

Abstract
Bullous pemphigoid (BP) is an autoimmune disease associated with autoantibodies directed against the hemidesmosomal antigens anti-BP230 and anti-B180. Neonatal mice injected with rabbit anti-mouse BP180 (mBP10) IgG develop a BP-like disease. Complement, immune complexes, mast cells, and neutrophils play a key role in subepidermal blistering in this animal model. In this study we investigated the role of β2 integrins in experimental BP. Wild-type (WT) mice pretreated with neutralizing antibody against CD11a (LFA-1), CD11b (Mac-1), CD11a plus CD11b, or CD18 alone failed to develop BP when injected with pathogenic anti-mBP180 IgG. This was associated with a significant reduction in neutrophil accumulation in neutralizing antibody-treated mice. Mac-1-deficient (Mac-1 knockout [KO]) mice were resistant to experimental BP despite normal complement deposition and mast cell and neutrophil degranulation. Neutrophil infiltration in Mac-1 KO mice was severely impaired at 24 hours. However, more neutrophils accumulated in the skin of Mac-1 KO mice compared with WT mice at early time points (2-4 hours), which was associated with an increase in their survival as determined by apoptosis markers. These data suggest that β2 integrins play differential roles in experimental BP: LFA-1 is required for neutrophil recruitment, while Mac-1 mediates late neutrophil accumulation and apoptosis of infiltrating neutrophils.
Introduction
Bullous pemphigoid (BP) is a subepidermal blistering skin disorder of the elderly.1 Lesional skin of patients with BP is characterized by a detachment of the basal keratinocytes of the epidermis from the dermis resulting in tense, fluid-filled vesicles. Immunofluorescence (IF) studies demonstrated that these patients exhibit circulating and tissue-bound autoantibodies directed against antigens of the basement membrane zone (BMZ).2-7
BP autoantibodies recognize 2 antigens, BP230 and BP180, which are localized in the hemidesmosome, the main epidermal structure involved in dermal-epidermal adhesion.5-13 The BP230 antigen is restricted to the cytoplasmic plaque of the hemidesmosome and appears to interact directly with the intermediate filament network.8,9 The BP180 antigen is a type II transmembrane protein that spans the lamina lucida, projecting into the lamina densa of the basement membrane.10-16 The ectodomain of human BP180 harbors major extracellular antigenic sites recognized by sera from patients with BP.17,18 Administration of rabbit antibody to mBP180 in neonatal mice results in the binding of pathogenic antibody to its target on basal keratinocytes, resulting in neutrophil accumulation, complement deposition at the BMZ junction, and subepidermal blistering reminiscent of human BP lesions.19-25 The early phase of neutrophil infiltration is dependent on complement activation leading to mast cell degranulation.20,21 Upon the initial neutrophil-mediated tissue injury, skin inflammation is amplified by the recruitment of additional neutrophils, resulting in clinical blistering.25 This later (amplification) stage of neutrophil infiltration is mediated through local tissue damage of the basement membrane by released neutrophil elastase (NE) and matrix metalloproteinase-9 (MMP-9) by activated neutrophils.23-25 It is noteworthy that a threshold of neutrophil accumulation is required for clinical blistering with only a 30% reduction in neutrophil influx resulting in the inhibition of subepidermal blisters.21 Thus, the tissue burden of infiltrating neutrophils and their cytotoxic products are key predictors of disease progression in this model.
Neutrophil accumulation in tissues is a balance of neutrophil recruitment and neutrophil clearance. Recruitment of inflammatory cells requires firm integrin-mediated adhesion and diapedesis.26,27 The β2 integrin family members LFA-1 (CD11a/CD18), Mac-1 (CD11b/CD18), and p150,95 (CD11c/CD18) are expressed on neutrophils and have diverse functions, including leukocyte recruitment to sites of inflammation, host defense, and immune-mediated tissue injury.28,29 Neutrophil clearance occurs upon neutrophil apoptosis followed by engulfment of the apoptotic cells via tissue macrophages. A deficiency in Mac-1 alone results in increased neutrophil accumulation at inflamed sites that is associated with enhanced survival of extravasated neutrophils.30 Finally, Mac-1, also known as complement receptor 3 (CR3), is a major receptor for complement C3bi, and its interaction with this ligand may be important for promoting neutrophil cytotoxic functions.31 In this study, we investigated the role of β2 integrins and their relative contributions to neutrophil accumulation and disease development in experimental BP.
Materials and methods
Laboratory animals
Breeding pairs of wild-type (WT) C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). C57BL/6J Mac-1-deficient (Mac-1 knockout [KO]) mice were generated using standard gene-targeting approaches with a targeting vector in which the 64-bp exon encoding the translational initiation codon was replaced with a 1.7-kb neomycin gene cassette driven by a phosphoglycerate kinase (PGK) promoter. Homozygous animals were shown to be negative for Mac-1 expression on peripheral blood neutrophils and peritoneal macrophages, thus confirming knockdown of Mac-1 protein levels in these animals.30 The mice were maintained at the University of North Carolina-Chapel Hill Animal Center. Neonatal mice (24 to 36 hours old, with body weights between 1.4 and 1.6 g) were used for passive transfer experiments. Animal care and animal experiments were in accordance with the Animal Care Committee at the University of North Carolina at Chapel Hill and NIH guidelines.
Preparation of pathogenic anti-BP180 IgG
The preparation of recombinant murine BP180 and the immunization of rabbits were performed as previously described.19 Briefly, a segment of the ectodomain of the murine BP180 antigen was expressed as a glutathione S-transferase (GST) fusion protein using the pGEX prokaryotic expression system (Pharmacia LKB Biotechnology, Piscataway, NJ).32 The murine BP180 fusion protein, designated GST-mBP180ABC, was purified to homogeneity by affinity chromatography.33 New Zealand White rabbits were immunized with the purified mBP180 fusion protein and the IgG fraction from the serum (designated R530) was purified as previously described.19 The IgG fractions were concentrated, sterilized by ultrafiltration, and the protein concentrations determined by OD280 [E (1%, 1 cm) = 13.6]. The titers of anti-murine BP180 antibodies in both the unfractionated rabbit serum and in the purified IgG fraction were assayed by indirect IF using mouse skin cryosections as the substrate. These antibody preparations were also tested by immunoblotting against the GST-mBP180ABC fusion protein. The IF and immunoblotting techniques have been reported elsewhere.19 The pathogenicity of these IgG preparations was tested by passive transfer experiments as described below.
