

Abstract
Serum transferrin reversibly binds iron in each of two lobes and delivers it to cells by a receptor-mediated, pH-dependant process. The binding and release of iron results in a large conformational change in which two subdomains in each lobe close or open with a rigid twisting motion around a hinge. We report the structure of human serum transferrin (hTF) lacking iron (apo-hTF) which was independently determined by two methods: (1) the crystal structure of recombinant non-glycosylated apo-hTF was solved at 2.7 Å resolution using a MAD phasing strategy, by substituting the nine methionines in hTF with selenomethionine and (2) the structure of glycosylated apo-hTF (isolated from serum) was determined to a resolution of 2.7 Å by molecular replacement using the human apo-N-lobe and the rabbit holo-C1-subdomain as search models. These two crystal structures are essentially identical. They represent the first published model for full-length human TF and reveal that, in contrast to family members (human lactoferrin and hen ovotransferrin), both lobes are almost equally open: 59.4° and 49.5° rotations are required to open the N- and C-lobe, respectively, (compared to closed pig TF). Availability of this structure is critical to a complete understanding of the metal binding properties of each lobe of hTF; the apo-hTF structure suggests that differences in the hinge regions of the N- and C-lobes may influence the rates of iron binding and release. In addition, we evaluate potential interactions between apo-hTF and the human transferrin receptor.
The transferrins are a family of bilobal iron-binding proteins that play the crucial role of binding ferric iron and keeping it in solution, thereby controlling the levels of this important metal in the body (1;2). Human serum transferrin (hTF) is synthesized in the liver and secreted into the plasma; it acquires Fe (III) from the gut and delivers it to iron requiring cells by binding to specific transferrin receptors (TFR) on their surface. The entire hTF-TFR complex is taken up by receptor-mediated endocytosis culminating in iron release within the endosome (3). Essential to the reutilization of hTF, iron free hTF (apo-hTF) remains bound to the TFR at low pH. When the apo-hTF-TFR complex is returned to the cell surface, apo-hTF is released to acquire more iron.
Strong homologies exist, both between TF family members, and between the two lobes of any given TF (4;5). Each N- and C-lobe is divided into two subdomains (designated N1 and N2, and C1 and C2) connected by a hinge that gives rise to a deep cleft containing the iron-binding ligands. Iron is coordinated by four highly conserved amino acid residues: an aspartic acid (the sole ligand from the N1- or C1-subdomain), a tyrosine in the hinge at the edge of the N2- or C2-subdomain, a second tyrosine within the N2- or C2-subdomain, and a histidine at the hinge bordering the N1- or C1-subdomain. In addition, the iron atom is bound by two oxygen atoms from the synergistic anion (carbonate) which is itself stabilized by a conserved arginine residue (6).
A key feature of iron binding and release by TF family members is the large conformational change involving not only opening of the two subdomains in each lobe but also a twist between the N1- and N2-, or C1- and C2-subdomains (7;8). Although the N- and C-lobes of hTF share 56% sequence similarity, many studies show that the rate of iron release from the C-lobe is considerably slower than the rate of release from the N-lobe, particularly at the putative endosomal pH of ~5.6 (9–15). At least some of the difference is attributed to the presence of a “dilysine trigger” in the N-lobe, which is replaced by a triad of residues in the C-lobe (14). The dilysine trigger is composed of Lys206 in the N2-subdomain and Lys296 in the N1-subdomain which reside on opposite sides of the iron binding cleft and are oriented with side chains extending toward one another allowing them to share a hydrogen bond in the iron bound (closed) conformation (16;17). In the C-lobe, a triad of residues (Lys534 in the C2-subdomain, Arg632 and Asp634 in the C1-subdomain) replaces the lysine pair (14;18).
The release of iron from hTF depends upon a number of factors including pH, a chelator (physiologically relevant chelators include citrate, pyrophosphate and ATP), and ionic strength, as well as the specific TFR (19–21). Raymond et al. (22) suggest that a complete model must explain the differences in the rate of iron release from the two lobes, the observed variable chelator concentration dependence, and the effect of anions, as well as, the presence or absence of cooperativity between the lobes.
A 7.5 Å cryo-electron microscopy (cryo-EM) model of diferric hTF bound to TFR was created by docking a human TF N-lobe structure and a rabbit TF C-lobe structure into the electron density map of the complex (since there is no full length human TF structure available) (23). This model offers a preliminary view of the regions of hTF and TFR which interact; it is suggested that both the N1- and N2- subdomains of the hTF N-lobe contact the TFR, while only the C1-subdomain of the hTF C-lobe appears to be involved in the interaction. Interestingly, a ~9 Å translation of the ferric N-lobe (relative to the ferric C-lobe) is required to dock the two lobes into the cryo-EM density. Of relevance, at pH 7.4, the TFR discriminates between diferric, the two monoferric species, and apo-hTF although the basis of this discrimination has not been explained (24–26). Significantly, our studies with authentic monoferric hTF constructs established that each lobe contributes equally (and non-additively) to the binding energy of this interaction with the TFR (15). Clearly a structure of apo-hTF is required to determine whether a change in orientation of the two lobes could provide both a rationale for discrimination and further insight into the receptor interaction.
Here we report the structure of full-length apo-hTF that has been independently determined by two methods; both a non-glycosylated recombinant form of hTF (pH 6.5) and a glycosylated native form of hTF (pH 7.0) were solved to a resolution of 2.7 Å. These two structures, which are identical within the limits of the resolution, find both the N- and C-lobes in the open conformation. This work represents the first mammalian TF structure with an apo C-lobe, the first published structure of full-length hTF, and the first report of a baby hamster kidney (BHK) expression system to substitute the methionine residues in hTF with selenomethionine (SeMet). The apo-hTF structure allows comparisons to other relevant structures including those for diferric pig (2.15 Å, 72% identical) and rabbit TF (2.6 Å, 79% identical) (27), and an unpublished model for an unrefined monoferric hTF with iron in the C-lobe (3.3 Å) (28).
