

Abstract
We show that multiple myeloma (MM), the second most commonly diagnosed hematologic malignancy, is responsive to hsp90 inhibitors in vitro and in a clinically relevant orthotopic in vivo model, even though this disease does not depend on HER2/neu, bcr/abl, androgen or estrogen receptors, or other hsp90 chaperoning clients which are hallmarks of tumor types traditionally viewed as attractive clinical settings for use of hsp90 inhibitors, such as the geldanamycin analog 17-AAG. This class of agents simultaneously suppresses in MM cells the expression and/or function of multiple levels of insulin-like growth factor receptor (IGF-1R) and interleukin-6 receptor (IL-6R) signaling (eg, IKK/NF-κB, PI-3K/Akt, and Raf/MAPK) and downstream effectors (eg, proteasome, telomerase, and HIF-1α activities). These pleiotropic proapoptotic effects allow hsp90 inhibitors to abrogate bone marrow stromal cell-derived protection on MM tumor cells, and sensitize them to other anticancer agents, including cytotoxic chemotherapy and the proteasome inhibitor bortezomib. These results indicate that hsp90 can be targeted therapeutically in neoplasias that may not express or depend on molecules previously considered to be the main hsp90 client proteins. This suggests a more general role for hsp90 in chaperoning tumor- or tissue-type-specific constellations of client proteins with critical involvement in proliferative and antiapoptotic cellular responses, and paves the way for more extensive future therapeutic applications of hsp90 inhibition in diverse neoplasias, including MM.
Introduction
Hsp90 is the central component of a ubiquitous chaperone complex that interacts with a variety of intracellular client proteins to facilitate their proper folding, prevent misfolding or aggregation, and preserve their 3-dimensional conformation to a functionally competent state.1 Unlike other heat shock proteins, hsp90 interacts with a more restricted set of client proteins,1 including cell-surface receptors (eg, HER2/neu),2,3 nuclear receptors for androgens (AR)4,5 and estrogens (ER),6 or chimeric kinases (eg, bcr/abl).7,8 Although hsp90 interacts with diverse client proteins (reviewed in Isaacs et al1) that may be important for proliferation/survival of tumor cells, clinical trials of hsp90 inhibitors (eg, the geldanamycin analog 17-allylamino-17-demethoxy-geldanamycin [17-AAG]) have originally focused on tumor types hallmarked by specific hsp90 chaperoning targets (eg, breast cancer [HER2/neu and ER], prostate cancer [AR], or bcr/abl-positive leukemias).
Hsp90 client proteins lack specific hsp90 binding motif(s) and vary in terms of intracellular localization, structure, and function. Their main currently recognizable common denominator is their role in promoting cell proliferation and protection from apoptosis. We thus hypothesized that hsp90 may interact with a broader spectrum of proliferative/antiapoptotic molecular targets and consequently, that hsp90 inhibition may have potent antitumor effects, even against malignancies that are not critically dependent upon conventional hsp90 targets. We specifically tested this hypothesis in the setting of multiple myeloma (MM) cells, which do express hsp90, but lack or do not critically depend upon the aforementioned traditional hsp90 client proteins. Additional impetus for studying hsp90 inhibitors was our finding (C.S.M., N.M., C.J.M., T.H., D.C., D.R., Carrasco, N.C.M., P.G.R., M.J., T.A.L., A.L.K., and K.C.A., manuscript submitted) that hsp90 can be up-regulated in MM cells interacting (in vitro or in vivo) with stromal cells, suggesting that hsp90 may mediate or regulate signaling events conferring the protective effects of bone marrow stromal cells (BMSCs) on MM cells.9 We indeed observed that small-molecule hsp90 inhibitors (geldanamycin and its analogs) suppress proliferation and survival of MM cells both in vitro and in vivo, via pleiotropic molecular sequelae, which converge to suppress signaling events triggered by insulin-like growth factors (IGFs) and interleukin-6 (IL-6). Hsp90 inhibition circumvents resistance to conventional or other investigational antitumor agents and overcomes protective effects conferred on MM cells by their interaction with BMSCs. These findings indicate that hsp90 can function as a central regulator of proliferative/antiapoptotic signal transduction, and suggest that hsp90 inhibition may have antitumor activities against a broader spectrum of neoplasias than previously appreciated. Importantly, our findings highlight a critical role for hsp90 and its chaperoning targets in the pathophysiology of MM and have direct implications for the development of novel anti-MM therapeutic strategies.
Materials and methods
Detailed information pertinent to cell lines and primary tumor specimens10; transfections and retroviral transductions11-15; ex vivo drug sensitivity assays16; whole-body real-time fluorescence imaging17; flow cytometry16,18,19; immunoblotting analyses16; functional assays for telomerase20,21 proteasome22 and transcription factor11,18,23,24 activities; in vitro and in vivo gene expression profiling11,21,24,25; and proteomic analyses of signaling state of MM cells24-26 has been previously published and is also included as supplemental data on the Blood website; see the Supplemental Data Set link at the top of the online article.
The in vivo anti-MM activity of 17-AAG was evaluated in a previously established model of diffuse GFP+ MM lesions in severe combined immunodeficient/nonobese diabetic (SCID/NOD) mice by serially monitoring with whole-body fluorescence imaging.17 Briefly, 40 male (6- to 8-week old) SCID/NOD mice were obtained from Jackson Laboratories (Bar Harbor, ME); housed and monitored in the Animal Research Facility of the Dana-Farber Cancer Institute; γ-irradiated (3 Gy [300 rad]) using cesium 137 (137Cs) γ-irradiator source; and received (24 hours after irradiation) tail IV injections of 5 × 106 RPMI-8226/S-GFP+ cells suspended in total volume of 100 μL of phosphate-buffered saline (PBS) per mouse. Mice were monitored daily for changes in body weight, signs of infection, or paralysis, and with thrice-weekly fluorescence imaging. In accordance with institutional guidelines, mice were humanely killed by CO2 inhalation in the event of paralysis or major compromise in their quality of life. All experimental procedures and protocols had been approved by the Animal Care and Use Committee of the Dana-Farber Cancer Institute. Overall survival (defined as time between IV injection of tumor cells and death) was compared in control versus 17-AAG-treated mice by Kaplan-Meier method.