Induction of experimental BP and clinical evaluation of animals
Neonates were given on the back 1 intradermal (ID) injection of a sterile solution of IgG in phosphate-buffered saline (PBS; 50 μL IgG, 2.64 mg IgG/g body weight) as described elsewhere.19 The skin at the IgG injection site of the mice from the test and control groups was examined at different time points after the IgG injection. The extent of cutaneous disease was scored as follows: - indicates no detectable skin disease; 1+, mild erythematous reaction with no evidence of the epidermal detachment sign (this sign was elicited by gentle friction of the mouse skin which, when positive, produced fine, persistent wrinkling of the epidermis); 2+, intense erythema and epidermal detachment sign involving 10% to 50% of the epidermis in localized areas; and 3+, intense erythema with frank epidermal detachment sign involving more than 50% of the epidermis in the injection site.
After clinical examination, the animals were killed, and skin and serum specimens were obtained. Skin sections were used for routine histologic examination by light microscopy (hematoxylin and eosin [H/E] staining) to localize the lesional site, and neutrophil infiltration and toluidine blue staining were used to quantify mast cells and mast cell degranulation. Direct IF of skin sections were performed to detect rabbit IgG and mouse C3 deposition at the BMZ. Skin protein extracts were analyzed by myeloperoxidase (MPO) enzymatic assay to quantify the neutrophil accumulation at the skin injection site described in the next section. The sera of injected animals were tested by indirect IF techniques to determine the titers of rabbit anti-murine BP180 antibodies. Direct and indirect IF studies were performed as previously described using commercially available FITC-conjugated goat anti-rabbit IgG (Kirkegard & Perry Laboratories, Gaithersburg, MD).19 Monospecific goat anti-mouse C3 IgG was purchased from Cappel Laboratories (Durham, NC).
IF slides were mounted in Citifluor Mounting Medium (Ted Pella, San Jose, CA) and were examined by Leitz Orthoplan fluorescence microscopy (Enrst Leitz Canada, Midland, ON, Canada) using a 20×/0.5 NA objective. H/E slides were mounted in Cytoseal XYL Mounting Medium (Richard-Allan Scientific, Kalamazoo, MI) and were examined by Leica DMLB light microscopy (McBain Instruments, Chatsworth, CA) using a 20×/0.5 NA objective. Images were captured using a 1.1.0 Spot CCD microscope camera (Diagnostic Instruments, Sterling Heights, MI) and processed using Photoshop (Adobe Systems, San Jose, CA).
Quantification of PMN accumulation at antibody injection sites
Tissue MPO activity was used as an indicator of polymorphonuclear neutrophils (PMNs) within skin samples of experimental animals, as described elsewhere.34 We previously showed that clinical skin blistering is directly correlated with the number of infiltrating neutrophils in the IgG injection site.22 A standard reference curve was first established by obtaining activity levels on aliquots of known amounts of purified MPO. The mouse skin samples were extracted by homogenization in extraction buffer containing 0.1 M Tris-Cl (pH 7.6), 0.15 M NaCl, and 0.5% hexadecyltrimethylammonium bromide. MPO activity levels in supernatant fractions were determined by the change in optical density at 460 nm resulting from decomposition of H2O2 in the presence of o-dianisidine. MPO content was expressed as relative MPO activity (OD460nm/mg protein). Protein concentrations were determined by the Bio-Rad dye-binding assay (Hercules, CA) using bovine serum albumin (BSA) as a standard.
Quantification of mast cells and mast cell degranulation
Mast cells (MCs) and MC degranulation in skin samples were quantified according to Wershil et al, with modification.21,35 Briefly, lesional and nonlesional skin sections of IgG-injected mice were fixed in 10% formalin. Paraffin sections (5-μm thick) were prepared and stained with toluidine blue and H/E. Toluidine blue-stained slides were mounted in Cytoseal XYL Mounting Medium (Richard-Allan Scientific) and were examined with a Leica DMLB light microscope (McBain Instruments) using a 20×/__NA objective. Images were captured using a 3.2.0 Spot digital camera (Diagnostic Instruments) and processed using Photoshop (Adobe Systems). Total number of MCs was counted and classified as degranulated (> 10% of the granules exhibiting fusion or discharge) or normal in 5 fields under a light microscope as described previously. The results were expressed as percentage of MC degranulating.
PMN isolation
Mouse PMNs were isolated from heparinized blood by dextran sedimentation followed by separation on a density gradient as described.36 Red blood cells were removed from the cell preparation by hypotonic lysis in 0.2% NaCl. PMNs were washed and resuspended in cold PBS/10 mM glucose, counted in a hemocytometer, and adjusted to a concentration of 1 × 107 cells/mL. PMN purity of the final cell preparation was consistently greater than 96% as determined by cell-cytospin and LeukoStat staining (Fisher Diagnostics, Middletown, VA). The viability of the neutrophils was greater than 96% as determined by trypan blue exclusion.
Quantification of NE and MMP-9 in vitro and in vivo
To quantify neutrophil activation in vitro, purified neutrophils from WT or Mac-1 KO mice were incubated with purified rabbit anti-mBP180 IgG in the presence of purified mBP180 antigen at 37°C for 30 minutes.37 NE and MMP-9 in the supernatant were quantified by NE activity assay and MMP-9 colorimetric assay.37 NE activity was measured using the NE-specific substrate methoxysuccinyl-Ala-Ala-Pro-Val-P-nitroanalide (Met-O-Suc-Ala-Ala-Pro-Val-pNA; Enzyme Systems Products, Dublin, CA) as described previously.24 The NE activity was expressed as relative NE activity (OD420nm/min/mg protein). Levels of MMP-9 were measured with a MMP colorimetric assay kit following manufacturer's instructions (BIOMOL Research Laboratories, Plymouth Meeting, PA) with minor modification.36 Briefly, supernatant samples were treated with AMPA to activate MMP-9 and then incubated with the MMP colorimetric substrate Ac-PLG[2-mercapto-4-methyl]-LG-OC2H5 in reaction buffer (final substrate concentration = 100 μM) at 37°C. MMP activity in protein extracts was measured by the change in optical density at 412 nm and was expressed as relative MMP activity (OD412nm reading/min/mg protein of mouse skin injected with pathogenic IgG - OD412nm reading/min/mg protein of mouse skin injected with normal control IgG).
To quantify infiltrating neutrophil activation at the IgG injection site, 100:1 of PBS was injected into the skin of WT and Mac-1 KO mice at 4 and 24 hours after pathogenic IgG injection and withdrawn 1 minute later. The “washout” PBS was centrifuged at low speed (1000g) for 5 minutes to remove cells and then at high speed (12000g) for 5 minutes to remove cell debris.24 The supernatant was analyzed by MMP-9 and NE enzyme assays as given earlier in this section.
Intradermal injection of PMNs
PMNs were purified from WT and Mac-1 KO mice. Mac-1 KO mice were injected intradermally with pathogenic anti-mBP180 IgG (2.64 mg/g body weight/50 μL PBS). Two hours later, these mice were injected with 5 × 105 PMNs intradermally (in 50 μL PBS/10 mM glucose) at the same site.21 The animals were analyzed 24 hours after the IgG injections, as described in “Induction of experimental BP and clinical evaluation of animals.”