Experimental Procedures
Production of hTF-NG
To produce recombinant non-glycosylated hTF (hTF-NG) with SeMet substituted for methionine, BHK cells transfected with the pNUT plasmid containing the sequence of the N-His tagged hTF-NG were placed into four expanded surface roller bottles (13). Addition of culture media containing SeMet results in a significant deterioration of the cells within 24–48 h. In order to maximize the incorporation, medium containing SeMet was added when production of hTF was at a maximum as determined by a competitive immunoassay (29). Briefly, adherent BHK cells were grown in DMEM-F12 containing 10% FBS. This medium was changed twice at two day intervals, followed by addition of DMEM-F12 containing 1% Ultroser G and 1 mM butyric acid (BA). After one or two changes in this medium, 200 mL of SeMet containing DMEM-F12 (lacking normal methionine), with BA and Ultroser G was added to each roller bottle. Following a 4 h wash-in period this media was discarded and replaced with 250 mL of the same medium for an additional 48 hours of incubation. The recombinant hTF-NG was purified from the medium as described in detail (30). In two production runs between 8 and 16 mg of SeMet containing N-His hTF-NG was produced, of which approximately half was recovered. Electrospray mass spectrometry analysis indicated a mass consistent with incorporation of 8–9 SeMet residues (data not shown).
Purification of apo-hTF-Gly
In independent experiments, lyophilized human serum TF lacking iron (apo-hTF-Gly) was obtained from Sigma-Aldrich and reconstituted in 50 mM Tris-HCl, pH 8.0 and 20 mM sodium carbonate at a protein concentration of 5 mg/mL. The protein was applied to a 10 mL Q-Sepharose High Performance column (GE Heathcare) equilibrated with 50 mM Tris-HCl, pH 8.0 and 20 mM sodium carbonate. The apo-hTF was eluted using a linear gradient from 0 to 150 mM NaCl over 3 column volumes. Peak fractions were pooled and dialyzed overnight into 20 mM Tris-HCl, pH 8.0, 20 mM sodium carbonate and 200 mM sodium chloride. Crystallization- Recombinant diferric hTF-NG (with or without a His tag) at a concentration of 15 mg/mL in 0.1 M ammonium bicarbonate, was mixed with an equal volume of reservoir solution composed of 0.3 M ammonium citrate (pH 6.5) and 16–18% PEG 3350 at 20°C. The SeMet N-His hTF-NG required slightly lower levels of PEG 3350 and streak seeding using the non-SeMet labeled hTF-NG crystals. Clear crystals (0.2 mm x 0.3 mm x 0.4 mm) formed in 3–5 weeks. Crystals of the SeMet-labeled protein were essentially isomorphous with those of the native protein, showing similar cell dimensions and crystallizing in the orthorhombic space group P212121 with two molecules in the asymmetric unit (Table 1). All crystals were cryoprotected by addition to the hanging drop of 0.5 μL of a solution of 25% PEG 3350/30% ethylene glycol.
Table 1.
Data collection, crystallographic refinement and model statistics
| Native 1 | Native 2 | Inflection | Peak | Remote | |
|---|---|---|---|---|---|
| Protein | hTF-Gly | hTF-NG | hTF-NG | hTF-NG | hTF-NG |
| Cell parameters | |||||
| space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| a, b, c (Å) | 88.32 | 88.99 | 88.49 | 88.28 | 88.47 |
| 103.26 | 102.16 | 103.35 | 103.39 | 103.55 | |
| 200.36 | 197.04 | 199.17 | 198.94 | 198.384 | |
| Data collection statistics | |||||
| location | APS | Chess A1 | BNL | BNL | BNL |
| 22ID | BNL X26 | X25 | X26C | X26C | |
| wavelength (Å) | 1.0000 | 0.935 | 0.9794 | 0.9790 | 0.9641 |
| 1.100 | |||||
| resolution range (Å) | 30 – 2.7 | 50 – 2.7 | 50 – 2.9 | 25 – 3.2 | 50 – 3.3 |
| unique reflections | 49,871 | 47,968 | 37,498 | 31,124 | 28,343 |
| completeness (%) | 96.7 (90.5) | 95.9 (83.6) | 99.9 (98.9) | 99.4 (93.8) | 100 (99.9) |
| redundancy | 6.6 (6.5) | 6.0 (4.5) | 6.0 (5.3) | 7.1 (6.5) | 7.3 (7.5) |
| Rmerge (%)a | 6.8 | 7.1 | 8.2 | 11.6 | 12.7 |
| I/σ | 22.8 (2.0) | 41.0 (5.3) | 12.6 (0.9) | 23.6 (3.4) | 22.8 (4.0) |
| detector | Mar300 | Q210 | Q315 | Q4 | Q4 |
| Q4 | |||||
| integration software | HKL2000 | HKL v1.98.2 | HKL2000 | HKL2000 | HKL2000 |
| Molecular replacement | 1A8E (N-lobe) | ||||
| search models | 1H76 (C-lobe) | ||||
| MAD phases | |||||
| Figure of Merit (FOM) | 100 – 2.9 Å | ||||
| acentrics | 0.41 | ||||
| centrics | 0.38 | ||||
| density-mod. mean FOM | 0.80 | ||||
| Model statistics | |||||
| Resolution (Å) | 15 – 2.7 | 15 – 2.7 | |||
| Rworkb | 23.2 | 27.3 | |||
| Rfreec | 29.3 | 32.3 | |||
| Refined model | |||||
| # amino acid residues | 1352 | 1352 | |||
| # citrate molecules | 7 | 4 | |||
| # glycerol molecules | 4 | 2 | |||
| # Atoms, non-hydrogen | |||||
| protein | 10486 | 10488 | |||
| ligand | 115 | 64 | |||
| r.m.s. deviation, bonds (Å) | 0.009 | 0.009 | |||
| r.m.s. deviation, angles (°) | 1.57 | 1.33 | |||
| Ramachandran plot (%) | |||||
| most favored regions | 77.0 | 78.2 | |||
| additionally allowed | 20.6 | 19.4 | |||
| generously allowed | 1.9 | 1.9 | |||
| disallowed regions | 0.5 | 0.5 | |||
| Average B-factor (Å2) | 75.2 | 67.4 | |||
| Range of B-factors (Å2) | 32 – 125 | 14 – 135 | |||
Rmerge = ∑ ∑ I(h)j − <I(h)>/∑I(h), where I(h)j is the jth measurement of diffraction intensity of reflection h and <I(h)> is the average intensity of reflection h for all j measurements.
