

Abstract
Acquired mutations in the FLT3 receptor tyrosine kinase are common in acute myeloid leukemia and result in constitutive activation. The most frequent mechanism of activation is disruption of the juxtamembrane autoregulatory domain by internal tandem duplications (ITDs). FLT3-ITDs confer factor-independent growth to hematopoietic cells and induce a myeloproliferative syndrome in murine bone marrow transplant models. We and others have observed that FLT3-ITD activates STAT5 and its downstream effectors, whereas ligand-stimulated wild-type FLT3 (FLT3WT) does not. In vitro mapping of tyrosine phosphorylation sites in FLT3-ITD identified 2 candidate STAT5 docking sites within the juxtamembrane domain that are disrupted by the ITD. Tyrosine to phenylalanine substitution of residues 589 and 591 in the context of the FLT3-ITD did not affect tyrosine kinase activity, but abrogated STAT5 activation. Furthermore, FLT3-ITD–Y589/591F was incapable of inducing a myeloproliferative phenotype when transduced into primary murine bone marrow cells, whereas FLT3-ITD induced myeloproliferative disease with a median latency of 50 days. Thus, the conformational change in the FLT3 juxtamembrane domain induced by the ITD activates the kinase through dysregulation of autoinhibition and results in qualitative differences in signal transduction through STAT5 that are essential for the transforming potential of FLT3-ITD in vivo.
Introduction
FMS-like tyrosine kinase-3 (FLT3) is a member of the type III receptor tyrosine kinase family that also includes c-kit receptor tyrosine kinase, FMS, and platelet-derived growth factor receptor (PDGFR).1,2 The members of this family share structural characteristics, including an extracellular domain with 5 immunoglobulin-like domains, a single transmembrane domain, a juxtamembrane domain, and a cytoplasmic tyrosine kinase domain split into 2 subdomains.3 Activation of FLT3 by its ligand (FL) results in receptor dimerization, intrinsic tyrosine kinase activity, and autophosphorylation of tyrosine residues.4 FLT3 is primarily expressed on the surface of CD34+ hematopoietic progenitor cells and plays an important role in their proliferation and differentiation.5-7 There are a spectrum of signal transduction pathways that are activated in response to stimulation of FLT3 with ligand, including Grb2-mediated activation of the RAS-MAPK pathway, Src, PI3K, Vav, CBL, and phospholipase C gamma.8-10
Mutations in FLT3 are commonly detected in patients with acute myeloid leukemia (AML). The most common of these mutations are internal tandem duplications (ITDs), which occur in approximately 30% of these patients as well as in 5% to 10% of patients with myelodysplastic syndrome (MDS).11 FLT3-ITDs consist of a duplication in the juxtamembrane domain that is always in frame but can vary in length. These mutations result in constitutive activation of the FLT3 receptor through disruption of an autoinhibitory domain,12 leading to factor-independent growth of murine hematopoietic cells and the development of a myeloproliferative phenotype in a murine bone marrow transplant model.13
Although FLT3-ITD activates many of the same signal transduction pathways as the native receptor, there are conflicting data suggesting that there may be qualitative differences in signal transduction between wild-type and mutant receptor.14-20 We have observed that, in contrast to the wild-type receptor, FLT3-ITD is a potent activator of STAT5. In vitro mapping of tyrosine autophosphorylation sites in an FLT3-ITD mutant identified 2 candidate STAT5 SH2 docking phosphorylation sites in the juxtamembrane domain, Y589 and Y591. This finding suggested the possibility that Y589 and Y591 might be occult STAT5 binding sites and that disruption of juxtamembrane structure by ITDs exposed these residues and enabled activation of STAT5. Consistent with this hypothesis, tyrosine to phenylalanine (Y→F) substitution of these residues abrogated activation of STAT5 and its downstream effector, Pim-1. Furthermore, these residues were critical for transformation of primary murine bone marrow cells in vivo. Thus, FLT3-ITD mutations not only activate the tyrosine kinase but enable activation of STAT5 through residues within the juxtamembrane domain that correlates with transformation in vivo.
Materials and methods
DNA constructs
Tyrosine to phenylalanine mutations Y591F, Y726F, Y955F, Y969F, Y591/726F, Y591/955F, Y726/955F, Y589/591F, Y955/969F, Y591/726/955F (Tri), and Y591/726/955/969F (4Y) were generated using the QuickChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. All mutations were confirmed by DNA sequencing. The full-length wild-type FLT3 (FLT3WT), FLT3-ITD (N51), and FLT3-D835Y cDNA as well as the FLT3-ITD Y→F mutant cDNAs were subcloned into the retroviral destination vectors MSCV-Gateway-neoEB and MSCV2.2-Gateway-IRESGFP as described.21 The MSCV2.2-Gateway-IRESGFP vector was used for retroviral transduction of bone marrow, and the MSCV-Gateway-neoEB vector was used for the transduction of Ba/F3 and 32D cell lines.