Immunostaining of active caspase 3
Apoptotic neutrophils in the skin of IgG-injected mice were identified by indirect IF with antibody directed against the active form of caspase 3 as described with some modification.38 Briefly, mouse skin biopsies were fixed in 10% PBS-buffered formalin overnight and embedded in paraffin. The cut skin sections (6-μm thick) were stained by routine H/E and total neutrophils were counted under light microscope. To quantify apoptotic neutrophils, the paraffin-embedded skin sections were deparaffinized and hydrated by series of xylene and ethanol, followed by boiling for 10 minutes in 10 mM sodium citrate buffer (pH 6.0) for antigen unmasking. The unmasked skin sections were incubated with rabbit anticleaved caspase-3 monoclonal antibody at 1:100 dilution (Cell Signaling Technology, Beverly, MA) in Tris-buffered saline (TBS) containing 5% BSA overnight at 4°C, followed by incubation with FITC-conjugated secondary goat anti-rabbit antibody at 1:100 dilution (Jackson ImmunoReseach Lab, West Grove, PA) for 1 hour at room temperature. The apoptotic neutrophils were visualized under a fluorescent microscope. Neutrophil apoptosis was expressed as relative neutrophil apoptosis (% apoptotic neutrophils/field).
Statistical analysis
The data were expressed as mean plus or minus SEM and were analyzed using Student t test. A P value less than .05 was considered significant.
Results
Inhibition of β2 integrins abolishes experimental BP
To determine whether β2 integrins are involved in subepidermal blister formation in experimental BP, neonatal C57BL/6J mice were pretreated with PBS or neutralizing antibodies against different β2 integrin subunits, were injected with control IgG or pathogenic anti-mBP180 IgG R530 2 hours afterward, and then analyzed 24 hours later. Mice receiving R530 alone developed significant blistering (Figure 1Aii; Table 1). Mice injected with control IgG (Figure 1Ai), R530 plus anti-CD11a neutralizing antibody (Figure 1Aiii), R530 plus anti-CD11b neutralizing antibody (Figure 1Aiv), R530 plus both anti-CD11a and anti-CD11b neutralizing antibodies (Figure 1Av), or R530 plus anti-CD18 neutralizing antibody (Figure 1Avi) showed no skin lesions. MPO activity assay of skin protein extracts at the injection site revealed that β2 integrin-neutralizing antibody treatments significantly inhibited neutrophil accumulation induced by pathogenic anti-mBP180 IgG (Figure 1B). These results demonstrated that neutrophil accumulation and subsequent subepidermal blistering require the β2 integrins LFA-1 and Mac-1.
Figure 1.
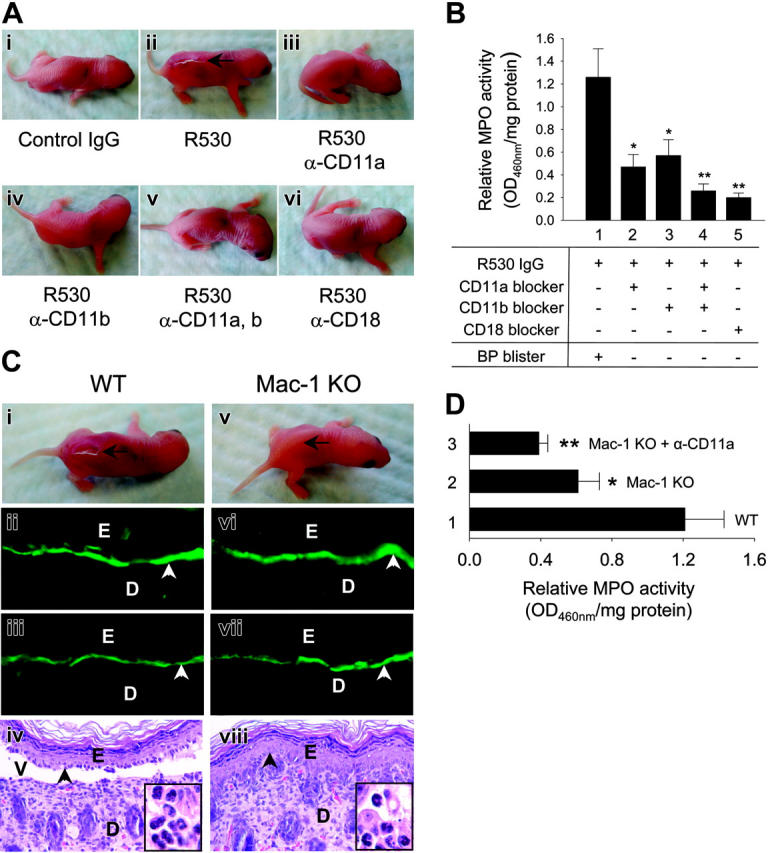
LFA-1 and Mac-1 are involved in experimental BP. WT and Mac-1 KO mice were pretreated with PBS or β2 integrin-neutralizing antibody and 2 hours later were injected intradermally with control or pathogenic IgG R530. The mice were examined 24 hours after R530 injection. (A) WT mice injected with R530 (ii), but not injected with control (i) or pretreated with antibody against CD11a (iii), CD11b (iv), both CD11a and CD11b (v), or CD18 (vi) showed no skin lesions. (B) MPO assay showed significantly higher levels of neutrophil infiltration in WT mice (bar 1) than WT mice pretreated with antibody against CD11a (bar 2), CD11b (bar 3), both CD11a and CD11b (bar 4), or CD18 (bar 5). Data shown are the mean ± SEM. n = 8 for each group. (C) R530-injected WT mice developed skin blisters and showed linear deposition of rabbit IgG (ii) and mouse C3 (iii) at the BMZ by direct IF. H&E-stained section from these mice revealed a subepidermal vesicle with neutrophilic infiltrate (iv, inset). In contrast, R530-injected Mac-1 KO mice showed no clinical (v) nor histological (viii) blisters, although direct IF showed rabbit IgG and mouse C3 deposition at the BMZ (vi-vii). Position of the skin lesion (arrow), site of basal keratinocytes (arrowhead), dermis (D), epidermis (E), vesicle (V) (original magnification, × 200). Higher magnifications of H&E-stained sections exhibit infiltrating neutrophils in the dermis (insets), original magnification, × 920. (D) MPO assay revealed a significant decrease in neutrophil accumulation in Mac-1 KO (bar 2) and Mac-1 KO mice pretreated with anti-CD11a antibody (bar 3) compared with WT mice (bar 1). Data shown are the mean ± SEM. n = 9. *P < .01; **P < .001. P < .05 (bar 2 vs bar 3).