Rwork = ∑ (|Fo| − |Fc|)/∑ |Fo|.
Rfree is calculated using a test set of 5% of the reflection excluded from refinement.
Native apo-hTF-Gly was concentrated in a Centriprep 30 concentrator (Millipore) to 30 mg/mL and screened against commercially available 96-condition screens using a Mosquito crystallization robot (TTP Labtech) with a hanging drop format (drop size 200 nL protein plus 200 nL well solution). Conditions yielding the best crystals were then further refined using 24-well VDX plates (Hampton Research). The crystals used for native data collection were obtained from hanging drops with a well solution of 0.2 M ammonium citrate pH 7.0, 20% PEG-3350, and 15% glycerol, incubated at 21°C. Drop sizes varied from 2 to 16 μL and consisted of equal parts of protein and well solution. Crystals grew in ~24 hours and were flash frozen in propane cooled to −170°C.
Data collection and refinement
For the apo-hTF-NG crystals which contained SeMet, multiple wavelength anomalous dispersion (MAD) data were collected at −170°C on beamlines X26C (peak and remote) and X25 (inflection) at Brookhaven National Laboratory. The data was reduced, scaled, and merged using DENZO/SCALEPACK (31) (Table 1). In order to find the selenium sites, MAD data were prepared with XPREP and analyzed with ShelxD (32). The data sets were combined and refinement of the selenium sites was carried out using autoSHARP (33;34). Profess (35) was used to find NCS, which revealed two molecules in the asymmetric unit. Following a round of density modification, the structure of apo-hTF N-lobe (PDB 1BP5) was used as a search model for a phased translation and rotation function using MOLREP (36). Subsequent model building of the C-lobe was done using O (37) in a stepwise manner by incorporating fragments of pig holo-TF (converted to the human sequence). Iterative rounds of density modification and phase recombination were performed with SOLOMON (38). Refinement was accomplished using both CNS (39) and REFMAC (35;40).
Native diffraction data from an apo-hTF-Gly crystal were collected at the Advanced Photon Source on SER-CAT beamline 22ID at −170°C. The crystal belonged to the orthorhombic space group P212121, with two molecules in the asymmetric unit and a solvent content of 59.3%. The images were reduced, scaled and merged using HKL2000 (31) (Table 1) and the structure was solved using the molecular replacement program Phaser (41). A search model consisting of the human apo-N-lobe structure (1BP5) and the rabbit holo-C1-subdomain (residues 342 – 424 and 579 – 676 from 1JNF) was used, leading to a single solution containing two copies of each component. After a single round of rigid body refinement using the program REFMAC (35;40) the rabbit C2-subdomain was fit into the electron density. The C1- and C2-subdomains were then mutated to the human sequence and the linker regions between the N- and C-lobes and between the C1- and C2-subdomains were built into the electron density. This structure was further refined with REFMAC using medium NCS restraints initially, followed by release of NCS restraints. After building in the glycerol and citrate molecules, final rounds of refinement were performed in CNS (39).
RESULTS & DISCUSSION
Comparison of (recombinant) apo-hTF-NG and (native) apo-hTF-Gly
Both protein preparations crystallized in the orthorhombic space group P212121, having almost identical unit cell dimensions and two molecules per asymmetric unit (Table 1). The recombinant and native structures had an r.m.s. deviation of only 0.73 Å for 1352 equivalent Cα positions and therefore we are, for the most part, presenting and discussing them as a single structure (apo-hTF). As in all other serum TF structures, no electron density for residues 1–3 is visible. Even the His- tagged recombinant apo-hTF-NG, with 14 extra amino acids at the N-terminus, showed no density in this region, implying that the amino terminus is very flexible. Additionally, no density is found in the vicinity of Asn413 and Asn611 in the native apo-hTF-Gly, indicating that the glycan moieties are flexible and/or present in multiple conformations. Clearly our recombinant hTF lacks carbohydrate due to the mutation of the asparagine linkage sites to aspartic acid residues. Significantly, the presence or absence of carbohydrate has no effect on either iron release or interaction with the TFR (42;43).
Quality of Final Model
Data collection, refinement, and model statistics are summarized in Table 1. No breaks in the main chain density are observed in apo-hTF and, as shown in Table 1, the geometry is good. The Ramachandran plot of the main chain torsion angles shows that 97.6% of the residues lie in most favored or allowed regions and that only six of the 1352 residues of the two non-crystallographic symmetry related molecules reside in the disallowed region. Four of these residues are Leu294 and Leu630 in each of the molecules, comprising the central residues in classic γ -turns (Leu-Leu-Phe) with phi and psi angles of approximately 77° and −46°, respectively (44). This structural feature was first noted in the hTF N-lobe (45), and subsequently has been observed in each lobe of all mammalian and avian TF molecules and in the C-lobe of all fish TFs (5;6). Interestingly, the Leu-Leu-Phe sequence is not conserved in any insect TF N-lobe and is only partially conserved in the insect C-lobe sequences (5;6). However, since the function of insect TF remains unknown, the importance of this finding is unclear. While the role of the γ -turn in mammalian TFs has not been definitively established, we believe that it is significant that it immediately precedes the dilysine trigger residue Lys296 in the N-lobe and triad residue Arg632 in the C-lobe (45). Within this context, the γ -turn may help to stabilize the orientation of these two residues to provide better repulsion or a triggering action, aiding in opening of the cleft.
Overall organization
As with all TF family members, the structure of hTF illustrates the bilobal nature of the molecule (Figure 1) with an amino-terminal lobe (N-lobe, residues 1–331) and a carboxyl-terminal lobe (C-lobe, residues 339–679). The lobes are connected by a linker peptide (residues 332–338) that is unstructured, but visible in our model (although missing in the diferric pig TF structure (27)). Each of the lobes is further separated into two subdomains: the N1- (1–92, 247–331) and C1- (339–425, 573–679) subdomains are each composed of two discontinuous sections of the polypeptide chain, while the N2- (93–246) and C2- (426–572) subdomains are each composed of a single region of continuous polypeptide (Figure 1 and Supplemental Figure 1). Though kinetically distinct, the fold of the C-lobe is equivalent to the fold of other N- and C-lobes (Table 2).