Cell culture and retrovirus production
Ba/F3 and 32D cells were maintained in RPMI 1640 media with 10% FCS and either interleukin-3 (IL-3) (0.5 ng/mL; R&D Systems, Minneapolis, MN) or WEHI-conditioned media as a source of IL-3. The 293T cells were maintained in Dulbecco modified eagle media (DMEM) with 10% FCS. Retroviral stocks were generated from transfection of 293T cells, and viral titers were determined as previously described.13 Cells were transduced with each of the respective MSCV retroviral constructs and were grown in the presence of IL-3 for 2 days. G418 selection was performed for an additional 14 days in the presence of IL-3. For cell growth assays of stable Ba/F3 and 32D cell lines, cells were washed twice in phosphate-buffered saline (PBS) and seeded at density of 1 × 105/mL in 24-well plates in RPMI with 10% FCS in the absence of IL-3. Cells were counted daily by trypan blue exclusion in a standard hemocytometer. Recombinant human FLT3 ligand was obtained from Peprotech (Rocky Hill, NJ).
In vitro mapping of tyrosine phosphorylation sites in FLT3
A cDNA encoding residues 580 to 993 of the cytoplasmic domain of wild-type human FLT3 was cloned into a pFastBac (Invitrogen, Carlsbad, CA) baculovirus expression vector. Production of FLT3 kinase protein in infected Sf9 insect cells was done according to the manufacturer's standard protocol. The FLT3 kinase protein was purified using Ni-NTA agarose. The purified FLT3 kinase domain was subjected to an in vitro autophosphorylation reaction followed by quenching the reaction with 30 mM EDTA. To maximize the recovery of peptides that account for the entire FLT3 protein sequence, aliquots of the in vitro kinase reaction were separately digested with trypsin and chymotrypsin at 37°C for 4 hours. After quenching the digestions by lowering the pH to about 2 to 3 with acetic acid, the proteolytic-digested samples were subsequently analyzed by liquid chromatography–coupled tandem MS (LC/MS/MS) using electrospray ionization on a Finnigan LCQ Deca XP ion trap mass spectrometer (Thermo, San Jose, CA) as described previously.22
Immunoprecipitation and Western analysis
The Ba/F3 and 32D cells were grown to a density of approximately 1 × 106/mL, collected by centrifugation, and washed in 10 mL PBS supplemented with 0.4 mM Na3VO4 at 4°C. Cells were then lysed in 1.0 mL lysis buffer: 20 mM Tris-HCl (pH 7.4), 1% Triton X-100, 0.5 mM EDTA, 150 mM NaCl, 1 mM Na3VO4, 25 mM NaF, and 20 μM phenylarsine oxide supplemented with Protease Arrest cocktail (GBiosciences, St Louis, MO). Lysates were incubated for 5 minutes at 4°C and were then cleared by centrifugation at 14 000g for 10 minutes at 4°C.
Fresh lysates were used for all immunoprecipitations. Immunoprecipitations were performed by incubating 1000 μg total cell lysate on a rocker at 4°C overnight with either polyclonal anti-FLT3 (Santa Cruz Biotechnology, Santa Cruz, CA) or anti-STAT5 (Santa Cruz Biotechnology). Immunoprecipitates were collected with protein A–Sepharose (Amersham Biotech, Piscataway, NJ). Immunoprecipitates were washed 3 times in lysis buffer and boiled for 5 minutes in sodium dodecyl sulfate (SDS) sample buffer.
Samples were separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred by electrophoresis to polyvinylidene difluoride (PVDF) membranes (Invitrogen). Membranes were blocked with 1% bovine serum albumin (Sigma Fraction V; Sigma Chemicals, St Louis, MO) in wash buffer (Tris-buffered saline and 0.1% Tween 20) and incubated overnight at 4°C with mouse monoclonal 4G10 (Upstate Biotechnology, Lake Placid, NY). Membranes were then washed and incubated with either HRP-conjugated anti–rabbit IgG or HRP-conjugated anti–mouse IgG (Amersham Biotech). Blots were rinsed and visualized by enhanced chemiluminescence. Western blot analysis for Pim-1 was conducted with a monoclonal antibody from Santa Cruz Biotechnology. Western blot analysis of phospho-p44/42 MAP kinase and p44/42 MAP kinase was conducted with antibodies from Cell Signaling (Beverly, MA).
Murine bone marrow transplant assay
BALB/c mice were purchased from Taconic (Germantown, NY). BM transplant assays were carried out as described previously.13 Briefly, 4- to 6-week-old male donor mice were primed with intraperitoneal injection of 5′-fluorouracil (150 mg/kg; Sigma Chemicals) and subsequently killed after 6 days by CO2 asphyxiation. Bone marrow was flushed from femurs and tibias, and red blood cells were lysed (Red Blood Cell Lysis; Gentra, Minneapolis, MN). Cells were cultured overnight with IL-3 (6 ng/mL; R&D Systems), IL-6 (10 ng/mL; R&D Systems), and stem cell factor (10 ng/mL; Peprotech) in RPMI with 10% FCS (transplant medium). Cells were transduced by 2 rounds of spin infection, at 24 hours and 48 hours after harvesting. Centrifugation of viral supernatant and 4 × 106 cells in 3 mL transplant media containing 5 μg/mL Polybrene and 7.5 mM HEPES buffer was carried out for 90 minutes at 1800g. Equivalent viral titers were used for each retroviral construct, and equal transduction efficiencies for various constructs in primary cells were confirmed by flow cytometric analysis for EGFP after transduction. Cells were washed in PBS, resuspended in Hank balanced salt solution, and injected (5 × 105 cells per 0.5 mL) into the lateral tail vein of lethally irradiated (2 × 450 cGy) female recipient mice. Mice were housed in microisolator cages with autoclaved chow and acidified water.