Table 1.
The role of the β2 integrins in experimental BP
| IgG-injected | Treatment | No. mice | Mean disease activity* |
|---|---|---|---|
| Wild-type | |||
| R50 | – | 8 | 0.00 ± 0.00 |
| R530 | – | 22 | 2.59 ± 0.10 |
| R530 | Anti-CD11a | 12 | 0.08 ± 0.06 |
| R530 | Anti-CD11b | 8 | 0.25 ± 0.09 |
| R530 | Anti-CD11a + CD11b | 8 | 0.06 ± 0.06 |
| R530 | Anti-CD18 | 8 | 0.12 ± 0.08 |
| Mac-1 KO | |||
| R50 | – | 8 | 0.00 ± 0.00 |
| R530 | – | 16 | 0.00 ± 0.00 |
| R530 | 5 × 105 WT PMNs | 6 | 3.00 ± 0.00 |
| R530 | 5 × 105 Mac-1 KO PMNs | 6 | 3.00 ± 0.00 |
Neonatal WT and Mac-1-deficient (Mac-1 KO) mice were pretreated with isotype control IgG, anti-CD11a, anti-CD11b, anti-CD11a plus anti-CD11b, or anti-CD18. Two hours later, the mice were injected intradermally with either control rabbit IgG R50 or pathogenic rabbit anti-mBP180 IgG R530. For PMN reconstitution, Mac-1 KO mice were injected intradermally with R530 and 2 hours later locally reconstituted with 5 × 105 PMNs from Wt or Mac-1 KO mice. The animals were examined 24 hours after the pathogenic IgG injection.
The extent of cutaneous disease was scored as follows:–indicates no detectable skin disease; 1+, mild erythematous reaction with no evidence of the “epidermal detachment sign” `this sign was elicited by gentle friction of the mouse skin which, when positive, produced fine, persistent wrinkling of the epidermis'; 2+, intense erythema and epidermal detachment sign involving 10% to 50% of the epidermis in localized areas; and 3+, intense erythema with frank epidermal detachment sign involving more than 50% of the epidermis in the injection site. The clinical disease severity is expressed as mean ± SE and analyzed by paired Student t test. P < .001 (WT injected with R530 vs WT injected with R530 plus neutralizing antibody). Three to 4 independent experiments were done for each group of mice
CD11b-deficient mice are resistant to experimental BP
To further establish the role of CD11b in experimental BP, we evaluated the BP model in CD11b/CD18 (Mac-1) KO mice. WT and Mac-1 KO mice were injected intradermally with pathogenic anti-mBP180 IgG (2.6 mg/g body weight) and examined 24 hours later. As expected, WT mice developed intense blisters (Figure 1Ci; Table 1). Direct IF of perilesional skin showed in vivo deposition of pathogenic rabbit IgG and mouse C3 (Figure 1Cii-iii) and H/E-stained skin sections revealed dermal-epidermal separation with neutrophilic infiltration (Figure 1Civ). In contrast, Mac-1 KO mice exhibited no blisters 24 hours after injection with pathogenic IgG (Figure 1Cv). Direct IF of the skin of these mice showed in situ deposition of rabbit IgG and mouse C3 (Figure 1Cvi-vii) at the BMZ. Histologic examination of the skin of the Mac-1-/- mice showed no signs of dermal-epidermal detachment (Figure 1Cviii). Indirect IF demonstrated that the titers of circulating rabbit anti-mBP180 IgG in the Mac-1 KO mice were comparable to those of the WT mice (1:2560 in both groups).
Neutrophil accumulation in the skin of the pathogenic IgG-injected Mac-1 KO mice was significantly attenuated relative to the diseased WT mice (Figure 1D). Mac-1 KO mice pretreated with anti-CD11a neutralizing antibody (Figure 1D, bar 3) prior to pathogenic IgG injection resulted in a further reduction in neutrophil infiltration to levels observed in WT mice pretreated with neutralizing antibodies against both CD11a and CD11b (compare with Figure 1B, bar 4). These results further suggest that LFA-1 and Mac-1 are required for neutrophil accumulation in experimental BP.
Mac-1-deficient mice show normal mast cell degranulation but reduced neutrophil degranulation
Experimental BP depends on MC and neutrophil degranulation.21,22 To determine if these functions are impaired in Mac-1-deficient mice, the number of MCs and MC degranulation in the skin were evaluated, and neutrophil degranulation in vitro was assayed. Toluidine blue staining revealed that the number of MCs and percentage of degranulating MCs in the pathogenic IgG-injected Mac-1 KO mice were comparable with those in diseased WT animals (Figure 2). In vitro, neutrophils from WT and Mac-1 KO mice, stimulated with pathogenic anti-mBP180 IgG plus mBP180 antigen, released similar levels of NE (Figure 3A) and MMP-9 (Figure 3B). In vivo, however, extracellular levels of NE and MMP-9 released by infiltrating neutrophils in the IgG-injected skin of Mac-1 KO mice were significantly reduced compared with the diseased WT mice at 4- and 24-hour time points (Figure 3C-D). These results suggest that reduced neutrophil activation in Mac-1 KO mice results in below-threshold levels of MMP-9 and NE, which are responsible for the amplification of neutrophil infiltration and subsequent blistering.
Figure 2.
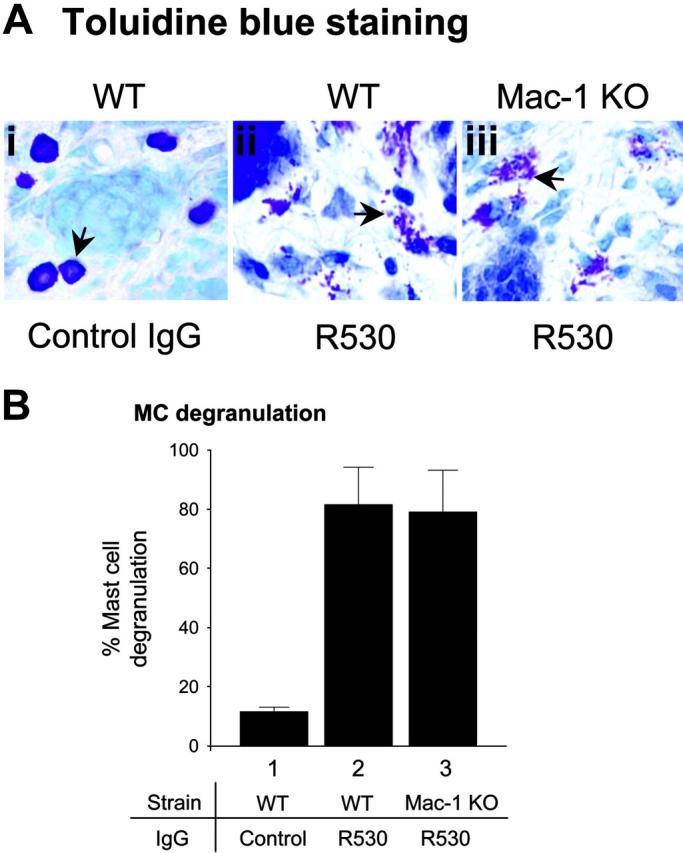
Activation of mast cells in Mac-1 KO mice. WT and Mac-1 KO mice (n = 6) were injected intradermally with pathogenic IgG (R530). At 2 hours after injection when MC degranulation peaked, skin sections were stained with toluidine blue and MCs in the dermis were counted and classified as degranulated (> 10% of the granules exhibiting fusion or discharge) or normal as previously described.21 (A) Toluidine blue staining showed similar degrees of MC degranulation (arrows) in pathogenic IgG-injected WT and Mac-1 KO mice (panels Ai vs Aiii). (B) Quantitation of degranulated MCs revealed no significant difference in MC degranulation between these 2 groups of mice (expressed as percentage of MC degranulation, mean ± SE).