Fig. 1.
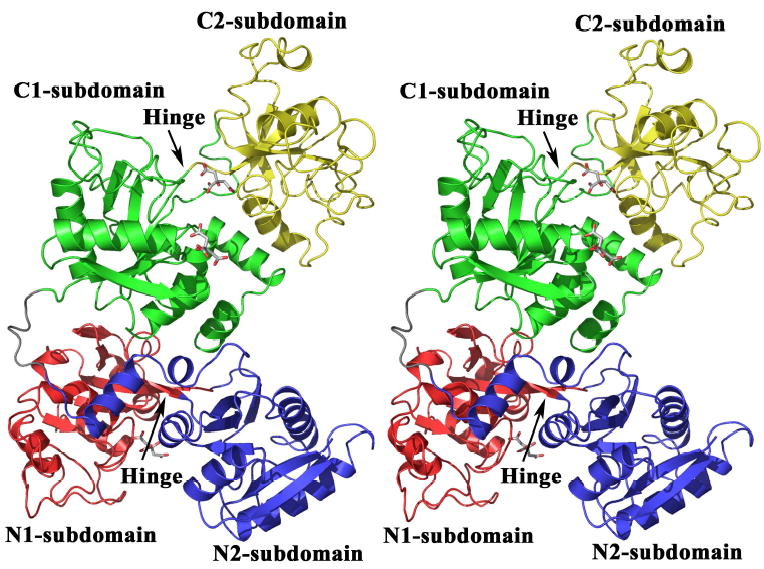
Stereo image of the crystal structure of apo-hTF with subdomain N1 in red, N2 in blue, C1 in green and C2 in yellow. The linker region between the lobes is colored grey and the hinge between the N1- and N2- subdomains and the C1- and C2-subdomains is identified. Four of the citrate molecules associated with apo-hTF are shown by stick representation. All figures were made with PYMOL (89).
Table 2.
Comparison of apo-hTF secondary structure to holo-pig TF and holo-chicken oTF.
|
α -Helices |
||||||
|---|---|---|---|---|---|---|
|
N-lobe |
C-lobe |
|||||
| Nomenclature | hTFa | pTFb | oTFc | hTF | pTF | oTF |
| 1 | 13–25 | 12–29 | 13–28 | 349–362 | 353–366 | 351–365 |
| 2 | 45–53 | 43–53 | 41–53 | 374–383 | 378–387 | 376–388 |
| 3 | 64–71 | 62–70 | 60–68 | 393–401 | 396–406 | 395–405 |
| 418–420 | ||||||
| 4 | 109–111 | 108–110 | 105–109 | 441–443 | 444–448 | |
| 5 | 129–135 | 127–134 | 121–136 | 461–466 | 463–478 | 460–476 |
| 5a | 137–138 | 135–137 | 468–470 | |||
| 6 | 146–153 | 145–152 | 147–156 | 476–478 | 481–486 | 480–483 |
| 6a | 168–170 | 166–169 | 167–174 | 502–504 | 499–501 | 496–499 |
| 6b | 175–177 | |||||
| 7 | 187–196 | 191–201 | 189–201 | 516–526 | 525–536 | 523–534 |
| 7a | 537–540 | |||||
| 8 | 210–213 | 212–217 | 211–217 | 549–551 | 545–550 | 543–549 |
| 8a | 217–220 | 223–225 | 220–223 | 565–567 | ||
| 8a1 | 556–557 | |||||
| 8b | 235–240 | 239–244 | 572–576 | 580–585 | ||
| 9 | 260–273 | 263–278 | 259–275 | 594–608 | 600–617 | 600–616 |
| 10 | 311–315 | 314–319 | 315–321 | 647–651 | 655–659 | 652–658 |
| 11 | 317–328 | 321–331 | 321–332 | 653–662 | 661–673 | 658–669 |
| 12 | 669–677 | 677–682 | 674–686 | |||
|
β -Stands
|
||||||
|
N-lobe
|
C-lobe
|
|||||
| Nomenclature | hTFa | pTFb | oTFc | hTF | pTF | oTF |
| a | 5–10 | 4–10 | 6–11 | 342–346 | 346–352 | 344–351 |
| b | 36–41 | 35–41 | 33–39 | 366–370 | 369–375 | 368–374 |
| c | 60–61 | 59–61 | 54–59 | 388–391 | 393–395 | 389–394 |
| d | 77–84 | 76–84 | 74–82 | 405–411 | 409–415 | 407–415 |
| e | 94–102 | 91–102 | 91–100 | 428–433 | 431–439 | 430–439 |
| f | 116–119 | 114–119 | 112–116 | 447–451 | 454–458 | 451–455 |
| g | 156–158 | 154–159 | 157–161 | 482–484 | 489–492 | 485–489 |
| h | 202–206 | 205–211 | 204–210 | 530–533 | 539–544 | 536–542 |
| i | 223–225 | 226–231 | 223–228 | 559–562 | 568–572 | 565–570 |
| ia | 235–238 | 232–234 | 568–570 | 575–579 | 574–576 | |
| j | 244–247 | 247–259 | 244–255 | 588–591 | 586–597 | |
| ja | 249–254 | 586–588 | 595–598 | |||
| k | 301–304 | 304–309 | 305–310 | 637–640 | 645–649 | 642–647 |
When we superimpose our apo-hTF structure on the diferric pig TF structure, the N1- and C1-subdomains align closely (r.m.s. deviation = 1.18 Å) (Figure 2; Table 3). This finding suggests that the twist and opening of each lobe when iron is released is restricted to the N2- and C2-subdomains which clearly has implications with regard to how the TFR discriminates between diferric, monoferric and apo-conformations.
Fig. 2.