Histopathology
Murine tissues were fixed for at least 72 hours in 10% neutral buffered formalin (Sigma Chemicals), dehydrated in alcohol, cleared in xylene, and infiltrated with paraffin on an automated processor (Leica, Bannockburn, IL). The tissue sections (4 μm thick) from paraffin-embedded tissue blocks were placed on charged slides and deparaffinized in xylene, rehydrated through graded alcohol solutions, and stained with hematoxylin and eosin.
Histologic images were obtained on a Nikon Eclipse E400 microscope (Nikon, Tokyo, Japan) equipped with a SPOT RT color digital camera model 2.1.1 (Diagnostic Instruments, Sterling Heights, MI). The microscope was equipped with a 10×/22 ocular lens. Low-power images (×100) were obtained with a 10×/0.25 numeric aperture (NA) objective lens. High-power images (×600) were obtained with a 60×/1.4 NA objective lens with oil (Trak 300; Richard-Allan Scientific, Kalamazoo, MI). Images were analyzed in Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA).
Results
FLT3-ITD, but not FLT3WT, is a potent activator of STAT5
Because of disparities in the literature, we first compared the ability of FLT3-ITD (N51) and ligand-stimulated FLT3WT to activate STAT5. Analysis of STAT5 phosphorylation by immunoblot analysis in Ba/F3 lysates following 4 hours of IL-3 deprivation showed robust ligand-independent activation of STAT5 by FLT3-ITD. This phenomenon was not observed in FLT3WT-expressing Ba/F3 cells that were either unstimulated or were stimulated with FL (100 ng/mL) for 5, 10, 15, or 20 minutes (Figure 1A). FL stimulation of FLT3WT-expressing cells demonstrated FLT3 receptor phosphorylation (Figure 1B) and p44/42 MAP kinase phosphorylation (Figure 1C). No significant activation of p44/42 MAP kinase was observed in FLT3-ITD–expressing Ba/F3 cells (Figure 1C).
Figure 1.
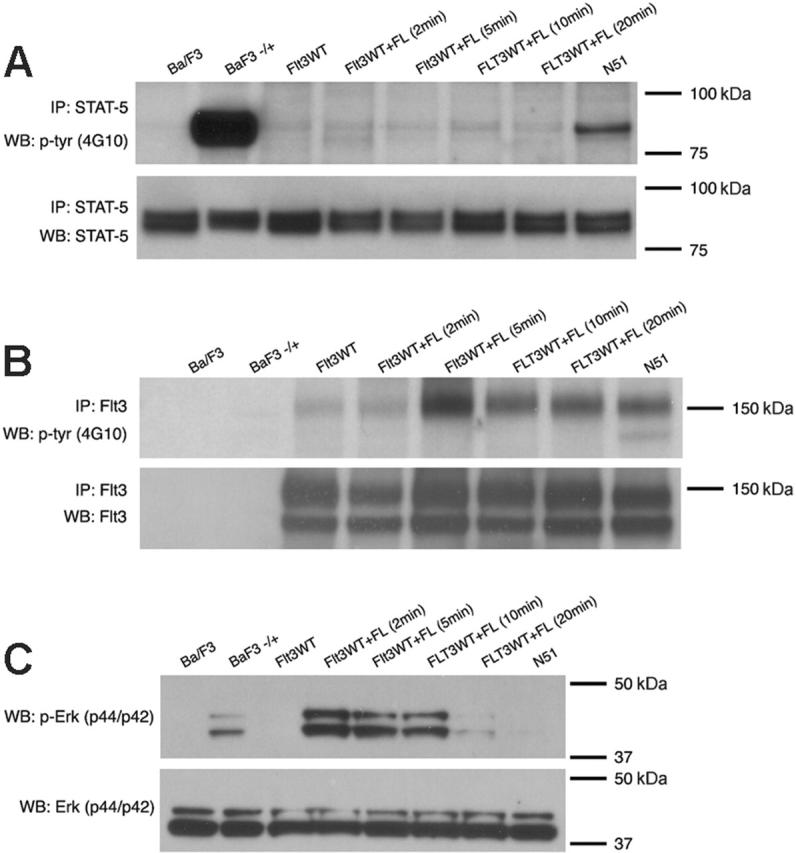
Differences in STAT5 activation by FLT3-ITD and ligand-stimulated Flt3WT. STAT5 (A) or FLT3 (B) activation was examined by immunoblot analysis of Ba/F3 cell lysates starved of IL-3 for 4 hours. For FL stimulation, starved cells were treated with 100 ng/mL FL and incubated for 2, 5, 10, or 20 minutes at 37°C. For stimulation with IL-3, starved cells were treated with 10 ng/mL IL-3 and incubated for 10 minutes at 37°C Total cell lysates were immunoprecipitated with an anti-STAT5 antibody or anti-FLT3 antibody and separated by SDS-PAGE. Immunoblot analysis was conducted with an antiphosphotyrosine (4G10) antibody (top rows). The blots were then stripped and reprobed with anti-STAT5 or anti-FLT3 antibody (bottom rows). The same lysates were also analyzed for p44/42 MAP kinase (Erk) activation (C) by immunoblotting with an anti–phospho-Erk antibody (top row) and anti-Erk antibody (bottom row).