Figure 3.
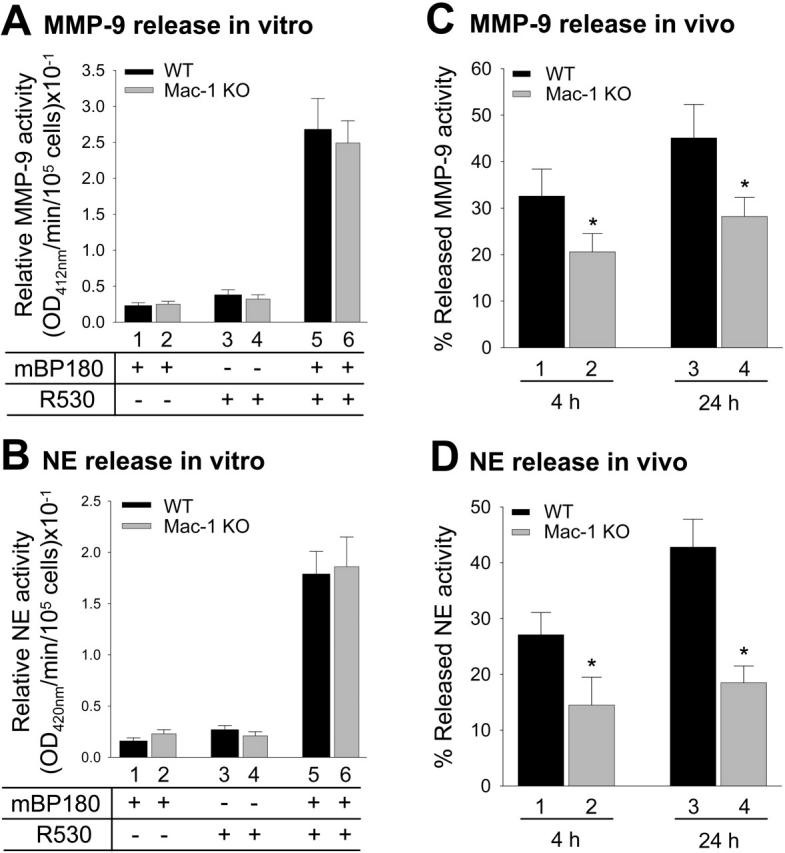
Activation of neutrophils in vitro and in vivo. To examine anti-mBP180 IgG-induced neutrophil activation in vitro, mouse neutrophils from WT and Mac-1 KO mice were stimulated with pathogenic anti-mBP180 IgG (R530) plus mBP180 antigen, and the supernatant was then assayed for MMP-9 (A) and NE (B) by MMP-9 colorimetric and NE enzymatic activity assay, respectively. Mac-1 KO and WT neutrophils showed no difference in neutrophil release of these proteinases. To examine anti-mBP180 IgG-induced neutrophil activation in vivo, WT and Mac-1 KO mice were injected intradermally with pathogenic anti-mBP180 IgG R530, and extracellular levels of released MMP-9 (C) and NE (D) in the IgG injection sites at 4- and 24-hour time points were quantified by MMP-9 colorimetric and NE enzymatic activity assays. Data shown are the mean ± SEM.
Pathogenic anti-mBP180 IgG induces subepidermal blistering in Mac-1-deficient mice reconstituted with mouse neutrophils
To further evaluate if the tissue damaging functions of Mac-1-deficient neutrophils accumulated in tissue is intact, Mac-1 KO mice were injected with pathogenic IgG and then directly reconstituted in the dermis with purified mouse PMNs to circumvent the neutrophil recruitment defect observed in these animals. The Mac-1 KO mice reconstituted with either WT or Mac-1 KO mice developed similar subepidermal blisters 24 hours after IgG injection (Figure 4; Table 1). Significantly higher numbers of neutrophils were seen in the lesional skin of Mac-1 KO mice reconstituted with Mac-1 KO PMNs compared with WT PMNs (1.23 ± 0.24 vs 1.83 ± 0.50 OD460reading/mg protein; P = .02) (Figure 4), which may reflect increased neutrophil survival as addressed in later sections. These results demonstrate that neutrophils in Mac-1 KO mice retain their tissue-damaging activities and that the reduction in BP development in these mice is likely attributable to the reduction in later stages of anti-BP induced neutrophil accumulation.
Figure 4.

In vivo reconstitution of neutrophils at the tissue site restores experimental BP in Mac-1-deficient mice. Mac-1 KO mice were injected intradermally with either pathogenic anti-mBP180 IgG R530 alone (bar 1), pathogenic IgG plus 5 × 105 WT (bar 2), or Mac-1 KO neutrophils (bar 3). The animals were examined 24 hours after IgG injection. The Mac-1 KO mice reconstituted with neutrophils from WT (bar 2) or Mac-1 KO mice (bar 3) developed blisters and the lesional skin of the diseased mice had a significantly higher number of neutrophils than the nonlesional skin of the Mac-1 KO mice without neutrophil reconstitution (bar 1). Data shown are the mean ± SEM. n = 6 for each group. *P < .05 (bar 1 vs bar 2; bar 2 vs bar 3); **P < .001 (bar 1 vs bar 3).