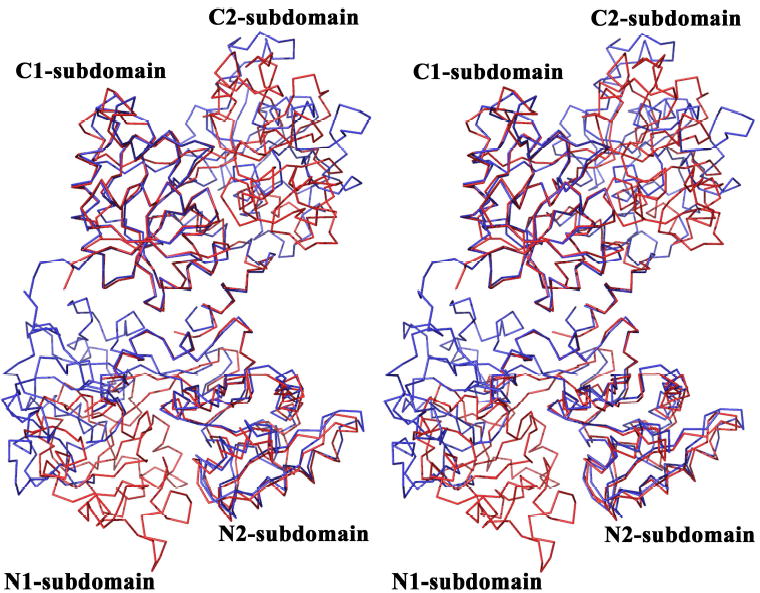
Stereo image of the structural superposition of apo-hTF with holo-pig TF created by aligning the C1-subdomains of the two structures and then superimposing the full molecules on their own C1-subdomains. Apo-hTF is blue and holo-pig TF is red. Note that when the C1-subdomains are superimposed, the N1-subdomains fall into register, but the N2- and C2-subdomains are misaligned. The overlay used SPDBV (90)
Table 3.
Structural alignmentsa of the subdomains of apo-hTF with the subdomains of holo-pig TF.
| Apo hTF N1- | Apo hTF N2- | Apo hTF C1- | Apo hTF C1- | |
|---|---|---|---|---|
| Pig TFb N1- | 1.175 Å(371 AA) | |||
| Pig TF N2- | 1.006 Å (162 AA) | |||
| Pig TF C1- | 1.308 Å (151 AA) | 1.175 Å (371 AA) | ||
| Pig TF C2- | 1.097 Å (139 AA) | 1.033 Å (171 AA) |
B-factors
The regions with the lowest B factors include core regions in the N1- and N2-subdomains as well as regions comprised of residues 343–359, 368–407, 627–632, and 667–674 of the C1-subdomain (Supplemental Figure 2). The regions with the highest B factors include a loop in the beginning of the N1-subdomain (residues 29–34), the hinge between the N- and C-lobes (residues 332–338), residues 415–420 and 611–618 in the C1-subdomain and the majority of the C2-subdomain. This finding is consistent with an electrospray ionization mass spectrometry study indicating that the apo-N-lobe is significantly less flexible than the apo-C-lobe (43).
Hinge
The extent of the movement of the subdomains can be estimated by superimposing the N1- or C1-subdomains and determining the rotation and translation functions required to superimpose the N2- or C2-subdomains on the respective N2- or C2-subdomains from an open or closed molecule (46). By this analysis an average rotation of 59.4° has occurred in the apo-hTF N-lobe relative to the diferric pig structure, similar to the average 63.2° rotation (range of 62.9°–64.8° for the four molecules in the asymmetric unit) reported for the isolated apo-hTF-N-lobe structure (46). In the case of apo-hTF C-lobe, a subdomain rotation of 51.8° has occurred relative to the closed monoferric C-lobe (28), and a 49.5° rotation is required relative to the diferric pig TF structure (27) (see Supplemental Figure 3).
In contrast to other family members, the N- and C-lobes of apo-hTF are open to almost the same degree. For example, in the human apo-lactoferrin (LTF) structure the iron-binding cleft in the N-lobe opens 53.4°, whereas the C-lobe is closed (similar to the diferric structure but lacking iron) (47). Two alternative crystal forms of LTF have been reported by Baker et al. featuring C-lobes open by 18° or o27° (11). The chicken apo-ovotransferrin (oTF) C-lobe opens 35° compared to a 53° opening for the apo-oTF N-lobe (8); thus chicken oTF is intermediate between the hTF and LTF structures. Nevertheless, duck oTF and camel LTF have equivalent open N- and C-lobes, 51.6° and 49.9° and 57.8° and 57.3°, respectively, so the patterns are not consistent or predictable (48;49).
Jeffrey et al. (46) delineated the hinge residues in the N-lobe of hTF as Thr93, Val246, and Pro247 due to the ~36° psi changes between the ferric and apo forms of these residues. As expected from the close structural alignment of our N-lobe with the isolated apo N-lobe, we observe similar changes in the same residues when compared to the iron-containing pig TF N-lobe. In contrast in the C-lobe the hinge appears larger, comprised of Ala424 and Gly425 preceding strand e, and Arg581, Ala582 & Pro583 which begin strand j (see Supplemental Figure 1 for helix and strand designations). On average these residues change ~36° in psi between apo-hTF and the diferric pig TF structure. Inspection of the secondary structure of the hinge regions of the N-lobe and C-lobe of apo-hTF reveals a striking difference. In the N-lobe, the hinge lies adjacent to an anti-parallel β-sheet formed from strands e and j. However, in the C-lobe, strand e is shorter (starting 3 residues from the hinge) and strand j is entirely absent (Figure 3). We suggest that this difference in the hinge regions of the N- and C-lobes may contribute to differences in overall flexibility between the lobes (43) and could therefore play an important role in the differing rates of iron binding and release.
Fig. 3.