Results
Expression of hsp90 and client proteins in MM cells
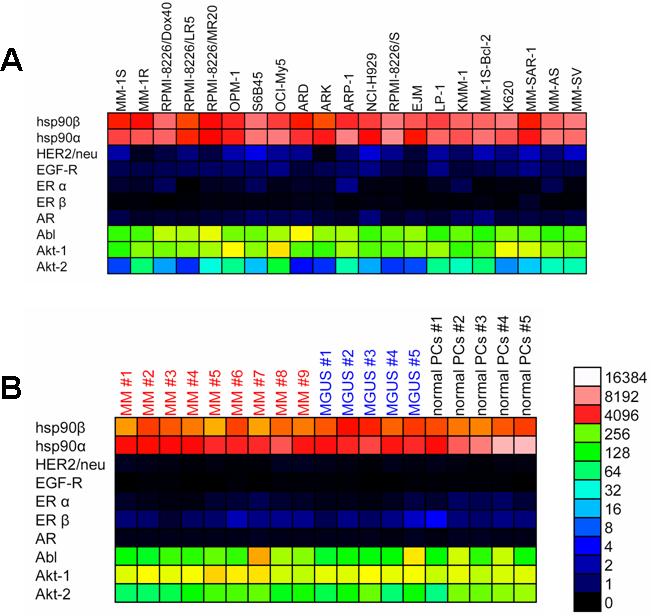
We studied in human MM cells the expression of transcripts for hsp90 and those of its chaperoning clients (including HER2/neu, EGF-R, AR, and ER), which are hallmarks of tumor types viewed as attractive clinical settings for hsp90 inhibitor treatment. Transcriptional profiles were generated from human MM cell lines (Supplemental Figure S1A), primary tumor cells purified from newly diagnosed MM patients, and CD138+ plasma cells (PCs) from healthy donors or individuals with monoclonal gammopathy of undetermined significance (MGUS), a premalignant clonal expansion of the PC population (Supplemental Figure S1B). These gene expression profiles confirmed universal expression of hsp90 transcripts in MM cell lines and primary specimens, without significant differences in expression between MM cells versus normal PCs or MGUS PCs. MM cells and normal PCs or PCs from MGUS patients had minimal, if any, expression of hsp90 client proteins such as HER2/neu (erbB2), other members of the EGF-R superfamily, ER-α or -β, or AR. Inhibition of Abl kinase with imatinib mesylate had minimal effect on MM cell viability at clinically relevant nanomolar concentrations that selectively target abl (data not shown), indicating that Abl kinase function is redundant for MM cell viability.
Activity of hsp90 inhibitors against drug-resistant MM cells
To address the hypothesis that hsp90 function is critical for MM cell proliferation and survival, we first tested the effect of hsp90 inhibitors on the viability of a panel of human MM cell lines. MTT colorimetric survival assays showed that hsp90 inhibitors of the ansamycin family, including geldanamycin (NSC122750) and 3 of its analogs (NSC255109, NSC330507, and NSC683664), potently suppress the survival of MM cells in vitro (Figure 1). Importantly, the clinically developed agent 17-allylamino-17-demethoxygeldanamycin (17-AAG; NSC 330507) (at concentrations relevant to the levels achieved in the peripheral blood of cancer patients enrolled in phase 1 clinical trials of 17-AAG27) is active even against MM cell lines resistant to conventional agents (eg, dexamethasone [MM-1R], doxorubicin [RPMI-8226/Dox40], melphalan [RPMI-822/LR5], and mitoxantrone [RPMI-8226/MR20]) or others which have low sensitivity to investigational anti-MM agents, including immunomodulatory thalidomide derivatives (S6B45, RPMI-8226/S), TRAIL/Apo2L (MM-1S-TR15), or the proteasome inhibitor bortezomib (MM-SA-1) (Figure 1D). Furthermore, 17-AAG significantly suppressed the survival of CD138+ purified primary tumor cells from patients with MM who were resistant to conventional or high-dose cytotoxic chemotherapy, as well as novel therapies such as immunomodulatory thalidomide derivatives (IMiDs, eg, lenalidomide) or the proteasome inhibitor bortezomib (also known as PS-341) (Figure 1C).
Figure 1.

In vitro anti-MM activity of hsp90 inhibitors. (A) In vitro anti-MM activity of geldanamycin against both drug-sensitive and drug-resistant MM cell lines, including the Dex-resistant MM-1R and the doxorubicin-resistant RPMI-8226/Dox40 cells. (B) Comparative in vitro activity of geldanamycin and its analogs against MM-1S myeloma cells. (C) In vitro activity of the geldanamycin analog 17-AAG (NSC 330507) against primary MM tumor cells isolated from patients resistant to conventional or investigational therapies (including conventional or high-dose chemotherapy, thalidomide, IMiDs, or the proteasome inhibitor PS-341). For each sample, red bars represent the percent survival (mean ± SD) of drug-treated cells; blue bars, the survival of their respective controls. (D) Dose-response matrix of human MM cell lines (as well as cells from other B-cell malignancies, such as IM-9 and ARH-77) to 17-AAG in vitro (0-5 μM for 72 hours). The survival of 17-AAG-treated cells (expressed as percent of the vehicle-treated control) is visualized in color format according to their values on a linear scale (0-100%).
In vivo anti-MM activity of hsp90 inhibitors
We next evaluated the in vivo anti-MM activity of 17-AAG in a SCID/NOD model of diffuse GFP+ fluorescent MM lesions established by IV injections of RPMI-8226/S-GFP+ cells, and serially monitored by whole-body real-time fluorescence imaging to allow for external visualization of GFP+ tumors.17 In this model, SCID/NOD mice (2 groups of 20 mice each) were randomly assigned to respectively receive 50 mg/kg intraperitoneally of 17-AAG (daily IP injections for 5 consecutive days followed by 2 days off therapy in each cycle, for a total of 4 cycles), or an equal volume of vehicle on identical schedule. The primary end point of this study was overall survival, defined as time between injection of tumor cells and time of killing (for paralysis or moribund state) or death. Control mice had diffuse GFP+ lesions, detected by fluorescent imaging (Figure 2A) and confirmed by histopathology, primarily involving the axial skeleton (thoracic/lumbar spine, skull, pelvis, thoracic cage) and extremities. Soft-tissue (eg, subcutaneous) plasmacytomas were also observed, but visceral metastases (eg, in liver, lung, spleen, or kidneys) were rare (Supplemental Table S1). The primary cause of morbidity in control mice was paralysis associated with development of bone vertebral lesions. In contrast, 17-AAG was well tolerated, without vital organ tissue damage evident in histopathologic analyses. Kaplan-Meier survival analysis showed that 17-AAG treatment significantly prolongs median overall survival of mice (> 250 days, with 14 of 20 of 17-AAG-treated mice surviving at the last analysis, versus 29 days median overall survival for control mice, with 0 of 20 mice surviving) (P < .001, log-rank test) (Figure 2B).
Figure 2.