LFA-1 and Mac-1 have differential roles in experimental BP
Lack of LFA-1 or Mac-1 activity leads to a significant reduction in neutrophil accumulation in the skin of mice at 24 hours after IgG injection. To assess the contribution of LFA-1 and Mac-1 in the early stages of neutrophil recruitment during the disease development, a time-course of anti-mBP180 IgG-induced neutrophil accumulation was performed in WT, Mac-1 KO, and WT pretreated with CD11a neutralizing antibody. As shown previously,21,22 pathogenic anti-mBP180 IgG triggered a continuous increase in neutrophil infiltration starting at 2 hours, which reached a peak at 12 hours after IgG injection (Figure 5). The mice injected with LFA-1-neutralizing antibody showed minimal levels of neutrophil infiltration across all time-course points tested. In contrast, the Mac-1 KO mice exhibited a significantly increased level of neutrophil accumulation at the 4-hour time point compared with the WT mice (0.41 ± 0.10 vs. 0.56 ± 0.12 OD460reading/mg protein; P = .03); however, after 4 hours, no further increase in neutrophil infiltration was observed, resulting overall in a markedly reduced level of neutrophil infiltration at 12 and 24 hours relative to the WT mice. These results demonstrated that LFA-1 plays a major role in neutrophil infiltration in experimental BP while Mac-1 is required for later but not early stages of neutrophil accumulation.
Figure 5.

Time course of neutrophil infiltration. Neonatal WT, Mac-1 KO, and WT mice pretreated with CD11a-blocking antibody were injected intradermally with pathogenic anti-mBP180 IgG R530. Levels of infiltrating neutrophils at the IgG injection site were examined at the indicated time points after IgG injection. Data shown are the mean ± SEM. n = 6. *P < .05 and **P < .01 between R530-injected WT and Mac-1 KO mice.
The increased numbers of tissue accumulated neutrophils in Mac-1 KO mice and Mac-1 KO mice reconstituted with Mac-1 KO neutrophils led to the hypothesis that Mac-1 regulates survival of infiltrating neutrophils in experimental BP. To test this, lesional skin sections of the WT and nonlesional skin sections of the Mac-1 KO mice harvested at 2, 4, and 24 hours after anti-BP antibody injection were stained for the active form of caspase-3, a cell marker for apoptosis. Immunostaining identified apoptotic neutrophils in the dermis of both WT and Mac-1 KO mice that increased over time (Figure 6A). Notably, the number of apoptotic neutrophils in the Mac-1 KO mice was significantly lower than the WT mice at all 3 time points tested (Figure 6B). This suggests that Mac-1 deficiency results in an increase in the survival of tissue accumulated neutrophils in experimental BP.
Figure 6.

Neutrophil apoptosis in pathogenic IgG-injected Mac-1-deficient mice. WT and Mac-1 KO mice were injected intradermally with pathogenic anti-mBP180 IgG R530. The animals were examined 2, 4, and 24 hours after IgG injection for neutrophil apoptosis by detection of active caspase 3, an apoptosis effector enzyme. (A) Indirect IF showed significantly lower numbers of apoptotic neutrophils in the skin of Mac-1 KO mice than in WT mice. (B) Quantification revealed a significant decrease in the number of active caspase 3-stained neutrophils in the skin of Mac-1 KO relative to WT mice. Data shown are the mean ± SEM. n = 6, *P < .01.
Discussion
The aim of this paper was to investigate the role of the β2 integrins in experimental BP. Both Mac-1 and LFA-1 were essential for disease development, with a functional loss of either resulting in the ablation of clinical blistering at both the gross and microscopic level. Disease development is dependent on neutrophil accumulation. LFA-1 (CD11a/CD18) was essential for both early and late (or amplification) phases of anti-BP induced neutrophil recruitment. Mac-1 (CD11b/CD18) was important for anti-BP antibody-induced neutrophil accumulation at later stages, while early neutrophil accumulation in Mac-1-deficient mice was enhanced compared with WT mice. The latter may reflect the observed increase in survival of tissue accumulated neutrophils. The critical role for LFA-1 in anti-BP-induced neutrophil recruitment and associated pathology is consistent with the described role of this integrin in leukocyte recruitment in several models of inflammation39,40 and autoimmune lupus and arthritis.41,42 For example, in MRL/MpJ-Faslpr lupus model, mice deficient in LFA-1, but not Mac-1, showed significantly increased survival, decreased anti-DNA autoantibody formation, and reduced glomerulonephritis. In K/BxN serum transfer inflammatory arthritis model, both WT and Mac-1-null mice demonstrated extensive arthritic changes histologically, including synovial hyperplasia, leukocytic infiltration, joint effusions, and erosive pannus formation, while the synovia of LFA-1-null mice showed a normal-appearing synovial lining and sublining with little evidence of inflammatory change.
A limited number of studies have shown that Mac-1 is required for neutrophil recruitment and subsequent disease development in models of inflammation, with the contribution of LFA-1 exceeding that of Mac-1 in most models.30,39,40,43 Mac-1 KO mice have been shown to exhibit decreased accumulation of leukocytes and a reduction in intimal proliferation and thickening after angioplasty-induced endothelial denudation of the vessel wall.44 Mac-1 is also required for sustained neutrophil accumulation and proteinuria in antiglomerular basement membrane nephritis.45 Mac-1 deficiency led to a partial decrease in neutrophil accumulation and associated brain injury following stroke.46,47 The critical requirement for Mac-1 in BP is one of the few demonstrations that this β2 integrin is essential in disease development. The similar protection against clinical blistering observed in Mac-1 KO mice and mice given functional blocking Mac-1 antibody further strengthens this conclusion. The mechanism for protection in Mac-1 KO mice is likely the absence of the amplification of neutrophil accumulation following anti-BP injection. There are several possible mechanisms for why this step does not occur. One possibility is that Mac-1 interaction with immune complexes and/or complement results in extracellular cytotoxicity31 that could potentially result in the initial tissue damage required for subsequent amplification of neutrophil accumulation. Our data that Mac-1 KO neutrophils in skin site show a reduced degranulation may support this speculation. The second possibility is that Mac-1-deficient neutrophils fail to release, or fail to induce expression of, mediators that promote subsequent neutrophil accumulation.48,49 The third possibility is that LFA-1 and Mac-1 synergize to reach a maximal neutrophil recruitment and subsequent subepidermal blistering in this antibody-mediated disease model. Indeed, previous reports suggest that neutrophil accumulation may require a cooperative process of CD11a-dependent capture and stabilization of the tethering phase of rolling followed by CD11b-mediated stabilization of adhesion and emigration.44,50
Mac-1 deficiency resulted in enhanced anti-BP-induced neutrophil accumulation in the tissue at early time points. This correlated with an increase in neutrophil survival at these and later time points, with significantly more apoptotic cells present in the dermis of WT mice compared with Mac-1 KO mice at all time points tested. This provides in vivo evidence that Mac-1 regulates the survival of infiltrating neutrophils. This could either be through mechanisms directly or indirectly dependent on Mac-1. Mac-1 has been shown to be important for promoting apoptosis of neutrophils. In vitro, Mac-1 engagement of cytokine-stimulated neutrophils results in apoptosis, and Mac-1-dependent phagocytosis of complement opsonized particles induces reactive oxygen species and caspase-dependent apoptosis referred to as phagocytosis-induced cell death.51 These mechanisms of Mac-1-dependent neutrophil apoptosis may be operative in stimulating apoptosis, and thus clearance of neutrophils in BP. It is also possible that a switch in the balance of pro-versus antiapoptotic cytokines in the affected skin of Mac-1 KO mice results in the observed increase in neutrophil survival. However, this is unlikely since dermal reconstitution of Mac-1 KO mice with Mac-1 KO neutrophils resulted in enhanced neutrophil accumulation (inferring increased neutrophil survival) compared to reconstitution with WT neutrophils.