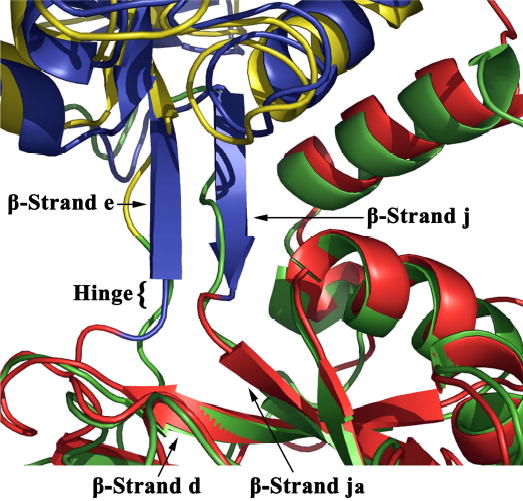
A superimposition of the N-lobe and C-lobe of apo-hTF illustrating the difference in secondary structure of the region surrounding the hinge residues between the two subdomains of each lobe. In the N-lobe (N1- blue and N2- red), the hinge is adjacent to an anti-parallel β-sheet formed from β-strands e and j. In the C-lobe (C1- green and C2- yellow) the hinge is located within an unstructured region, as β-strand e is shortened and β-strand j is entirely absent. The overlay used SPDBV (90)
In our apo-structure, crystal packing could influence opening of the cleft in the N-lobe because the N-lobe of one molecule in the asymmetric unit intercalates into the cleft of the N-lobe of the other molecule (Figure 2). In fact the side chain of Gln20 in the A molecule lies within hydrogen bonding distance of Cys177 and Cys179 in the B molecule. Additionally, the side chain of Glu13 in molecule A interacts with the side chain of Arg124 in molecule B. This is of interest since Arg124 is the critical anion binding residue in the N-lobe of hTF, whose important role in iron uptake and release has been extensively studied (6;19;50–52). Nevertheless, we conclude, that crystal packing is not a factor given the similarity of the degree of opening of the isolated hTF N-lobe (63.2°) to the current structure (59.4°).
Metal and anion binding sites
Seven molecules of citrate were found bound to apo-hTF-Gly and four citrate molecules were found in apo-hTF-NG (Table 1). The presence and location of citrate in the structure is notable due to the physiological relevance of citrate as both an anion and a chelator (53–55). Six of the seven citrates bound to the two NCS-related molecules are found within the open clefts of the N- and C-lobes (see Supplemental Table 1). As shown in Supplemental Figure 4, one citrate molecule (9207) located within the N-lobe cleft of apo-hTF-Gly molecule A is hydrogen-bonded to the side chains of dilysine trigger residues Lys206 and Lys296 as well as coordinating the side chain of Arg124 (normally bound to the synergistic carbonate anion). Since it is thought that binding of an anion sets up the site for the iron to be captured this is an interesting finding. Given that the concentration of citrate within serum is 100 μM (56), it is plausible that citrate could be the anion that binds to the hTF and prepares the site to receive the iron. However, in the N-lobe of apo-hTF-Gly molecule B, the citrate (9206) that lies in a similar position does not coordinate the dilysine trigger residues, contacting only the backbone nitrogens of residues 125–127.
In the C-lobes of both molecules in the asymmetric unit of apo-hTF-Gly, two citrate molecules are present. In molecule A, one citrate (9202), coordinates the side chains of iron-binding ligand His585 and triad member Arg632, as well as the backbone nitrogen of Thr457. The corresponding citrate (9204) in molecule B contacts Arg632 and Thr457. The second citrate (9203 in A and 9205 in B) in both molecules contacts the backbone nitrogens of the ligand Tyr517 as well as Thr518. The differences in the positions of the citrates in each molecule emphasize the flexible nature of the clefts within the two lobes.
The seventh citrate interacts with the backbone nitrogens of two His residues (349 and 350) at the beginning of α helix 1 (residues 349–362) of the C-lobe in molecule B with the same residues in a symmetry-related molecule, facilitating crystal packing. This feature of the structure is especially interesting because of the evidence that these two histidines, particularly His349, are important in interacting with the TFR to bring about the release of iron from the C-lobe at endosomal pH (57). Notably only citrates equivalent to 9201, 9202 9204 and 9207 are observed in apo-hTF-NG.
Triad
Lys534, Arg632, and Asp634 have been implicated in the pH dependent conformational change in the C-lobe during iron release (14;16), making their positions in the apo-C-lobe of great interest. Arg632 and Asp634 (in the C2-subdomain) are adjacent to one another on one side of the open C-lobe cleft. A salt bridge exists between the guanidinium group of Arg632 and the side chain carboxyl group of Asp634. Lys534 lies in the C1-subdomain ~10 Å across the cleft, a relationship highly reminiscent of that observed for the dilysine pair in the N-lobe (46). Previously we demonstrated that Arg632 could serve as a kinetically significant anion binding site (18). We note that the guanidinium group of Arg632 is solvent accessible in the apo-structure and is found bound to citrate, emphasizing both its reactivity and accessibility.
Interactions between lobes
A co-operative effect of one lobe on the other lobe has been convincingly documented in a number of studies using a variety of techniques which include: NMR of hTF (58), absorption spectra of oTF (59), pH-dependent iron release of LTF (60), calorimetric studies of both hTF and oTF (61–63) electrospray ionization mass spectrometry studies of hTF (43), iron release from monoferric LTF (64), analysis by urea gels (22) and chemical relaxation studies (65–67). Significantly, a number of these studies specifically attribute cooperative effects between the two lobes of hTF to participation of helix 12 from the C-lobe (2;68). However, this helix undergoes no discernable movement between the apo, monoferric, or diferric states of TF (using pig serum TF as a model for the diferric form). The absence of a significant change in the position of this helix between the conformations may simply reflect the absence of the TFR which we believe may be critical in inducing such a change (see below) (69;70).
Inspection of the interface between the lobes suggests a loose, predominantly hydrophobic interface in which only a small amount (550 Å2) of the total accessible surface area (~8800 Å2) is buried (Table 4). This is similar to what has been observed in other TF structures (7;8;71–73). Although we assumed that since the diferric structure is more compact than the apo-structure, it would sequester more surface area at the interface, the opposite trend prevails. Diferric TF family members tend to have smaller interfaces (~421 Å2) than the apo molecules (~569 Å2), although both are quite modest compared to the overall surface area of the molecule. In addition to the hydrophobic interface between the lobes, two residues from the N-lobe form salt bridges with residues in the C1-subdomain (Figure 4). Asp240 at the end of α-helix 8b in the N2-subdomain interacts with Arg678 adjacent to the C-terminal helix (helix 12) in the C1-subdomain, and Arg308 in the N1-subdomain (in the loop prior to helix 10) interacts with Asp376 in helix 2 of the C1-subdomain. These observations may be relevant to interaction with the TFR (see below)
Table 4.