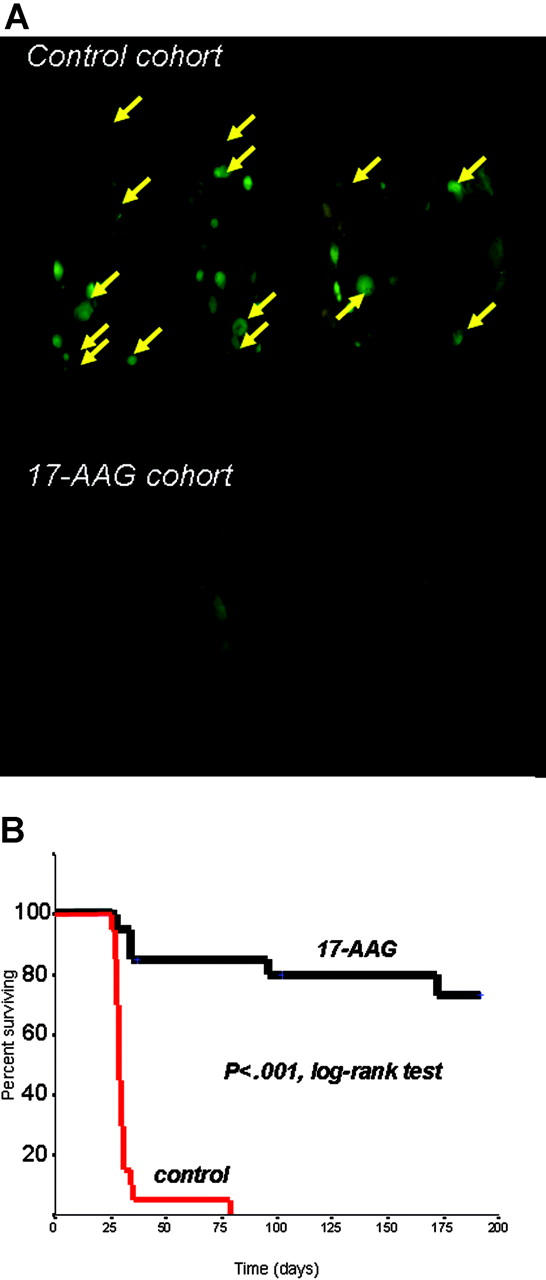
In vivo anti-myeloma activity of hsp90 inhibition. (A) Representative results of whole-body fluorescence imaging of SCID/NOD mice injected intravenously with 5 × 106 RPMI-8226/S-GFP+ human MM cells and receiving either 17-AAG treatment or vehicle. Imaging results obtained 4 weeks after intravenous injection of GFP+ MM cells indicate extensive diffuse GFP+ bone lesions (arrows) in the spine, skull, lower extremities, and pelvis, as well as subcutaneous GFP+ lesions in the control cohort, in marked contrast to the absence of MM lesions in 17-AAG-treated mice. (B) Kaplan-Meier survival curve of 17-AAG-treated vs. control SCID/NOD mice injected intravenously with RPMI-8226/S-GFP+ cells. 17-AAG treatment significantly prolonged median overall survival of mice (> 250 days, with 14/20 of 17-AAG-treated mice surviving at the last interim analysis, vs 29 days median overall survival for control mice, with 0/20 mice surviving) (P < .001, log-rank test).
Ex vivo molecular profiling of hsp90 inhibition in MM cells
The favorable profile of in vitro and in vivo anti-MM activity of hsp90 inhibitors prompted us to investigate the entire spectrum of their molecular sequelae, using oligonucleotide microarrays and proteomic profiling, with the intent to rationally identify potential combinations of hsp90 inhibitors with other antitumor agents. To screen for proliferative/antiapoptotic signaling molecules regulated by hsp90, we performed proteomic analyses of the signaling state of hsp90 inhibitor-treated MM cells, using multiplex immunoblotting arrays,24-26 which allowed for assessment of expression and/or phosphoryation status of kinases and kinase targets implicated in cell proliferation, survival, transcription/translation regulation, etc. These analyses showed that hsp90 inhibitors deplete the intracellular levels of Akt (Supplemental Figure S3) and Raf (Supplemental Figure S3B and Figure 3A), as well as several other proliferative/antiapoptotic signaling mediators, including IKK-α, a kinase downstream of Akt and responsible for IκB phosphorylation and transcriptional activation of NF-κB;28 p70S6K, which is activated by Akt and is implicated in regulation of protein translation;29 src; RhoA kinase; Bmx (bone marrow-X kinase), a PI-3K-stimulated kinase mediating PI-3K-dependent, Akt-independent proliferative/antiapoptotic signals in B-lineage cells;30 and DNA-PK, which is involved in regulation of DNA-damage repair (Supplemental Figure S3B).24
Figure 3.

Functional sequelae of hsp90 inhibition. (A) Immunoblotting analyses of MM-1S cells treated with 17-AAG (500 nM for 0-24 hours) confirms that hsp90 inhibition suppresses the intracellular levels of multiple downstream effectors of the IGF-1R and IL-6R pathways, including the Raf-1 and IKK-α kinases (coupled with decreased constitutive phosphorylation of MEK1/2), the antiapoptotic proteins FLIP, XIAP, A1/bfl-1, cIAP2, and the pro-osteoclastogenic stimulator RANKL. In contrast, hsp90 inhibition by 17-AAG does not confer a significant effect on the intracellular levels of MEK1/2, for example. (B) Telomerase activity assay (by densitometric evaluation of the telomeric repeat amplification protocol (TRAP) method indicates that 17-AAG (500 nM, 0-24 hours) suppresses both constitutive and IGF-1 (200 ng/mL)-induced activation of the catalytic subunit of telomerase (hTERT). (C) 17-AAG treatment (750 nM for 0-36 hours) suppresses the NF-κB activity of MM-1S, as evidenced by NF-κB DNA binding enzyme-linked immunosorbent assay (ELISA). (D) 17-AAG treatment (750 nM for 0-36 hours) suppresses the activity of the 20S proteasome, as evidenced by 20S chymotryptic activity assay. (B-D) Error bars indicate SD.
Hsp90 inhibition targets cell-surface or intracellular receptors that promote proliferation/survival in various solid tumor models, and our analyses indicated that hsp90 regulates the expression of members of the PI-3K/Akt/p70S6K pathway in MM cells. We therefore focused on effects of hsp90 inhibitors on cell-surface expression and downstream signaling cascades of IGF-1R and IL-6R, which are upstream regulators of PI-3K/Akt function and play important roles in MM pathophysiology.12,32 Flow cytometry revealed that 17-AAG-induced cell death is preceded by down-regulation of IGF-1R and IL-6R cell surface expression (in contrast, there was no significant decrease in surface expression of CD40, another receptor with potential role in MM cell proliferation) (Figure 4). Hsp90 inhibition abrogated constitutive and cytokine-induced signaling events in MM cells, including Akt kinase activity; phosphorylation, and cytoplasmic levels of proapoptotic FKHRL-1 Forkhead transcription factor (data not shown), a known phosphorylation target of Akt in IGF-induced signaling12,25,31; NF-κB transcriptional activity (Figure 3); phosphorylation of MEK1/2, a key component in Ras/Raf/MAPK pathway function (Figure 3A); constitutive and IGF-induced activity of telomerase (Figure 3B); and IGF-1-induced up-regulation of chymotryptic 20S proteasome activity (Figure 3D). Hsp90 inhibition also suppressed the levels of several antiapoptotic molecules (Figure 3A), which are up-regulated by IGF-1R activation,12,25 including caspase-8 inhibitors FLIP and cIAP-2, caspase-9 inhibitor XIAP (Figure 3), as well as the osteoclastogenic stimulator RANKL (Figure 3), and the transcription factor XBP-1 (data not shown), a known downstream target of IL-6 signaling in MM cells33 and antiapoptotic regulator of protective responses against endoplasmic reticulum stress.33
Figure 4.

Hsp90 inhibition targets IGF-1R and IL-6R signaling. Flow cytometric analysis of 17-AAG-treated (750 nM for 24 hours) MM-1S cells indicates complete abrogation of cell surface expression of IGF-1R (CD221) and suppression of surface IL-6R (CD126). The lack of significant suppression of cell surface expression of CD40, another receptor implicated in regulation of MM cell pathophysiology, supports the notion of specificity of the role of hsp90 on surface expression and function of certain growth/survival receptors. Filled curves correspond to staining with anti-IGF-1R, anti-IL-6R, or anti-CD40 antibody; open curves, staining with the respective isotype control antibody.