In summary, we have demonstrated that the β2 integrins play critical but differential roles in subepidermal blistering in experimental BP. LFA-1 is absolutely required for neutrophil recruitment, while Mac-1 is primarily responsible for the amplification of neutrophil accumulation and the apoptosis of tissue-accumulated neutrophils. These findings may shed new light on the pathogenic mechanisms responsible for BP and other subepidermal blistering diseases with an autoimmune response to BP180, such as herpes gestationis, mucous membrane pemphigoid, pemphigoid gestationis, and lichen planus pemphigoides.52-55 Furthermore, in contrast to LFA-1, which has important functions in T cells, Mac-1 blockade should have no direct effect on the adaptive immune response and has not been shown previously to have global effects on neutrophil trafficking into tissues. Thus, a blockade of this receptor may be an attractive therapy for human autoimmune blistering diseases because there would be fewer predicted side effects on other immune functions in patients.
Acknowledgments
We thank Dr Simon Warren for routine histology.
The authors have declared that no conflict of interest exists.
Prepublished online as Blood First Edition Paper, October 18, 2005; DOI 10.1182/blood-2005-08-3123.
Supported in part by U.S. Public Health Service National Institutes of Health (NIH) grant nos. AI40768 and AI61430 (Z.L.), AR32599 and AR32081 (L.A.D.), and HL065095, AR50800, and DK51643 (T.N.M.).
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Lever WF. Pemphigus. Medicine. 1953;32: 1-123. [DOI] [PubMed] [Google Scholar]
- 2.Jordon RE, Beutner EH, Witebsky E, Blumental G, Hale WC, Lever WF. Basement zone antibodies in bullous pemphigoid. JAMA. 1967;200: 751-758. [PubMed] [Google Scholar]
- 3.Jordon RE. Complement activation in pemphigus and bullous pemphigoid. J Invest Derm. 1976;67: 366-371. [DOI] [PubMed] [Google Scholar]
- 4.Katz SI, Hertz KC, Yaoita H. Herpes gestationis. Immunopathology and characterization of the HG factor. J Clin Invest. 1976;57: 1434-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanley JR, Hawley-Nelson P, Yuspa SH, Shevach EM, Katz SI. Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified epithelia. Cell. 1981;24: 897-903. [DOI] [PubMed] [Google Scholar]
- 6.Mutasim DF, Takahashi Y, Labib RS, Anhalt GJ, Patel HP, Diaz LA. A pool of bullous pemphigoid antigen(s) is intracellular and associated with the basal cell cytoskeleton-hemidesmosome complex. J Invest Dermatol. 1985;84: 47-53. [DOI] [PubMed] [Google Scholar]
- 7.Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA. Molecular heterogeneity of bullous pemphigoid antigens as detected by immunoblotting. J Immunol. 1986;136: 1231-1235. [PubMed] [Google Scholar]
- 8.Tanaka T, Parry DAD, Klaus-Kovtun V, Steinert PM, Stanley JR. Comparison of molecularly cloned bullous pemphigoid antigen to desmoplakin I confirms that they define a new family of cell adhesion junction plaque proteins. J Biol Chem. 1991;266: 12555-12559. [PubMed] [Google Scholar]
- 9.Sawamura D, Li K, Chu M-L, Uitto J. Human bullous pemphigoid antigen (BPAG1): amino acid sequences deduced from cloned cDNAs predict biologically important peptide segments and protein domains. J Biol Chem. 1991;266: 17784-17790. [PubMed] [Google Scholar]
- 10.Diaz LA, Ratrie H III, Saunders WS, et al. Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera: immunolocalization of this protein to the hemidesmosome. J Clin Invest. 1990;86: 1088-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giudice GJ, Squiquera HL, Elias PM, Diaz LA. Identification of two collagen domains within the bullous pemphigoid autoantigen, BP180. J Clin Invest. 1991;87: 734-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giudice GJ, Emery DJ, Diaz LA. Cloning and primary structural analysis of the bullous pemphigoid autoantigen, BP-180. J Invest Dermatol. 1992;99: 243-250. [DOI] [PubMed] [Google Scholar]
- 13.Hopkinson SB, Riddelle KS, Jones JRC. Cytoplasmic domain of the 180-kD bullous pemphigoid antigen, a hemidesmosomal component: molecular and cell biologic characterization. J Invest Dermatol. 1992;99: 264-270. [DOI] [PubMed] [Google Scholar]
- 14.Bedane C, McMillan JR, Balding SD, et al. Bullous pemphigoid and cicatricial pemphigoid auto-antibodies react with ultrastructurally separable epitopes on the BP180 ectodomain: evidence that BP180 spans the lamina lucida. J Invest Derm. 1997;108: 901-907. [DOI] [PubMed] [Google Scholar]
- 15.Hirako Y, Usukura J, Nishizawa Y, Owaribe K. Demonstration of the molecular shape of BP180, a 180-kDa bullous pemphigoid antigen and its potential for trimer formation. J Biol Chem. 1996;271: 13739-13745. [DOI] [PubMed] [Google Scholar]
- 16.Balding SD, Diaz LA, Giudice GJ. A recombinant form of the human BP180 ectodomain forms a collagen-like homotrimeric complex. Biochemistry. 1997;36: 8821-8830. [DOI] [PubMed] [Google Scholar]
- 17.Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Liu Z, Diaz LA. Bullous pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous site on the BP180 ectodomain. J Immunol. 1993;151: 5742-5750. [PubMed] [Google Scholar]
- 18.Zillikens D, Rose PM, Balding SD, et al. Tight clustering of extracellular BP180 epitopes recognized by bullous pemphigoid autoantibodies. J Invest Dermatol. 1997;109: 573-579. [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Diaz LA, Troy JL, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. 1993;92: 2480-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Giudice GJ, Swartz SJ, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest. 1995;95: 1539-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen R, Ning G, Zhao ML, et al. Mast cells play a key role on neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest. 2001;108: 1151-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z, Giudice GJ, Zhou X, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. 1997;100: 1256-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Shipley JM, Vu TH, et al. Gelatinase B-deficient mice are resistant to experimental BP. J Exp Med. 1998;188: 475-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Shapiro SD, Zhou X, et al. A critical role for neutrophil elastase in experimental bullous pemphigoid. J Clin Invest. 2000;105: 113-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z, Zhou X, Shapiro SD, et al. The serpin α1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102: 647-655. [DOI] [PubMed] [Google Scholar]