In vitro mapping of FLT3 autophosphorylation sites
To determine the in vitro tyrosine phosphorylation sites of FLT3, a histidine-tagged cDNA construct encoding residues 580 to 993 of the cytoplasmic domain of wild-type human FLT3 was expressed in baculovirus, and recombinant protein was purified using Ni-NTA agarose. The purified FLT3 kinase domain was subjected to an in vitro autophosphorylation reaction, and Western blotting using a polyclonal antiphosphotyrosine antibody demonstrated the phosphorylation of the FLT3 protein; tryptic or chymotryptic peptides were then analyzed by mass spectrometry. Tryptic peptides covering more than 80% of the FLT3 kinase domain were recovered, and 5 tyrosine residues (Y591, Y726, Y842, Y955, and Y969) within this region were phosphorylated. In addition, 10 tyrosine sites were nonphosphorylated (Y589, Y597, Y599, Y768, Y793, Y865, Y889, Y899, Y913, Y919), and the phosphorylation status of the remaining 6 tyrosine residues was indeterminate (Y630, Y688, Y693, Y695, Y702, Y875). Representative MS data for tyrosines 591, 726, and 955 are shown in Figure S1 (available at the Blood website; see the Supplemental Figures link at the top of the online article).
To understand the role of these tyrosine phosphorylation sites on FLT3-ITD–mediated signaling and transformation, a series of single and multiple tyrosine to phenylalanine (Y→F) mutations were generated in FLT3-ITD (N51) (Figure 2A) and cloned into MSCV retroviral vectors containing a neomycin resistance gene. Following retroviral transduction, Ba/F3 and 32D cells were selected with G418 (1 mg/mL) in the presence of IL-3 for 2 weeks.
Figure 2.

FLT3-ITD–mediated STAT5 activation in cells expressing Tyr→Phe mutants. (A) A schematic diagram showing the 5 identified tyrosine-phosphorylated sites and their localization within the FLT3 receptor. The duplicated residues of the N51 internal tandem duplication are also shown. The various Tyr→Phe substitutions created in FLT3-ITD are indicated. ECD indicates extracellular domain; TM, transmembrane domain; TK1 and TK2, tyrosine kinase domains; KI, kinase insert; ITD, internal tandem duplication; and CT, C-terminal tail. STAT5 activation was examined by immunoblot analysis of Ba/F3 (B) or 32D (C) cell lysates starved of IL-3 for 4 hours. Total cell lysates were immunoprecipitated with an anti-STAT5 antibody and separated by SDS-PAGE. Immunoblot analysis was conducted with an antiphosphotyrosine (4G10) antibody (top rows). The blots were then stripped and reprobed with an anti-STAT5 antibody (bottom rows). (D) Pim-1 expression was examined by immunoblot analysis of 32D cell lysates starved of IL-3 for 4 hours. The blot was stripped and reprobed with anti-GAPDH antibody to show equivalent protein loading. Pim-1 is expressed as both a 33 and 44 kDa form in mice due to the presence of an upstream alternative translational start site.23
Activation of downstream signaling pathways by FLT3-ITD and related mutants
We next assessed the effect of these mutants on the ability of FLT3-ITD to activate STAT5 signal transduction in the Ba/F3 and 32D cells. Analysis of STAT5 phosphorylation in Ba/F3 and 32D cell lysates following 4 hours of IL-3 deprivation showed that the Y→F mutant at sites 589 and 591 of FLT3-ITD (Y589/591F) had a significantly weaker activation of STAT5 as compared with FLT3-ITD in both cell types (Figure 2B-C). Each of the additional single and multiple tyrosine site mutations that we tested displayed a similar level of ligand-independent STAT5 phosphorylation as compared with FLT3-ITD. The activation of the STAT3, Erk, or Akt pathways by FLT3-ITD was minimal when compared with IL-3–stimulated cells (Figure 1 and data not shown).
STAT5 regulates the expression of several immediate early cytokine response genes, including Pim-1, a serine/threonine kinase that potentiates cell growth and survival.23 A previous study has shown that constitutively activated FLT3 signaling up-regulates Pim-1 expression in leukemic cells.25 Consistent with these findings, we demonstrated that Pim-1 protein was expressed at a significantly greater level in 32D cells stably expressing FLT3-ITD as compared with FLT3WT-expressing cells. In addition, 32D cells that expressed the N51-Y589/591F mutant demonstrated less Pim-1 expression than FLT3-ITD–expressing cells (Figure 2D).
Effects of substitution of phenylalanine for tyrosine sites on the constitutive activation and IL-3–independent growth of FLT3-ITD
Stable cell lines expressing each of the FLT3-ITD Y→F mutations were examined for the constitutive tyrosine phosphorylation of FLT3. FLT3 immunoprecipitation, followed by Western blotting with an antiphosphotyrosine antibody, was performed on lysates from stable Ba/F3 and 32D cell lines that were starved of IL-3 for 4 hours. FLT3-ITD as well as each of the Y→F mutants demonstrated robust ligand-independent autophosphorylation of FLT3 (Figure 3), whereas FLT3WT was only phosphorylated at basal levels. In addition, Western blot analysis confirmed that each stable cell line contained comparable amounts of FLT3 protein—both the glycosylated (155 kDa) and nonglycosylated (130 kDa) isoforms.
Figure 3.

FLT3 activation in cells expressing FLT3-ITD Tyr→Phe mutants. Tyrosine phosphorylation was examined in Ba/F3 (A) or 32D (B) cell lysates that were starved of IL-3 for 4 hours. Cell lysates were immunoprecipitated with an anti-FLT3 antibody and subjected to SDS-PAGE separation. Immunoblot analysis was conducted with an antiphosphotyrosine (4G10) antibody (top rows). The blots were then stripped and reprobed with anti-FLT3 antibody (bottom rows).