The malignant phenotype of MM cells is not driven by a single pathognomonic genetic lesion causative for all MM cases.34 Instead, MM cells encompass complex patterns of molecular lesions, which can confer multiple levels of possible resistance to conventional therapies. The multiplicity of antiapoptotic signaling effectors that are targeted by hsp90 inhibitors in MM cells suggests that it is unlikely for any single one of these effectors to be the sole target and mediator of antitumor effects of hsp90 inhibition. Indeed, overexpression of constitutively active Akt in MM-1S cells conferred only partial attenuation of in vitro antitumor effect of 17-AAG (Figure 1D). In addition, recent data show that selective inhibition of other single hsp90 targets, such as IL-6R25 or IKK,28 has per se modest in vitro anti-MM effects, which are in contrast to the marked effect of hsp90 inhibitors. Taken together these results indicate that the antitumor effect of hsp90 inhibition is likely multifactorial and is the composite result of simultaneous inhibition of multiple antiapoptotic client proteins of hsp90.
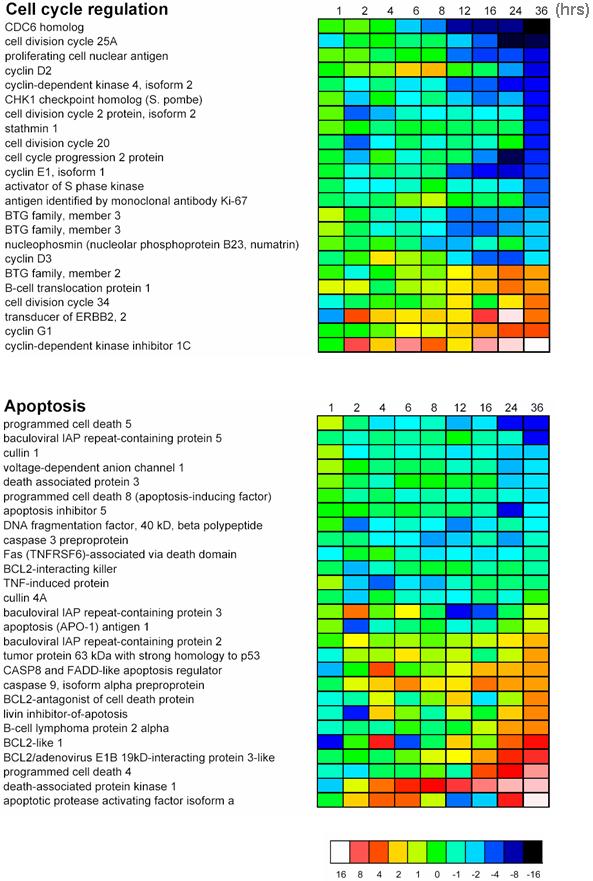
Hsp90 is mainly considered to affect protein function by direct posttranslational effect on protein confirmation rather than regulation at the transcriptional level. However, in settings of treatment with antitumor agents that regulate protein expression posttranslationally (eg, proteasome inhibitors), tumor cell transcriptional profiling may not only shed light into molecular mechanism of action of such drugs, but also provide clues for potential stress responses mounted by tumor cells to counteract proapoptotic effects of these therapeutics.11 Because constitutive or inappropriate up-regulation of such stress responses may contribute to attenuated clinical responses to hsp90 inhibition, we characterized the transcriptional profile of 17-AAG-treated cells, to charter a framework for specific therapeutic strategies that can suppress potential intrinsic mechanisms of attenuated response to hsp90 inhibition. Functional clustering analysis of transcriptional profiling data obtained with Affymetrix U133A microarrays of 17-AAG-treated MM-1S cells (Affymetrix, Santa Clara, CA; Supplemental Figures S4-S7) confirmed the impact of hsp90 inhibition on IGF- and IL-6-triggered signaling and its downstream effectors, including transcriptional suppression of known NF-κB target genes (including the caspase inhibitor cIAP-2), suppression of transcripts implicated in DNA synthesis and repair, positive regulation of cell-cycle activation, and production of 26S proteasome subunits. This latter effect correlated at a functional level with decreased 20S chymotryptic proteasome activity in 17-AAG-treated MM cells (Figure 3D). Furthermore, 17-AAG suppressed transcription of genes, such as transferrin receptor (CD71) and syndecan-1 (CD138), that contribute to MM cell proliferation and survival. Interestingly, however, other transcriptional events appeared to constitute stress responses mounted by tumor cells to counteract the effects of hsp90 inhibition. Indeed, 17-AAG treatment triggered up-regulation of transcripts for heat shock proteins (eg, hsp70) and some proteins suppressed by hsp90 inhibition (eg, IGF-1R, IL-6R, Raf-1, and XBP-1; Supplemental Figures S4-S7). The fact that these latter transcriptional changes are preceded by reduction in respective protein levels suggests that they result from a negative regulatory feedback loop, induced in an apparent stress response to hsp90 inhibition, which, however, cannot rescue MM cells from apoptosis.
In vivo molecular profile of hsp90 inhibition in MM cells
To confirm that molecular sequelae of hsp90 inhibition are consistent ex vivo and in vivo, we next characterized the impact of 17-AAG treatment of MM-bearing mice on expression of hsp90 targets in MM cells. SCID/NOD mice bearing diffuse bone lesions of MM-1S-GFP+/luc+ cells received single IP injections of either 17-AAG (50 mg/kg) or equal volumes of vehicle. Twenty-four hours after injections, mice were humanely killed by CO2 inhalation and necropsy was performed under the guidance of whole-body fluorescence imaging to identify the anatomic sites of GFP+ MM bone lesions and flush from the respective marrow cavities their cellular infiltrates, from which MM cells were purified (> 95% GFP+ CD138+) on the basis of their GFP+ status by flow cytometric cell sorting. These MM cells were then processed for further studies. Consistent with in vitro data, 17-AAG treatment in vivo decreased IGF-1R cell surface expression (evaluated by flow cytometry) on MM cells homing to the BM (Figure 5A) and suppressed the intracellular levels of Akt by immunoblotting analysis (data not shown). Unsupervised 2-dimensional hierarchical clustering analysis (Figure 5B) revealed that transcriptional profiles of MM-1S cells in 17-AAG-treated mice clustered together with the transcriptional profile of ex vivo 17-AAG-treated MM-1S (while control MM-1S cells homing to the bone clustered in a different branch of the hierarchical clustering dendrogram). These consistent in vitro and in vivo transcriptional profiles of hsp90 inhibition suggest that the molecular sequelae of hsp90 inhibition are not abrogated by interactions of MM cells with the BM milieu, suggesting a role for hsp90 inhibition in overcoming BM microenvironment-dependent mechanisms of drug resistance in MM cells.
Figure 5.

In vivo molecular profiling of hsp90 inhibition. MM-1S-GFP+/Luc+ cells injected intravenously in SCID/NOD mice led to development of diffuse bone lesions. Mice were randomly assigned to receive either a single dose of 17-AAG (50 mg/kg intraperitoneally) or vehicle; 24 hours after drug administration, all mice were humanely were killed and GFP+ MM bone lesions were visualized during necropsy by whole-body fluorescence imaging and harvested for further molecular analyses. (A) Flow cytometric analysis documents significant suppression of IGF-1R cell surface expression in MM tumor cells from 17-AAG-treated mice, compared to control mice. Filled curves correspond to staining with anti-IGF-1R antibody; open curves, staining with isotype control. (B) Hierarchical clustering analysis of in vivo gene expression profiles of MM-1S cells in 17-AAG-treated versus control mice.