- 26.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76: 301-314. [DOI] [PubMed] [Google Scholar]
- 27.Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. The leukocyte integrins. J Biol Chem. 2000;275: 23409-23412. [DOI] [PubMed] [Google Scholar]
- 28.Larson RS, Springer TA. Structure and function of leukocyte integrins. Immunol Rev. 1990;114: 181-217. [DOI] [PubMed] [Google Scholar]
- 29.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110: 673-687. [DOI] [PubMed] [Google Scholar]
- 30.Coxon A, Rieu P, Barkalow FJ, et al. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5: 653-666. [DOI] [PubMed] [Google Scholar]
- 31.Ross GD. Regulation of adhesion versus cytotoxic functions of the Mac-1/CR3/αMβ2-integrin glycoprotein. Crit Rev Immunol. 2000;20: 197-222. [PubMed] [Google Scholar]
- 32.Li K, Tamai K, Tan EML, Uitto J. Cloning of type XVII collagen: complementary and genomic DNA sequences of mouse 180-Kilodalton bullous pemphigoid antigen (BPAG2) predict an interrupted collagenous domain, a transmembrane segment, and unusual features in the 5′-end of the gene and the 3′-untranslated region of the mRNA. J Biol Chem. 1993;268: 8825-8834. [PubMed] [Google Scholar]
- 33.Liu Z, Diaz LA, Haas AL, Giudice GJ. cDNA cloning of a novel human ubiquitin carrier protein: an antigenic domain specifically recognized by endemic pemphigus foliaceus autoantibodies is encoded in a secondary reading frame of this human epidermal transcript. J Biol Chem. 1992;267: 15829-15835. [PubMed] [Google Scholar]
- 34.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78: 206-209. [DOI] [PubMed] [Google Scholar]
- 35.Wershil BK, Wang Z, Jordon JR, Galli SJ. Recruitment of neutrophils during IgE-dependent cutaneous late phase responses in the mouse is mast cell dependent: partial inhibition of the reaction with antiserum against tumor necrosis factor-alpha. J Clin Invest. 1991;87: 446-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metcalf JA, Gallin JI, Nauseef WM, Root RK. Laboratory Manual of Neutrophil Function. New York, NY: Raven Press; 1985: 2-10.
- 37.Liu Z, Li N, Diaz LA, Shipley M, Senior RM, Werb Z. Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest. 2005;115: 879-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bin Z, Hirahashi J, Cullere X, Mayadas TN. Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis. J Biol Chem. 2003;278: 28443-28454. [DOI] [PubMed] [Google Scholar]
- 39.Lu H, Smith CW, Perrard J, et al. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1-deficient mice. J Clin Invest. 1997;99: 1340-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding ZM, Babensee JE, Simon SI, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163: 5029-5038. [PubMed] [Google Scholar]
- 41.Kevil CG, Hicks MJ, He X, et al. Loss of LFA-1, but not Mac-1, protects MRL/MpJ-Fas(lpr) mice from autoimmune disease. Am J Pathol. 2004;165: 609-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watts GM, Beurskens FJ, Martin-Padura I, et al. Manifestations of inflammatory arthritis are critically dependent on LFA-1. J Immunol. 2005;174: 3668-3675. [DOI] [PubMed] [Google Scholar]
- 43.Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99: 336-341. [DOI] [PubMed] [Google Scholar]
- 44.Hendersona RB, Limb LHK, Tessiera PA, et al. The use of lymphocyte function-associated antigen (LFA)-1-deficient mice to determine the role of LFA-1, Mac-1, and α4 integrin in the inflammatory response of neutrophils. J Exp Med. 2001;194: 219-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang T, Rosenkranz A, Assmann KJ, et al. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fc receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. 1997;186: 1853-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soriano SG, Coxon A, Wang YF, et al. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30: 134-139. [DOI] [PubMed] [Google Scholar]
- 47.Arumugam TV, Salter JW, Chidlow JH, Ballantyne CM, Kevil CG, Granger DN. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am J Physiol Heart Circ Physiol. 2004;287: H2555-H2560. [DOI] [PubMed] [Google Scholar]
- 48.Walzog B, Weinmann P, Jeblonski F, Scharffetter-Kochanek K, Bommert K, Gaehtgens P. A role for β2 integrins (CD11/CD18) in the regulation of cytokine gene expression of polymorphonuclear neutrophils during the inflammatory response. FASEB J. 1999;13: 1855-1865. [DOI] [PubMed] [Google Scholar]
- 49.Kettritz R, Choi M, Rolle S, Wellner M, Luft FC. Integrins and cytokines activate nuclear transcription factor-kappaB in human neutrophils. J Biol Chem. 2004;279: 2657-2665. [DOI] [PubMed] [Google Scholar]
- 50.Hentzen ER, Neelamegham S, Kansas GS, et al. Sequential binding of CD11a/CD18 and CD11b/CD18 defines neutrophil capture and stable adhesion to intercellular adhesion molecule-1. Blood. 2000;95: 911-220. [PubMed] [Google Scholar]
- 51.Mayadas TN, Cullere X. Neutrophil β2 integrins: moderators of life or death decisions. Trends Immunol. 2005;26: 388-395. [DOI] [PubMed] [Google Scholar]
- 52.Morrison LH, Labib RS, Zone JJ, Diaz LA, Anhalt GJ. Herpes gestationis autoantibodies recognize a 180-kD human epidermal antigen. J Clin Invest. 1988;81: 2023-2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bernard P, Prost C, Durepaire N, Basset-Seguin N, Didierjean L, Saurat JH. The major cicatricial pemphigoid antigen is a 180-kD protein that shows immunologic cross-reactivities with the bullous pemphigoid antigen. J Invest Dermatol. 1992;99: 174-179. [DOI] [PubMed] [Google Scholar]
- 54.Tamada Y, Yokochi K, Nitta Y, Toshihiko I, Hara K, Owaribe K. Lichen planus pemphigoides: identification of 180 kD hemidesmosome antigen. J Am Acad Dermatol. 1995;32: 883-887. [DOI] [PubMed] [Google Scholar]
- 55.Zone JJ, Taylor TB, Meyer LJ, Peterson MJ. The 97 kDa linear IgA bullous disease antigen is identical to a portion of the extracellular domain of the 180 kDa bullous pemphigoid antigen, BPAg2. J Invest Dermatol. 1998;110: 207-210. [DOI] [PubMed] [Google Scholar]