Stable cell lines were next assessed for IL-3–independent growth. As described previously, FLT3-ITD but not FLT3WT conferred factor-independent growth to the cells. Each of the Y→F mutations tested were able to confer factor-independent growth to both Ba/F3 and 32D cells with a growth rate similar to that of FLT3-ITD (Figure 4). The 32D cell line expressing the Y589/591F mutation had a slower factor-independent growth rate as compared with FLT3-ITD alone; however, the cell number continued to increase when cells were counted up to 14 days following seeding (data not shown). Therefore, mutation of in vitro tyrosine phosphorylation sites did not have an appreciable effect on factor-independent cell growth or constitutive receptor activation mediated by FLT3-ITD.
Figure 4.

Growth rates of murine hematopoietic cells expressing FLT3-ITD Tyr→Phe mutants. Analysis of IL-3–independent growth in Ba/F3 (A) and 32D (B) cell lines stably transformed with each of the FLT3 mutants. Cells were washed twice in PBS, resuspended in RPMI and 10% FCS in the absence of IL-3, and seeded at a density of 1 × 105/mL. Viable cells were counted by trypan blue exclusion.
The role of tyrosines 589 and 591 in STAT5 activation by FLT3 tyrosine kinase domain mutants
Recent studies have suggested that the signaling differences between FLT3-ITD and FLT3 kinase domain mutants such as D835Y may account for the differing phenotypes observed when these mutations are examined in murine bone marrow transplant experiments. To examine the role of tyrosine residues 589 and 591 in D835Y-mediated signal transduction, we generated 32D cells that stably expressed D835Y and D835Y-Y589/591F. These stable cell lines were tested for ligand-independent FLT3 autophosphorylation and STAT5 activation. As reported by other groups, the D835Y mutant demonstrated significantly less STAT5 activation than FLT3-ITD in stably transformed 32D cells (Figure 5), and the level of STAT5 activation in the D835Y-expressing cells was similar to that of the cells expressing the N51-Y589/591F mutant. However, no further decrease in STAT5 signaling was observed in the cells expressing the D835Y-Y589/591F mutation as compared with D835Y alone. In addition, no significant differences were observed in FLT3 receptor activation of the D835Y-Y589/591F mutant.
Figure 5.

Role of tyrosines 589 and 591 in FLT3-D835Y–mediated FLT3 activation and STAT5 signaling. (A) STAT5 activation was examined in 32D cells stably expressing FLT3WT, FLT3-ITD (N51), N51-Y589/591F, D835Y, or D835Y-Y589/591F. Cells were starved of IL-3 for 4 hours. Cell lysates were immunoprecipitated with an anti-STAT5 antibody and separated by SDS-PAGE. Immunoblot analysis was conducted with an antiphosphotyrosine (4G10) antibody. The blot was then stripped and reprobed with an anti-STAT5 antibody. (B) FLT3 activation was also analyzed in the same cells by immunoprecipitation with anti-FLT3 antibody followed by immunoblot analysis with an antiphosphotyrosine antibody.
Analysis of the Y589/591F mutant on the development of FLT3-ITD–mediated myeloproliferative disease
Murine bone marrow transplant assays were performed to examine the effect of the Y589/591F mutant on the development of FLT3-ITD–induced myeloproliferative disease. Primary murine bone marrow cells were transduced with equivalent titers of MSCV-EGFP retrovirus expressing FLT3WT, FLT3-ITD, or FLT3-ITD–Y589/591F and injected into lethally irradiated syngeneic recipient mice. As observed previously, mice injected with FLT3-ITD–transduced bone marrow cells developed a myeloproliferative phenotype with a median latency of 40 to 60 days, whereas mice injected with FLT3WT-transduced bone marrow cells showed no evidence of disease with a follow-up of more than 240 days. Mice that received the Y589/591F-transduced bone marrow also did not show any signs of myeloproliferative disease and appeared grossly normal at 240 days after transplantation (Figure 6A). Mice that received transplants of FLT3-ITD developed leukocytosis with a white blood count (WBC) ranging from 14.7 × 109/L to 69.2 × 109/L (14.7 × 103/μL to 69.2 × 103/μL) and splenomegaly with spleen weights ranging from 363 to 610 mg. In comparison, mice that received transplants of Y589/591F had a relatively normal WBC, ranging from 1.9 × 109/L to 5.76 × 109/L (1.9 × 103/μL to 5.76 × 103/μL) and normal spleen weights of 60 to 90 mg (Table 1).
Figure 6.

Kaplan-Meier survival analysis and histopathology. (A) Mice receiving transplants of bone marrow transduced with FLT3WT, FLT3-ITD, or FLT3-ITD–Y589/591F. The percentage of surviving mice (y-axis) is plotted with respect to time in days (x-axis). The number of mice per group is indicated. (B) Bone marrow from a mouse that received FLT3-ITD–transduced bone marrow ([i] magnification, ×100; [ii] magnification, ×600) shows hypercellularity and myeloid hyperplasia. Bone marrow from a mouse that received N51-Y589/591F–transduced bone marrow ([iii] magnification, ×100; [iv] magnification, ×600) appears normal with open sinuses and fat spaces and a normal myeloid-erythroid ratio. Spleen sections from a mouse that received FLT3-ITD–transduced bone marrow ([v] magnification, ×100; [vi] magnification, ×600) demonstrate myeloid infiltration and effacement of splenic architecture by expansion of red pulp. Spleen sections from a mouse that received N51-Y589/591F–transduced bone marrow ([vii] magnification, ×100; [viii] magnification, ×600) show preserved areas of white pulp and no red pulp expansion. Hematoxylin and eosin was used in each panel.