Hsp90 inhibition targets the bone marrow microenvironment and its protective effects on MM cells
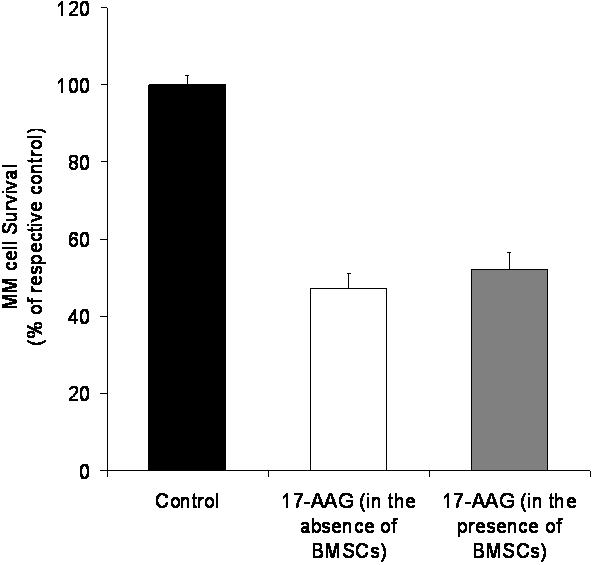
Binding of MM cells to BMSCs triggers NF-κB dependent up-regulation of IL-6 transcription and secretion to BMSCs,35 as well as increased autocrine (MM cell-derived) and/or paracrine (BMSC-derived) production of IGFs.25 These events contribute to MM cell proliferation, survival, and drug resistance in the BM milieu,9 and our recent data indicate that tumor-stromal interactions are associated with increased hsp90 expression in MM cells, (C.S.M. et al, manuscript submitted). We thus investigated whether hsp90 inhibition could also influence the interaction of MM cells with their BM microenvironment and its effect on protecting MM cells from apoptosis. Indeed, in ex vivo coculture assays of BMSCs and MM tumor cells, 17-AAG (750 nM for 3 days) induced selective cell death of MM cells (as evidenced by suppression of CD138+ population of primary MM tumor cells or GFP+/CD138+ population of MM-1S-GFP+ cells) despite the presence of BMSCs, suggesting that hsp90 inhibition can overcome protective effects conferred to MM cells by the BM milieu (Supplemental Figure S8) and providing additional functional confirmation for the significance of hsp90 up-regulation in MM cells upon their interaction with BMSCs (C.S.M. et al, manuscript submitted). Importantly, short-term exposures of MM cells alone, BMSCs alone, or their cocultures with 17-AAG (at concentrations and durations of exposure which did not affect viability of either MM or BMSCs) suppressed the constitutive and coculture-induced production of IGF-1, VEGF, and IL-6 (Supplemental Figure S9). These findings suggest that hsp90 inhibition can abrogate the protective effects conferred to MM by their interaction with the BM stroma, at multiple levels of the respective cytokine-mediated pathways, including not only suppression of activity or expression of signaling cascades required for response of MM cells to microenvironmental stimuli, but also abrogation of local production of these cytokines.
Because we have recently shown that IGF stimulates VEGF production in MM and other models,25,36 we next investigated the impact of hsp90 inhibition on survival of human microvascular endothelial cells, their bFGF- and VEGF-induced proliferation, and expression of cell-surface IGF-1R. 17-AAG suppressed proliferation of human microvascular endothelial cells (HMVECs; Figure 6A); abrogated its stimulation by bFGF and VEGF (Figure 6A); and down-regulated their cell-surface expression of IGF-1R (Figure 6C). Hsp 90 inhibition also suppresses HIF-1a transcriptional activity and IGF-1-induced VEGF secretion (Figure 6D-E). These data support the notion of hsp90 as regulator of tumor-associated neoangiogenesis, at the level of proangiogenic cytokine production by tumor cells and local BM milieu, and at the level of modulation of responses of tumor-associated endothelium to such stimuli.
Figure 6.
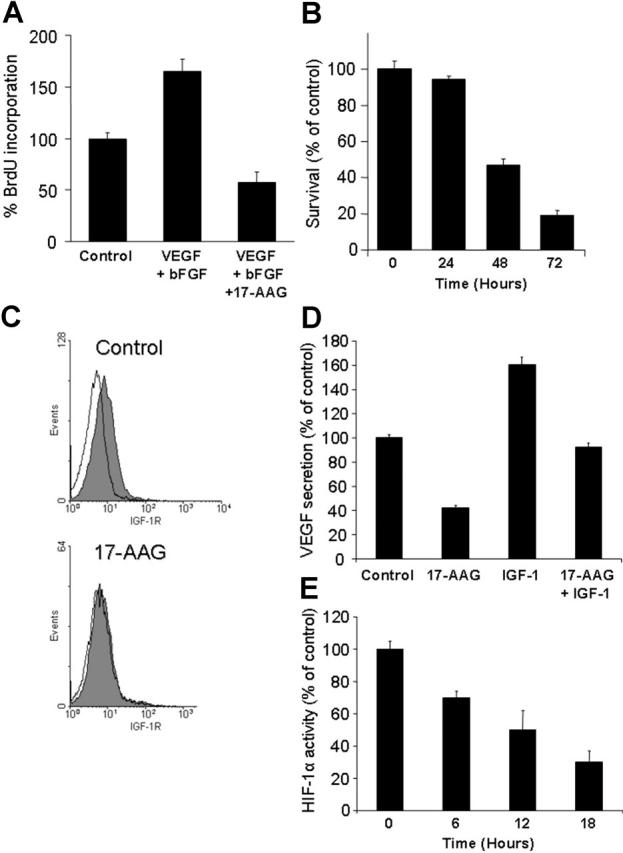
Antiangiogenic effects of hsp90 inhibiton. (A) 17-AAG abrogates VEGF + bFGF-induced endothelial cell proliferation. (B) 17-AAG (1 μM, 3-day incubation) suppresses the survival of human microvascular endothelial cells (HMVECs), as evidenced by MTT colorimetric survival assay. (C) 17-AAG (1 μM for 24 hours) suppresses the surface expression of IGF-1R in HMVECs. Filled curves correspond to staining with anti-IGF-1R antibody; open curves, staining with isotype control. (D) 17-AAG (500 nM for 24 hours) abrogates constitutive and IGF-1-induced VEGF production by MM cells. (E) 17-AAG suppresses the transcriptional activity of HIF-1α in primary MM cells. Error bars indicate SD.
Molecular profile-guided elucidation of therapeutic combinations of hsp90 inhibitors with conventional or other investigational anti-MM agents
The transcriptional and proteomic signatures of hsp90 inhibitor-treated cells provided a basis for rational combination therapies to enhance the anti-MM effect of hsp90 inhibition. The effect of hsp90 inhibition on DNA repair regulation (eg, down-regulation of DNA-PK and DNA repair enzymes) provided the basis for ex vivo combination of 17-AAG with DNA-damaging agents, such as doxorubicin, which revealed increased chemosensitivity of MM cells upon hsp90 inhibition (Figure 7 and Supplemental Figure S10). The 17-AAG-mediated suppression of caspase inhibitors (FLIP, XIAP, cIAP2) and antiapoptotic molecules such as IGF-1R and Akt provided the framework for combining hsp90 inhibitors with anti-MM agents that induce caspase-dependent apoptosis (Figure 7 and Supplemental Figure S10), such as IMiDs, Apo2L/TRAIL, and bortezomib.11,19,23
Figure 7.
