequency and defective macrophage apoptotic cell phagocytosis will be important directions for further investigation.
Acknowledgments
The authors thank the UNC Flow Cytometry Facility and Larry Arnold for their assistance and advice with the flow cytometry and cell sorting.
Prepublished online as Blood First Edition Paper, April 4, 2006; DOI 10.1182/blood-2005-12-006858.
Y.Q. and K.L.C. contributed equally to this work.
Supported by National Institutes of Health grants AI29576 and AI3587 to S.H.C. and AI050736 to G.K.M. Y.Q. was supported by an Arthritis Foundation Postdoctoral Fellowship, and K.L.C. was supported by an NIH training grant AI007273.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Kono DH, Theofilopoulos AN. Genetics of systemic autoimmunity in mouse models of lupus. Int Rev Immunol. 2000;19: 367-387. [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg RA, Tan EM, Dixon FJ. Presence of anti-Sm reactivity in autoimmune mouse strains. J Exp Med. 1978;147: 582-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kakkanaiah VN, Sobel ES, MacDonald GC, Cheek RL, Cohen PL, Eisenberg RA. B cell genotype determines the fine specificity of autoantibody in lpr mice. J Immunol. 1997;159: 1027-1035. [PubMed] [Google Scholar]
- 4.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356: 314-317. [DOI] [PubMed] [Google Scholar]
- 5.Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268: 1347-1349. [DOI] [PubMed] [Google Scholar]
- 6.Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81: 935-946. [DOI] [PubMed] [Google Scholar]
- 7.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37: 269-390. [DOI] [PubMed] [Google Scholar]
- 8.Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9: 243-269. [DOI] [PubMed] [Google Scholar]
- 9.Santulli-Marotto S, Retter MW, Gee R, Mamula MJ, Clarke SH. Autoreactive B cell regulation: peripheral induction of developmental arrest by lupus-associated autoantigens. Immunity. 1998;8: 209-219. [DOI] [PubMed] [Google Scholar]
- 10.Borrero M, Clarke SH. Low-affinity anti-Smith antigen B cells are regulated by anergy as opposed to developmental arrest or differentiation to B-1. J Immunol. 2002;168: 13-21. [DOI] [PubMed] [Google Scholar]
- 11.Culton DA, O'Conner BP, Conway KL, et al. Early preplasma cells define a tolerance checkpoint for autoreactive B cells. J Immunol. 2006;176: 790-802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santulli-Marotto S, Qian Y, Ferguson S, Clarke SH. Anti-Sm B cell differentiation in Ig transgenic MRL/Mp-lpr/lpr mice: altered differentiation and an accelerated response. J Immunol. 2001;166: 5292-5299. [DOI] [PubMed] [Google Scholar]
- 13.Xu H, Li H, Suri-Payer E, Hardy RR, Weigert M. Regulation of anti-DNA B cells in recombination-activating genedeficient mice. J Exp Med. 1998;188: 1247-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin F, Kearney JF. B-cell subsets and the mature preimmune repertoire: marginal zone and B1 B cells as part of a “natural immune memory.” Immunol Rev. 2000;175: 70-79. [PubMed] [Google Scholar]
- 15.Grimaldi CM, Michael DJ, Diamond B. Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J Immunol. 2001;167: 1886-1890. [DOI] [PubMed] [Google Scholar]
- 16.Thien M, Phan TG, Gardam S, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20: 785-798. [DOI] [PubMed] [Google Scholar]
- 17.Mackay F, Woodcock SA, Lawton P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190: 1697-1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Batten M, Groom J, Cachero TG, et al. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192: 1453-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watanabe N, Ikuta K, Nisitani S, Chiba T, Honjo T. Activation and differentiation of autoreactive B-1 cells by interleukin 10 induce autoimmune hemolytic anemia in Fas-deficient antierythrocyte immunoglobulin transgenic mice. J Exp Med. 2002;196: 141-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohan C, Morel L, Yang P, Wakeland EK. Genetic dissection of systemic lupus erythematosus pathogenesis: Sle2 on murine chromosome 4 leads to B cell hyperactivity. J Immunol. 1997;159: 454-465. [PubMed] [Google Scholar]