Table 1.
Analysis of mice that received bone marrow transplants of FLT3WT, FLT3-ITD, or FLT3-ITD-Y589/591F
| Construct | No. of mice | Peripheral WBC, × 109/L (median) | Spleen weight, mg (median) |
|---|---|---|---|
| FLT3WT | 12 | 4.34-14.48 (7.28) | 59.1-120.8 (87.6) |
| N51 | 13 | 14.7-81.66 (39.8) | 363.8-800 (580.9) |
| 589/591 | 14 | 1.9-10.24 (6.74) | 60-107.8 (82.7) |
Histopathologic analysis of the organs from FLT3-ITD–transduced mice demonstrated effacement of splenic architecture with significant expansion of the red pulp as well as extensive myeloid infiltration in the bone marrow, spleen, lung, and liver. In contrast, analysis of the Y589/591F mice demonstrated normal splenic architecture with preserved areas of red and white pulp as well as normal bone marrow containing open sinuses and fat spaces and normal liver with no signs of extramedullary hematopoiesis (Figure 6B). These findings indicate that the tyrosine residues 589 and 591 in the FLT3 kinase domain are required for the development of FLT3-ITD–mediated myeloproliferative disease.
Discussion
FLT3 mutations are among the most common genetic alterations in AML. ITD mutations occur in the juxtamembrane autoregulatory domain, whereas activation loop mutations occur in the catalytic domain. The ITD mutations appear to be loss of function alleles in that they are highly variable in length, ranging from a few to more than 70 amino acids, but are always in frame. Structural data strongly support the hypothesis that ITD mutations result in loss of structure of an autoinhibitory domain that interacts directly with the catalytic domain and maintains the kinase in an inactive state in the absence of ligand. Loss of structure of the autoinhibitory domain thus results in constitutive kinase activation. In contrast, the activation loop point mutations are thought to activate the kinase by affecting the stability of the activation loop using a mechanism that does not involve the juxtamembrane domain.
We were intrigued by the observation that FLT3-ITD, but not FLT3WT, was a potent activator of the STAT5 pathway. We sought to identify the residues that were required for STAT5 activation in FLT3-ITD. In vitro mapping of sites identified Y591 as a phosphorylated site as well as a number of others. Y591 is an attractive candidate for interacting with the STAT5 SH2 domain in that the homologous Y591 and Y589 residues have been implicated in STAT5 binding in the context of other type III tyrosine kinases, such as PDGFR-β. Other studies have suggested that juxtamembrane tyrosine residues may play a role in FLT3-ITD–mediated STAT5 activation; however, these studies have not conclusively determined a role for any specific tyrosine(s) required for the mediation of STAT5 activation. One study observed a decrease in STAT5 activation with tyrosine to phenylalanine mutation of one copy of tyrosine 589 and 591 when using an FLT3-ITD with a duplication in all of the juxtamembrane tyrosine residues.16 However, a study using a deletion mutant encompassing residues 589 to 599 of FLT3 showed no effect on the ITD-mediated STAT5 activation.19 These differing observations may be the result of different ITD constructs or cell culture systems that were analyzed.
The juxtamembrane tyrosine residues analogous to 589 and 591 in other members of related receptor tyrosine kinases have also been examined for their role in kinase activation. In PDGFR-β, the tyrosine to phenylalanine mutations of Y579 and Y581 resulted in a significant decrease in kinase activity to varying degrees dependent on cell type.26-29 In addition, phenylalanine substitutions of the corresponding juxtamembrane tyrosines in c-kit30 and CSF-1R31 also impaired kinase activity. In contrast, however, the corresponding substitutions in the context of TEL–PDGFR-β displayed no apparent decrease in kinase activity.32
Y589 and Y591 were also attractive candidate residues that might account for the signal transduction differences between FLT3-ITD and FLT3WT in that they are localized to the juxtamembrane domain that harbors the ITD mutation. Determination of the crystal structure of FLT3WT has indicated that the function of the JM switch motif (JM-S) is to provide a rigid and properly oriented framework for the interposition of tyrosines 589 and 591 between the JM-S and the C lobe of the kinase.12 These finding suggest that it may be possible that ITDs within the JM domain of FLT3 result in the loss of structure leading to constitutive activation of the kinase and exposure of Y589 and Y591 via release from the JM-S interface with the C loop to enable phosphorylation and engagement of STAT5.
Based on in vitro mass spectrometric determination of FLT3 tyrosine phosphorylation sites, we generated and tested a series of Y→F mutations alone and in various combinations. We only observed a robust effect on signal transduction with the Y589/591F mutants in the cell lines tested. There are several explanations for a lack of effect of the other substitutions, including the possibility that in vitro phosphorylation sites are different than in vivo phosphorylation sites. Additional mutational analysis will be required to assess the sites of engagement of other signal transduction pathways known to be activated by FLT3-ITD and FLT3WT. The mutant FLT3-ITD–Y589/591F does not activate STAT5 or its downstream effector Pim-1, which supports the role of these residues in STAT5 activation. Interestingly, the Y589F mutation alone demonstrated no significant difference in STAT-5 phosphorylation compared with FLT3-ITD (Figure S2). This finding suggests that the combination of tyrosine residues 589 and 591 is required for activation of STAT-5 signaling pathways.