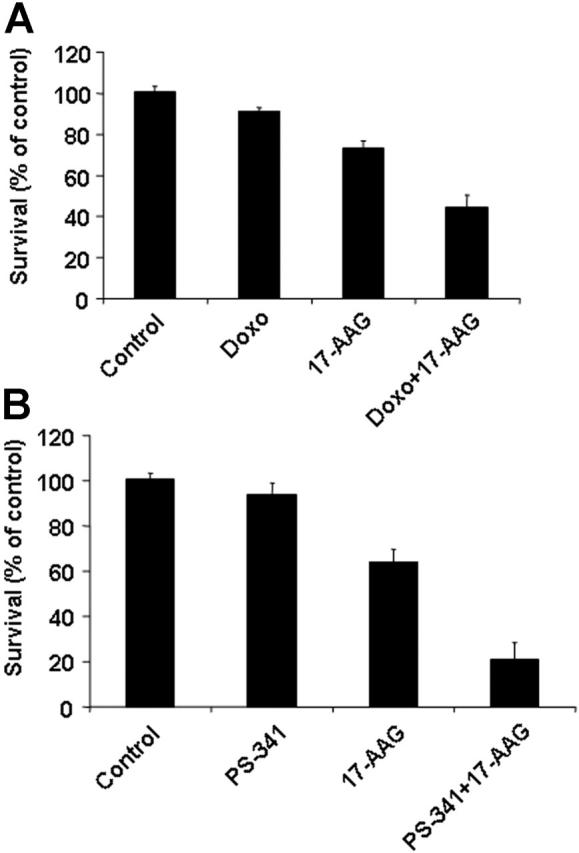
Hsp90 inhibition sensitizes tumor cells to other anticancer therapies. The in vitro anti-MM activities of doxorubicin (Doxo, 50 ng/mL, 48 hours, A) and bortezomib (PS-341, 5 nM, 24 hours, B) are enhanced by 17-AAG treatment (0.75 μM for the final 24 hours of each incubation) in primary MM tumor cells that are resistant to cytotoxic chemotherapy and bortezomib. Error bars indicate SD.
We have previously shown that the proteasome inhibitor bortezomib triggers pronounced stress response hallmarked by increased expression of heat shock proteins, such as hsp90,11 in an apparent effort to prevent misfolding of intracellularly accumulated undegraded proteins and rescue the cell from apoptosis. While the ability of 17-AAG to inhibit this stress response, at least in terms of its hsp90 part, can contribute to synergy of proteasome and hsp90 inhibitors, we also evaluated other potential molecular determinants of this interaction. We found that combination of bortezomib and 17-AAG triggers in bortezomib-resistant primary MM cells far more pronounced intracellular accumulation of ubiquitinated proteins than either drug alone (which is consistent with recent observations in breast cancer cells, albeit bortezomib-sensitive ones37) (Figure 8A). This effect can be mechanistically attributed to synergistic suppression of chymotryptic activity of the 20S proteasome by combination of bortezomib with 17-AAG (Figure 8B). In addition, because anti-MM effect of bortezomib has been associated with stimulation of endoplasmic reticulum stress,37,38 we evaluated the functional repercussions, at the endoplasmic reticulum level, of combined proteasome and hsp90 inhibition and found that it triggers more pronounced cleavage of caspase-12 than either drug alone (data not shown). Because caspase-12 activation and cleavage constitutes a critical event in endoplasmic reticulum stress-induced apoptosis, these data suggest that hsp90 inhibitors can counteract the resistance of tumor cells to bortezomib by further impairing their ability to withstand the endoplasmic reticulum stress generated by proteasome inhibition. This putative mechanism is further supported by the fact that the combination of 17-AAG and bortezomib also causes switch of the XBP-1 transcription factor from its spliced version (XBP-1s) to its unspliced one (XBP-1u) (data not shown). XBP-1u functions as dominant-negative suppressor of the transcriptional activity of XBP-1s, which in turn regulates the expression of endoplasmic reticulum chaperones that serve to counteract the consequences of endoplasmic reticulum stress-inducing agents.33 Therefore, our results, taken together, indicate that the marked antitumor activity of combined hsp90 and proteasome inhibitors is due to their mutually complementing abilities in simultaneously triggering intracellular accumulation of undegraded proteins (ie, inhibition by bortezomib of the catalytic site of chymotryptic proteasome activity and attenuation by hsp90 inhibitors of upstream cytokine-driven positive regulatory signals for proteasome activity) (Figure 3D) and preventing the function of mechanisms aiming at protecting the cells from the stress generated by such accumulation (eg, cytoplasmic and endoplasmic reticulum chaperones).
Figure 8.

Molecular basis of hsp90 inhibitor-induced sensitization of MM cells to proteasome inhibitor bortezomib. In vitro treatment of chemo- and bortezomib-resistant primary MM tumor cells with bortezomib (PS-341, 5 nM, 12 hours), 17-AAG (250 nM, 12 hours) or their combination indicates that the combination of the 2 drugs induces more pronounced (A) accumulation of ubiquitinated proteins, as shown by immunoblotting analysis, and (B) inhibition of proteasome activity, evidenced by 20S proteasome chymotryptic activity assay, than either drug alone. Error bars indicate SD.
Discussion
Hsp90 inhibitors are investigational anticancer agents with favorable profile of manageable and transient side effects in phase 1 clinical trials for solid tumors, and preliminary evidence of in vivo biological activity and objective responses.27,39-42 Our study documents in vitro and in vivo anti-MM activity of hsp90 inhibitors, which, when coupled with our mechanistic and molecular profiling studies, provided the framework for clinical trials of this novel class of agents in MM.
Our study illustrates potent anti-MM activity of hsp90 inhibitors and highlights the pleiotropic nature of their antitumor molecular sequelae. Indeed, hsp90 inhibitors abrogate cytokine (eg, IGF and IL-6)-induced signaling cascades at multiple molecular levels, including suppression of cell surface receptor expression (eg, IGF-1R); inhibition of downstream signaling mediators, including Akt, Raf, IKK-α, and p70S6K, with diverse ensuing proapoptotic sequelae, including decreased constitutive and IGF-induced activity of NF-κB, telomerase, and 20S proteasome; and suppressed expression of intracellular inhibitors of apoptosis (eg, FLIP, XIAP, cIAP-2).