- 21.Xu Z, Butfiloski EJ, Sobel ES, Morel L. Mechanisms of peritoneal B-1a cells accumulation induced by murine lupus susceptibility locus Sle2. J Immunol. 2004;173: 6050-6058. [DOI] [PubMed] [Google Scholar]
- 22.Qian Y, Wang H, Clarke SH. Impaired clearance of apoptotic cells induces the activation of autoreactive anti-Sm marginal zone and B-1 B cells. J Immunol. 2004;172: 625-635. [DOI] [PubMed] [Google Scholar]
- 23.Scott RS, McMahon EJ, Pop SM, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411: 207-211. [DOI] [PubMed] [Google Scholar]
- 24.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179: 1317-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold LW, Pennell CA, McCray SK, Clarke SH. Development of B-1 cells: segregation of phosphatidyl choline-specific B cells to the B-1 population occurs after immunoglobulin gene expression. J Exp Med. 1994;179: 1585-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian Y, Santiago C, Borrero M, Tedder TF, Clarke SH. Lupus-specific antiribonucleoprotein B cell tolerance in nonautoimmune mice is maintained by differentiation to B-1 and governed by B cell receptor signaling thresholds. J Immunol. 2001;166: 2412-2419. [DOI] [PubMed] [Google Scholar]
- 27.Mercolino TJ, Arnold LW, Hawkins LA, Haughton G. Normal mouse peritoneum contains a large population of Ly-1+ (CD5) B cells that recognize phosphatidyl choline: relationship to cells that secrete hemolytic antibody specific for autologous erythrocytes. J Exp Med. 1988;168: 687-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pop SM, Wong CP, Culton DA, Clarke SH, Tisch R. Single cell analysis shows decreasing FoxP3 and TGFbeta1 coexpressing CD4+CD25+ regulatory T cells during autoimmune diabetes. J Exp Med. 2005;201: 1333-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandik-Nayak L, Seo SJ, Sokol C, Potts KM, Bui A, Erikson J. MRL-lpr/lpr mice exhibit a defect in maintaining developmental arrest and follicular exclusion of anti-double-stranded DNA B cells. J Exp Med. 1999;189: 1799-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Li H, Ni D, Weigert M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med. 2002;196: 1543-1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn JH, Park SM, Cho HS, et al. Non-apoptotic signaling pathways activated by soluble Fas ligand in serum-starved human fibroblasts: mitogen-activated protein kinases and NF-kappaB-dependent gene expression. J Biol Chem. 2001;276: 47100-47106. [DOI] [PubMed] [Google Scholar]
- 32.Wajant H, Pfizenmaier K, Scheurich P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev. 2003;14: 53-66. [DOI] [PubMed] [Google Scholar]
- 33.Rothstein TL, Wang JK, Panka DJ, et al. Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature. 1995;374: 163-165. [DOI] [PubMed] [Google Scholar]
- 34.Rathmell JC, Cooke MP, Ho WY, et al. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 1995;376: 181-184. [DOI] [PubMed] [Google Scholar]
- 35.Seo SJ, Fields ML, Buckler JL, et al. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity. 2002;16: 535-546. [DOI] [PubMed] [Google Scholar]
- 36.Sakic B, Maric I, Koeberle PD, et al. Increased TUNEL staining in brains of autoimmune Fas-deficient mice. J Neuroimmunol. 2000;104: 147-154. [DOI] [PubMed] [Google Scholar]
- 37.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411: 342-348. [DOI] [PubMed] [Google Scholar]