To further understand the role of the ITD in generating this neomorphic function of STAT5 activation, we also tested the Y589/591F mutations in the context of FLT3 D835Y that activates FLT3 by a different mechanism. We observed that there was a lower level of STAT5 activation in the FLT3 D835Y, and this low level was not further reduced by the incorporation of Y589/591F. These findings indicate that FLT3 D835Y is qualitatively similar to FLT3WT in the ability to activate STAT5 and provide support for the hypothesis that the ITD mutation is responsible for enhanced STAT5 activation in the FLT3-ITD mutant.
What is the physiologic significance of these findings? There were modest effects of the FLT3-ITD–Y589/591F mutations in factor-dependent cells lines Ba/F3 and 32D that were still able to confer factor-independent growth. However, these are culture cell lines that certainly have additional mutations that might complement the FLT3-ITD–Y589/591F to engender growth factor–independent growth. As a more rigorous test for the role of STAT5 activation, we assessed the effect of the mutation in primary hematopoietic cells using a murine bone marrow transplant model. We observed that these residues were essential for the generation of a myeloproliferative disease. These findings suggest the intriguing possibility that the ITD simultaneously activates the FLT3 kinase and results in a neomorphic activation of STAT5 that is required for full transforming potential.
These observations also correlate with certain differences in the phenotypic manifestations of FLT3-ITD compared with activation loop alleles. In the murine model system, the FLT3-ITD causes a relatively rapid onset myeloproliferative disease, whereas the FLT3 D835Y mutant causes a long-latency lymphoid malignancy.33 In addition, although both types of mutations may be associated with myeloid malignancies, FLT3-ITDs are only very rarely identified in lymphoid malignancies, whereas FLT3 activation loop mutations are more common. There is a possibility that the mice receiving transplants with the Y589/591F-transduced bone marrow could have developed a very long–latency lymphoid disease with an onset of more that 240 days; however, the examples of lymphoid disease mediated by FLT3 tyrosine kinase domain mutants were observed within 177 days. These data suggest that FLT3-ITD–mediated activation of STAT5 may potentiate myeloid cell fate determination, whereas FLT3 activation loop mutants or activation of FLT3 by overexpression (as in mixed lineage leukemia [MLL]–rearranged leukemias of infancy) may favor development of a lymphoid lineage disease. These possibilities can be tested in part by retroviral transduction of purified progenitor populations or by generating knock-in alleles of the FLT3-ITD versus FLT3 D835Y.
FLT3 is not sufficient to cause leukemia alone, and it is most likely that additional cooperating mutations are required in addition to FLT3 mutations for the development of AML. FLT3 mutations confer a proliferative advantage to hematopoietic cells and frequently occur with genetic alterations that impair hematopoietic differentiation in leukemic patients, including AML1/ETO, CBFβ/SMMHC, PML/RARα, and MLL internal tandem repeat mutations.11 However, it is unknown which (if any) specific FLT3-ITD–mediated signaling pathways are required for the cooperative effects seen in the development of AML. It will be of interest to determine whether STAT-5 signaling mediated by tyrosine residues 589 and 591 is required for the cooperative effects of FLT3 with other chromosomal translocations that lead to the development of AML.
In conclusion, we have identified specific tyrosine residues that are required for the activation of STAT-5 signaling by internal tandem duplications of FLT3 and have demonstrated that this correlates with the FLT3-ITD phenotype observed in vivo. Our data provide a means for further analysis of FLT3 cooperativity in the development of AML as well as provide a potential target for therapeutics.
Supplementary Material
Prepublished online as Blood First Edition Paper, April 25, 2006; DOI 10.1182/blood-2005-11-011429.