Geldanamycin and its analogs specifically inhibit only hsp90 (and its endoplasmic reticulum homolog grp94),43,44 but not other chaperones or other intracellular molecular targets. This selectivity as well as the in vitro and in vivo effects of ansamycin hsp90 inhibitors delineate an important role for hsp90 in regulating diverse pathways mediating proliferation, survival, and drug resistance of tumor cells in the bone microenvironment. Hsp90 inhibitors thus illustrate an emerging paradigm in anticancer drug development, namely the simultaneous targeting of multiple intracellular pathways (and, preferably, of multiple targets within each individual pathway) regulating tumor cell proliferation and resistance to apoptosis. Despite recent successes of therapies neutralizing single molecular targets of major importance for tumor cell proliferation/survival (eg, imatinib mesylate in bcr/abl+ leukemias or solid tumors dependent upon c-kit or PDGF-R), resistance can eventually develop due to mutations/amplifications of respective target genes8 and/or compensatory activation of collateral/downstream pathways, which can sustain tumor cell survival even upon effective inhibition of the respective molecular target of these approaches. Therefore, the pleiotropic molecular sequelae of hsp90 inhibitors (Figure 9) may help minimize the development of such acquired resistance, and may account for their potent activity against a wide spectrum of MM cells, including those resistant to multiple conventional or novel anti-MM therapies. Furthermore, the effect of hsp90 inhibitors on multiple intracellular antiapoptotic effectors (eg, caspase inhibitors, PI-3K/Akt, and NF-κB pathways) and on pathways protecting tumor cells from anticancer treatments (eg, DNA damage repair genes protecting from chemotherapy-induced cell death, or hsp90 contributing to attenuated response of MM cells to proteasome inhibitors) may account for the ability of hsp90 inhibitors to sensitize MM cells to diverse antitumor agents, including cytotoxic chemotherapy, proteasome inhibitors, death receptor ligands (eg Apo2L/TRAIL), or immunomodulatory thalidomide analogs. It is also suggested that the clinical benefit derived from hsp90 inhibitors may be maximized by rational, molecular profiling-based design of combination therapies, whereby hsp90 inhibitors are used to sensitize MM cells to other antitumor agents and neutralize multiple signaling pathways conferring resistance to the other components of the combination therapy, allowing for dose adjustments in the individual components of these combinations, in an effort to minimize treatment-related adverse events.
Figure 9.

Schematic representation of diverse intracellular signaling cascades which can be impacted upon hsp90 inhibition, and of examples of multiple molecular levels where these effects can occur (through effects on expression/function of hsp90 client proteins and/or their downstream effectors). Solid arrows indicate activation and dashed lines indicate inhibitory events. Hsp90 inhibitors of the ansamycin family specifically inhibit hsp90, but the highly pleiotropic roles of their target confer to this class of agents the advantage of simultaneous blockade, by a single chemical entity, of diverse pathways implicated in tumor cell proliferation, survival, drug resistance, and microenvironmental interactions. This multifactorial activity of hsp90 inhibitors provides a framework for use of these agents to counteract mechanisms of resistance to multiple other agents, and increase tumor cell sensitivity to diverse conventional and novel agents.
Hsp90 inhibitors target MM cells not only directly, but also by perturbing their ability to respond to their local BM microenvironment and its protective stimuli. Multiple antiapoptotic pathways up-regulated in MM cells upon their interaction with BMSCs (manuscript submitted) involve hsp90 client proteins and are suppressed upon hsp90 inhibition, providing additional functional confirmation for a role of hsp90 in regulation of antiapoptotic signaling in MM cells residing in the BM milieu. 17-AAG can abrogate the up-regulation of local cytokine (IGF-1, IL-6, and VEGF) production which is triggered upon MM-BMSC binding. Furthermore, hsp90 inhibition exhibits antiangiogenic properties, by down-regulating the production of proangiongenic cytokines in the context of tumor-stromal interactions, but also by directly suppressing the ability of endothelial cells to respond to these cytokines. Therefore, our study highlights an intriguing role of hsp90 at multiple levels of interactions of MM cells with their local microenvironment and, coupled with data on hsp90 regulating MMP-mediated extracellular matrix remodeling in metastatic sites,45,46 suggests that hsp90 targeting may be therapeutically beneficial not only in terms of direct antiproliferative effects on tumor cells, but also by indirect effects on how they interact with their local milieu.
The in vivo anti-MM activity of hsp90 inhibitors may reflect their ability to target both MM tumor cells and their interactions with the local BM microenvironment. These latter microenvironmental effects of hsp90 inhibitors are not detectable in conventional ex vivo assays of tumor cell drug sensitivity and cannot be optimally assessed in traditional in vivo animal models of subcutaneous tumor xenografts. Instead, our in vivo mouse model of diffuse GFP+ MM bone lesions places MM cells in a microenvironmental context and anatomic distribution consistent with the pathophysiology and clinical presentation of the disease. Importantly, our in vivo molecular profiling studies indicate consistency of in vivo and ex vivo molecular effects of hsp90 inhibition in MM cells.
Although the pathophysiology of MM is not driven by classical client proteins of hsp90, its chaperoning function is critical for MM cells, and its inhibition confers pleiotropic sequelae against pathways (including IGF-1R, IL-6R, IKK-α, and p70S6K) that are not generally considered as major targets of hsp90-mediated regulation or contributors to the antitumor effects of ansamycins. Therefore, hsp90 may play a broader role in chaperoning tumor- or tissue-type-specific constellations of client proteins that are critically involved in tumor cell pathophysiology. This concept could provide the framework for further development of hsp90 inhibition as a therapeutic strategy for a broader spectrum of neoplasias, even those that may not express the currently characterized chaperoning targets of hsp90, and therefore may have not been previously recognized as potential areas for clinical development of hsp90 inhibitors. The critical role of hsp90 for the regulation of MM cell proliferation, survival, drug resistance, and microenvironmental interactions illustrates this disease as a setting for future mechanistic studies on hsp90, and provides the framework for phase 2 clinical trials of hsp90 inhibitors, either as monotherapy or in combination with other anti-MM agents, to improve patient outcome in MM.
Supplementary Material
Acknowledgments
We would like to acknowledge the expert assistance of Dr Roderick Bronson and the Rodent Histopathology Core of the Dana-Farber/Harvard Cancer Center. We apologize for the inability, due to space limitations, to reference all studies relevant to the scope of this article. Dr Rosen serves on the Scientific Advisory Board of Conforma Therapeutics (San Diego, CA), one of whose areas of interest is heat shock protein-90.
Prepublished online as Blood First Edition Paper, October 18, 2005; DOI 10.1182/blood-2005-03-1158.
Supported in part by the Multiple Myeloma Research Foundation (C.S.M., N.S.M.), International Waldenstrom Macroglobulinemia Foundation (C.S.M.), the National Institutes of Health (NIH) Specialized Program for Research Excellence (SPORE) Career Development Award (C.S.M.), the Fulbright Commission (C.J.M.), and NIH grant nos. RO-1 50947 and PO-1 78378.
C.S.M. is a Special Fellow of the Leukemia and Lymphoma Society.
C.S.M. and N.S.M. have equally contributed to this work.