Surface area (Å2)a of TFs sequestered by the lobe/lobe interface.
| Apo | Monoferricb | Diferric | |
|---|---|---|---|
| LTF | 660 (Human 1CB6) c | 454 (Humand 1B0L) | |
| 567 (Camel 1DTZ) | 253 (Human 1LFG) | ||
| oTF | 496 (Chicken 1AIV) | 430 (Duck 1DOT) | |
| 485 (Chicken 1OVT) | |||
| TF | 552 (Human) | 532 (Human) | 379 (Pig 1H76) |
| 524 (Rabbit 1JNF) |
Surface area calculated ((N-lobe + C-lobe) − TF) using the WHATIF (http://swift.cmbi.kun.nl/WIWWWI/) (86).
Structure from (28).
Delineates PDB accession numbers.
From the structure of recombinant human LTF.
Fig. 4.
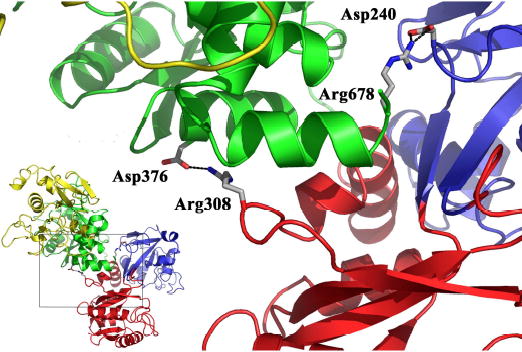
The interface between the two-lobes of apo-hTF is shown to illustrate the inter-lobe contacts. Two pairs of salt bridged side chains are present at the interface: Arg308 in the N1-subdomain to Asp376 in the C1-subdomain and Asp240 in the N2- subdomain to Arg678 in the C1-subdomain. The subdomains are colored as in Figure 1 and the salt bridging side chains are shown in stick representation. The inset shows the location of the inter-lobe contacts the context of the complete apo-hTF structure.
TFR
Of particular interest is how the conformational changes in hTF are facilitated and complimented by the TFR. It is well established that the TFR helps to modulate iron-release in a pH-dependent manner (74). Also, the receptor preferentially binds diferric hTF (compared to the monoferric and apo-hTF conformations) at pH 7.4, although, as mentioned above, the structural basis for this discrimination is unclear (69;75). Significantly, the extensive mutagenesis studies of Giannetti et al. (76) identified TFR mutants with differential effects in binding to apo- and diferric hTF, strongly suggesting differences in some of the contact regions in the two conformations of hTF. Thus, it is important to establish which region(s) of hTF interact with the TFR and how this association is manifested in a structural change that alters iron-release rates relative to hTF alone.
Tables 5 and 6 show residues in the N- and C-lobes of hTF that have been implicated in TFR binding by a variety of different approaches. Sequence alignments are shown for the regions of TF which bind human TFR with high affinity (human, rabbit and pig TF), low affinity (bovine TF) (77;78) or not at all (oTF and human LTF) (77). Clearly residues that are critical to the interaction should be conserved in the rabbit and pig TFs and not in the other three sequences. For the N-lobe, the sequence of residues Pro142 – Pro145 in the N2-subdomain is completely conserved in the TFs that bind the TFR, and is poorly conserved in the TFs that do not. Iron release from the N-lobe is accompanied by opening of the cleft formed by the two subdomains (45;46). This cleft opening occurs as a result of α-helix 5 (residues 129-134) pivoting on α-helix 11 (residues 317-328). Within the N2-subdomain, a loop comprised of residues 136-145 and sitting between two α-helices (5 and 6), undergoes considerable rearrangement attributed to its connection to α-helix 5. Additionally, this loop contains Cys137 which forms a disulfide bond with Cys331 lying just beyond α-helix 11. As a result of the pivoting action, the loop is pushed against the hinge region joining the two lobes. Four residues within this loop, Pro142-Arg143-Lys144-Pro145, appear to make contact with TFR residues Tyr123-Trp124-Asp125. The mobility of the loop in response to iron uptake and release therefore may have functional significance. Additionally, although this loop has moved by a distance of 10-12 Å in our apo structure compared to the iron containing N-lobe bound to the TFR (from the cryo EM complex model), it would still remain close enough to the TFR to participate in binding (23). In both the apo and diferric oTF structures, which do not bind to human TFR, there is a deviation from the hTF structures in which this region is slightly larger, and more mobile, and would clash with the TFR in the cryo-EM model of the complex.
Table 5.
Residues in the hTF N-lobe that may interact with the human TFR compared to residues at equivalent positions in rabbit TF and pig TF.
| N1-subdomain | N2-subdomaina | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bind TFRb | 71 | 72 | 73 | 74 | 142 | 143 | 144 | 145 | ||||||
| HUMAN TF | Y | L | A | P | P | R | K | P | ||||||
| RABBIT TF | G | L | T | P | P | R | K | P | ||||||
| PIG TF | G | L | A | P | P | R | K | P | ||||||
| Do not Bind TFR | ||||||||||||||
| BOVINE TF | G | L | K | P | P | Q | E | S | ||||||
| CHICKEN oTF | G | L | A | P | I | E | S | G | ||||||
| HUMAN LTF | G | L | A | P | P | P | E | P | ||||||
| Bind TFRc | 89 | 90 | 91 | 92 | 93 | 94 | 95 | 96 | 97 | 98 | 99 | 100 | 101 | 102 |
| HUMAN TF | E | D | P | Q | T | F | Y | Y | A | V | A | V | V | K |
| RABBIT TF | E | N | P | K | T | F | Y | Y | A | V | A | V | L | K |
| PIG TF | D | N | P | Q | T | H | Y | Y | A | V | A | V | V | K |
| Do not Bind TFR | ||||||||||||||
| BOVINE TF | D | N | P | Q | T | H | Y | Y | A | V | A | V | V | K |
| CHICKEN oTF | E | G | S | T | T | S | Y | Y | A | V | A | V | V | K |
| HUMAN LTF | R | Q | P | R | T | H | Y | Y | A | V | A | V | V | K |
Table 6.