Supported in part by National Institutes of Health (NIH) grants CA66996, DK50654, and UO1 CA04002 (D.G.G.) as well as the Leukemia and Lymphoma Society. D.G.G. is a Doris Duke Distinguished Clinical Investigator and an Investigator of the Howard Hughes Medical Institute.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Rosnet O, Marchetto S, deLapeyriere O, Birnbaum D. Murine Flt3, a gene encoding a novel tyrosine kinase receptor of the PDGFR/CSF1R family. Oncogene. 1991;6: 1641-1650. [PubMed] [Google Scholar]
- 2.Matthews W, Jordan CT, Gavin M, Jenkins NA, Copeland NG, Lemischka IR. A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to c-kit. Proc Natl Acad Sci U S A. 1991;88: 9026-9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosnet O, Birnbaum D. Hematopoietic receptors of class III receptor-type tyrosine kinases. Crit Rev Oncog. 1993;4: 595-613. [PubMed] [Google Scholar]
- 4.Hannum C, Culpepper J, Campbell D, et al. Ligand for FLT3/FLK2 receptor tyrosine kinase regulates growth of haematopoietic stem cells and is encoded by variant RNAs. Nature. 1994; 368: 643-648. [DOI] [PubMed] [Google Scholar]
- 5.Lyman SD, Jacobsen SE. c-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood. 1998;91: 1101-1134. [PubMed] [Google Scholar]
- 6.McKenna HJ, Stocking KL, Miller RE, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95: 3489-3497. [PubMed] [Google Scholar]
- 7.Muench MO, Roncarolo MG, Menon S, et al. FLK-2/FLT-3 ligand regulates the growth of early myeloid progenitors isolated from human fetal liver. Blood. 1995;85: 963-972. [PubMed] [Google Scholar]
- 8.Zhang S, Fukuda S, Lee Y, et al. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J Exp Med. 2000;192: 719-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S, Broxmeyer HE. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem Biophys Res Commun. 2000;277: 195-199. [DOI] [PubMed] [Google Scholar]
- 10.Lavagna-Sevenier C, Marchetto S, Birnbaum D, Rosnet O. FLT3 signaling in hematopoietic cells involves CBL, SHC and an unknown P115 as prominent tyrosine-phosphorylated substrates. Leukemia. 1998;12: 301-310. [DOI] [PubMed] [Google Scholar]
- 11.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100: 1532-1542. [DOI] [PubMed] [Google Scholar]
- 12.Griffith J, Black J, Faerman C, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13: 169-178. [DOI] [PubMed] [Google Scholar]
- 13.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99: 310-318. [DOI] [PubMed] [Google Scholar]
- 14.Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96: 3907-3914. [PubMed] [Google Scholar]
- 15.Spiekermann K, Bagrintseva K, Schwab R, Schmieja K, Hiddemann W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin Cancer Res. 2003;9: 2140-2150. [PubMed] [Google Scholar]
- 16.Murata K, Kumagai H, Kawashima T, et al. Selective cytotoxic mechanism of GTP-14564, a novel tyrosine kinase inhibitor in leukemia cells expressing a constitutively active Fms-like tyrosine kinase 3 (FLT3). J Biol Chem. 2003;278: 32892-32898. [DOI] [PubMed] [Google Scholar]
- 17.Hayakawa F, Towatari M, Kiyoi H, et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene. 2000;19: 624-631. [DOI] [PubMed] [Google Scholar]
- 18.Choudhary C, Schwable J, Brandts C, et al. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood. 2005;106: 265-273. [DOI] [PubMed] [Google Scholar]
- 19.Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. 2002;21: 2555-2563. [DOI] [PubMed] [Google Scholar]
- 20.Minami Y, Yamamoto K, Kiyoi H, Ueda R, Saito H, Naoe T. Different antiapoptotic pathways between wild-type and mutated FLT3: insights into therapeutic targets in leukemia. Blood. 2003;102: 2969-2975. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Williams IR, Lee BH, et al. Constitutively activated FGFR3 mutants signal through PLCgamma-dependent and -independent pathways for hematopoietic transformation. Blood. 2005;106: 328-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livingstone M, Ruan H, Weiner J, et al. Valosin-containing protein phosphorylation at Ser784 in response to DNA damage. Cancer Res. 2005;65: 7533-7540. [DOI] [PubMed] [Google Scholar]
- 23.Lilly M, Kraft A. Enforced expression of the Mr 33,000 Pim-1 kinase enhances factor-independent survival and inhibits apoptosis in murine myeloid cells. Cancer Res. 1997;57: 5348-5355. [PubMed] [Google Scholar]
- 24.Saris CJ, Domen J, Berns A. The pim-1 oncogene encodes two related protein-serine/threonine kinases by alternative initiation at AUG and CUG. EMBO J. 1991;10: 655-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim KT, Baird K, Ahn JY, et al. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005; 105: 1759-1767. [DOI] [PubMed] [Google Scholar]
- 26.Mori S, Ronnstrand L, Yokote K, et al. Identification of two juxtamembrane autophosphorylation sites in the PDGF beta-receptor; involvement in the interaction with Src family tyrosine kinases. EMBO J. 1993;12: 2257-2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baxter RM, Secrist JP, Vaillancourt RR, Kazlauskas A. Full activation of the platelet-derived growth factor beta-receptor kinase involves multiple events. J Biol Chem. 1998;273: 17050-17055. [DOI] [PubMed] [Google Scholar]
- 28.Vaillancourt RR, Heasley LE, Zamarripa J, et al. Mitogen-activated protein kinase activation is insufficient for growth factor receptor-mediated PC12 cell differentiation. Mol Cell Biol. 1995;15: 3644-3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drummond-Barbosa DA, Vaillancourt RR, Kazlauskas A, DiMaio D. Ligand-independent activation of the platelet-derived growth factor beta receptor: requirements for bovine papilloma-virus E5-induced mitogenic signaling. Mol Cell Biol. 1995;15: 2570-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimura Y, Jones N, Kluppel M, et al. Targeted mutations of the juxtamembrane tyrosines in the Kit receptor tyrosine kinase selectively affect multiple cell lineages. Proc Natl Acad Sci U S A. 2004; 101: 6015-6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rohde CM, Schrum J, Lee AW. A juxtamembrane tyrosine in the colony stimulating factor-1 receptor regulates ligand-induced Src association, receptor kinase function, and down-regulation. J Biol Chem. 2004;279: 43448-43461. [DOI] [PubMed] [Google Scholar]
- 32.Sternberg DW, Tomasson MH, Carroll M, et al. The TEL/PDGFbetaR fusion in chronic myelomonocytic leukemia signals through STAT5-dependent and STAT5-independent pathways. Blood. 2001;98: 3390-3397. [DOI] [PubMed] [Google Scholar]
- 33.Grundler R, Miething C, Thiede C, Peschel C, Duyster J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood. 2005; 105: 4792-4799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}