- 38.Kuan AP, Cohen PL. p53 is required for spontaneous autoantibody production in B6/lpr lupus mice. Eur J Immunol. 2005;35: 1653-1660. [DOI] [PubMed] [Google Scholar]
- 39.Botto M, Dell'Agnola C, Bygrave AE, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19: 56-59. [DOI] [PubMed] [Google Scholar]
- 40.Hanayama R, Tanaka M, Miyasaka K, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304: 1147-1150. [DOI] [PubMed] [Google Scholar]
- 41.Mahoney JA, Rosen A. Apoptosis and autoimmunity. Curr Opin Immunol. 2005;17: 583-588. [DOI] [PubMed] [Google Scholar]
- 42.Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41: 1241-1250. [DOI] [PubMed] [Google Scholar]
- 43.Baumann I, Kolowos W, Voll RE, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46: 191-201. [DOI] [PubMed] [Google Scholar]
- 44.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol. 2003;170: 3223-3232. [DOI] [PubMed] [Google Scholar]
- 45.Zurier RB. Reduction of phagocytosis and lysosomal enzyme release from human leukocytes by serum from patients with systemic lupus erythematosus. Arthritis Rheum. 1976;19: 73-78. [DOI] [PubMed] [Google Scholar]
- 46.Rescigno M, Piguet V, Valzasina B, et al. Fas engagement induces the maturation of dendritic cells (DCs), the release of interleukin (IL)-1beta, and the production of interferon gamma in the absence of IL-12 during DC-T cell cognate interaction: a new role for Fas ligand in inflammatory responses. J Exp Med. 2000;192: 1661-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo Z, Zhang M, Tang H, Cao X. Fas signal links innate and adaptive immunity by promoting dendritic-cell secretion of CC and CXC chemokines. Blood. 2005;106: 2033-2041. [DOI] [PubMed] [Google Scholar]
- 48.Kataoka T, Budd RC, Holler N, et al. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol. 2000;10: 640-648. [DOI] [PubMed] [Google Scholar]
- 49.Hu WH, Johnson H, Shu HB. Activation of NF-kappaB by FADD, Casper, and caspase-8. J Biol Chem. 2000;275: 10838-10844. [DOI] [PubMed] [Google Scholar]
- 50.Ogden CA, Pound JD, Batth BK, et al. Enhanced apoptotic cell clearance capacity and B cell survival factor production by IL-10-activated macrophages: implications for Burkitt's lymphoma. J Immunol. 2005;174: 3015-3023. [DOI] [PubMed] [Google Scholar]
- 51.Popi AF, Lopes JD, Mariano M. Interleukin-10 secreted by B-1 cells modulates the phagocytic activity of murine macrophages in vitro. Immunology. 2004;113: 348-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reap EA, Sobel ES, Cohen PL, Eisenberg RA. Conventional B cells, not B-1 cells, are responsible for producing autoantibodies in lpr mice. J Exp Med. 1993;177: 69-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wither JE, Roy V, Brennan LA. Activated B cells express increased levels of costimulatory molecules in young autoimmune NZB and (NZB x NZW)F(1) mice. Clin Immunol. 2000;94: 51-63. [DOI] [PubMed] [Google Scholar]
- 54.Hayakawa K, Hardy RR, Honda M, Herzenberg LA, Steinberg AD. Ly-1 B cells: functionally distinct lymphocytes that secrete IgM autoantibodies. Proc Natl Acad Sci U S A. 1984;81: 2494-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J Exp Med. 1983;157: 202-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murakami M, Tsubata T, Okamoto M, et al. Antigen-induced apoptotic death of Ly-1 B cells responsible for autoimmune disease in transgenic mice [see comments]. Nature. 1992;357: 77-80. [DOI] [PubMed] [Google Scholar]
- 57.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14: 617-629. [DOI] [PubMed] [Google Scholar]
- 58.Attanavanich K, Kearney JF. Marginal zone, but not follicular B cells, are potent activators of naive CD4 T cells. J Immunol. 2004;172: 803-811. [DOI] [PubMed] [Google Scholar]