The online version of this article contains a data supplement.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3: 213-217. [DOI] [PubMed] [Google Scholar]
- 2.Xu W, Mimnaugh E, Rosser MF, et al. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001;276: 3702-3708. [DOI] [PubMed] [Google Scholar]
- 3.Xu W, Mimnaugh EG, Kim JS, Trepel JB, Neckers LM. Hsp90, not Grp94, regulates the intracellular trafficking and stability of nascent ErbB2. Cell Stress Chaperones. 2002;7: 91-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohara-Nemoto Y, Nemoto T, Sato N, Ota M. Characterization of the nontransformed and transformed androgen receptor and heat shock protein 90 with high-performance hydrophobic-interaction chromatography. J Steroid Biochem. 1988;31: 295-304. [DOI] [PubMed] [Google Scholar]
- 5.Solit DB, Zheng FF, Drobnjak M, et al. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res. 2002;8: 986-993. [PubMed] [Google Scholar]
- 6.Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature. 1990;348: 166-168. [DOI] [PubMed] [Google Scholar]
- 7.An WG, Schulte TW, Neckers LM. The heat shock protein 90 antagonist geldanamycin alters chaperone association with p210bcr-abl and v-src proteins before their degradation by the proteasome. Cell Growth Differ. 2000;11: 355-360. [PubMed] [Google Scholar]
- 8.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293: 876-880. [DOI] [PubMed] [Google Scholar]
- 9.Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood. 2004;104: 607-618. [DOI] [PubMed] [Google Scholar]
- 10.Davies FE, Dring AM, Li C, et al. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis. Blood. 2003;102: 4504-4511. [DOI] [PubMed] [Google Scholar]
- 11.Mitsiades N, Mitsiades CS, Poulaki V, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A. 2002;99: 14374-14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitsiades CS, Mitsiades N, Poulaki V, et al. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene. 2002;21: 5673-5683. [DOI] [PubMed] [Google Scholar]
- 13.Costa GL, Benson JM, Seroogy CM, Achacoso P, Fathman CG, Nolan GP. Targeting rare populations of murine antigen-specific T lymphocytes by retroviral transduction for potential application in gene therapy for autoimmune disease. J Immunol. 2000;164: 3581-3590. [DOI] [PubMed] [Google Scholar]
- 14.Edinger M, Cao Y-A, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood. 2003;101: 640-648. [DOI] [PubMed] [Google Scholar]
- 15.Armstrong SA, Kung AL, Mabon ME, et al. Inhibition of FLT3 in MLL: validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3: 173-183. [DOI] [PubMed] [Google Scholar]
- 16.Mitsiades CS, Treon SP, Mitsiades N, et al. TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: therapeutic applications. Blood. 2001;98: 795-804. [DOI] [PubMed] [Google Scholar]
- 17.Mitsiades CS, Mitsiades NS, Bronson RT, et al. Fluorescence imaging of multiple myeloma cells in a clinically relevant SCID/NOD in vivo model: biologic and clinical implications. Cancer Res. 2003;63: 6689-6696. [PubMed] [Google Scholar]
- 18.Mitsiades N, Mitsiades CS, Poulaki V, et al. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002;99: 4079-4086. [DOI] [PubMed] [Google Scholar]
- 19.Mitsiades N, Mitsiades CS, Poulaki V, Anderson KC, Treon SP. Intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human multiple myeloma cells. Blood. 2002;99: 2162-2171. [DOI] [PubMed] [Google Scholar]
- 20.Akiyama M, Hideshima T, Hayashi T, et al. Cytokines modulate telomerase activity in a human multiple myeloma cell line. Cancer Res. 2002;62: 3876-3882. [PubMed] [Google Scholar]
- 21.Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci U S A. 2004;101: 540-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shringarpure R, Grune T, Mehlhase J, Davies KJ. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem. 2003;278: 311-318. [DOI] [PubMed] [Google Scholar]
- 23.Mitsiades N, Mitsiades CS, Poulaki V, et al. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood. 2002;99: 4525-4530. [DOI] [PubMed] [Google Scholar]
- 24.Mitsiades N, Mitsiades CS, Richardson PG, et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101: 2377-2380. [DOI] [PubMed] [Google Scholar]
- 25.Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell. 2004;5: 221-230. [DOI] [PubMed] [Google Scholar]
- 26.Mitsiades CS, Mitsiades N, Treon SP, Anderson KC. Proteomic analyses in Waldenstrom's macroglobulinemia and other plasma cell dyscrasias. Semin Oncol. 2003;30: 156-160. [DOI] [PubMed] [Google Scholar]
- 27.Ramanathan RK, Trump DL, Eiseman JL, et al. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clin Cancer Res. 2005;11: 3385-3391. [DOI] [PubMed] [Google Scholar]
- 28.Hideshima T, Chauhan D, Richardson P, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277: 16639-16647. [DOI] [PubMed] [Google Scholar]
- 29.Valentinis B, Navarro M, Zanocco-Marani T, et al. Insulin receptor substrate-1, p70S6K, and cell size in transformation and differentiation of hemopoietic cells. J Biol Chem. 2000;275: 25451-25459. [DOI] [PubMed] [Google Scholar]
- 30.Vihinen M, Smith CI. Structural aspects of signal transduction in B-cells. Crit Rev Immunol. 1996;16: 251-275. [DOI] [PubMed] [Google Scholar]
- 31.Hideshima T, Nakamura N, Chauhan D, Anderson KC. Biologic sequelae of interleukin-6 induced PI3-K/Akt signaling in multiple myeloma. Oncogene. 2001;20: 5991-6000. [DOI] [PubMed] [Google Scholar]
- 32.Chauhan D, Li G, Auclair D, et al. Identification of genes regulated by 2-methoxyestradiol (2ME2) in multiple myeloma cells using oligonucleotide arrays. Blood. 2003;101: 3606-3614. [DOI] [PubMed] [Google Scholar]
- 33.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23: 7448-7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson KC. Novel biologically based therapies for myeloma. Cancer J. 2001;7: S19-S23. [PubMed] [Google Scholar]
- 35.Chauhan D, Uchiyama H, Akbarali Y, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87: 1104-1112. [PubMed] [Google Scholar]
- 36.Poulaki V, Mitsiades CS, McMullan C, et al. Regulation of vascular endothelial growth factor expression by insulin-like growth factor I in thyroid carcinomas. J Clin Endocrinol Metab. 2003;88: 5392-5398. [DOI] [PubMed] [Google Scholar]
- 37.Mimnaugh EG, Xu W, Vos M, et al. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther. 2004;3: 551-566. [PubMed] [Google Scholar]
- 38.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100: 9946-9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banerji U, O'Donnell A, Scurr M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23: 4152-4161. [DOI] [PubMed] [Google Scholar]
- 40.Grem JL, Morrison G, Guo XD, et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23: 1885-1893. [DOI] [PubMed] [Google Scholar]
- 41.Chen EX, Bies RR, Ramanathan RK, et al. Population pharmacokinetic analysis of 17-(allylamino)-17-demethoxygeldanmycin (17AAG) in adult patients with advanced malignancies. Cancer Chemother Pharmacol. 2005;55: 237-243. [DOI] [PubMed] [Google Scholar]
- 42.Goetz M, Toft D, Reid J, et al. A phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23: 1078-1087. [DOI] [PubMed] [Google Scholar]
- 43.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91: 8324-8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chavany C, Mimnaugh E, Miller P, et al. p185erbB2 binds to GRP94 in vivo: dissociation of the p185erbB2/GRP94 heterocomplex by benzoquinone ansamycins precedes depletion of p185erbB2. J Biol Chem. 1996;271: 4974-4977. [DOI] [PubMed] [Google Scholar]
- 45.Eustace BK, Sakurai T, Stewart JK, et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol. 2004;6: 507-514. [DOI] [PubMed] [Google Scholar]
- 46.Picard D. Hsp90 invades the outside. Nat Cell Biol. 2004;6: 479-480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}