Residues in hTF C1-subdomain identified as interacting with the human TFRa
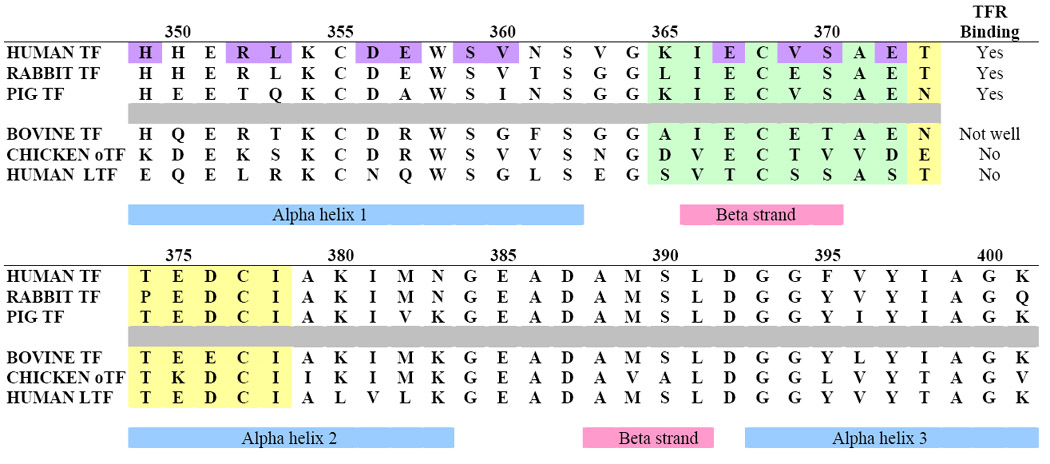 |
In contrast to the Pro141 – Pro145 sequence, residues 71-74 (identified in the cryo-EM model (31)) and residues 89-102 (identified by radiation foot printing (81)) do not show a convincing pattern of conservation. Likewise, one other region (residues 51-88) identified in the radiation foot printing studies (79) does not seem credible based on sequence comparisons. It is possible that proximity to the iron binding ligands (Asp63 in the 51-88 sequence and Tyr95 in the 89-102 sequence) could make these regions particularly susceptible to radiation damage; binding to the TFR might provide a shielding effect and explain the susceptibility differences.
In general there is better agreement between the regions identified by various techniques in the C-lobe of hTF. A monoclonal antibody (MAb) specific to a sequential epitope in the C-lobe of hTF blocks binding of hTF to the TFR on HeLa S3 cells (80). Interestingly, this same MAb binds to diferric hTF with two-fold higher affinity compared to apo-hTF. The epitope was mapped to the C1- subdomain of hTF (residues Lys365-Ile378). As shown in Table 6, 11 residues in the C1-subdomain that are in contact with the helical region of the TFR were identified in the cryo-EM model of hTF bound to TFR (23). These residues lie between His349 and Glu372, and include Glu367, Glu369 (Val in the human sequence), Ser370, and Glu372, all of which are part of the epitope recognized by the MAb. Thus, the mapping study confirms the identification of this particular region of the C-lobe in binding to the TFR and since the affinity of the MAb changes as a function of iron status, the results are consistent with a conformational change in this region of the C-lobe required for His349 to come into contact with the TFR. The only residue with a significantly different conformation in our apo-structure compared to the unrefined monoferric hTF is Arg352, whose side chain is oriented toward the iron-binding cleft, rather than away (28). This could be very important due to proximity of this residue to His349 (57). Radiation foot printing identified a region in the C2-subdomain (residues 457-470) that differed in susceptibility to radiation damage. Since the C2- subdomain makes no contact with the TFR in the model it is difficult to ascribe the effect to TFR interaction.
Understanding the basis by which the TFR discriminates between various family members and between the four conformers of hTF is a long standing pursuit in TF research. The cryo-EM model (23) was constructed with human TFR, human N-lobe TF and rabbit C-lobe TF. As described above, placement of the C1- subdomain is convincingly supported by all of the available data. We therefore superimposed the C1-subdomains of apo- and/or iron containing chicken oTF, human LTF, human TF, rabbit TF, and pig TF (Table 4). Two observations can be made: first, all of the sequences (oTF, LTF rabbit and pig TF) have either insertions or deletions in the loop formed by human TF Cys495 and Cys506, which lies near the C-terminus of the TFR. Second, the linker peptide which connects the N- and C-lobes closely approaches the TFR in the cryo-EM model. In LTF this region adopts a helical conformation which could constrain the independent movement of the lobes relative to each other and potentially restrict access to a TF receptor. In mammalian TFs the linker region is not structured but there is a disulfide bond between Cys339 and Cys596 (human numbering) which is missing in oTF and LTF. This feature would be predicted to constrain movement between the N- and C-lobes. In addition, although the unstructured linker of hTF is probably long enough to accommodate the 9 Å separation of the two lobes seen in the cryo-EM model, it would require disrupting two salt bridges formed between the N-lobe and C1- subdomain, as well as altering the regions of buried surface between the N- and C-lobes, seen in our apo-hTF structure. Perhaps the most logical explanation for the altered TF conformation observed in the cryo-EM structure is that there is a pH induced conformational change in the TFR itself to accommodate TF. Support for this idea comes from the observation of conformational changes in the TFR when going from neutral to acidic pH (81). Additionally, changes in the relative orientations of the TFR subdomains in response to binding HFE (hemochromatosis protein, which competes with hTF for binding to TFR), have been reported (82). Thus, a combination of pH and ligand (TF) binding may change the conformation of the TFR. A recent single particle reconstruction of apo-hTF (approximated by using human apo-TF N-lobe and duck apo-oTF C-lobe) in complex with TFR has provided a low resolution (16 Å) structure (83), and suggests that apo-hTF binds to the TFR in the same location as diferric hTF. However, it is possible that conformational changes occur in both hTF and the TFR upon complex formation and pH change; such changes could only be determined from higher resolution structure(s) of the hTF-TFR complex.
In summary, our structure reveals that apo-hTF adopts a similar molecular architecture to other TFs, although in our apo-hTF structure both the N-and C-lobes have a similar open conformation. Comparison of apo-hTF with holo-pig TF suggests that, as in other TFs, the two subdomains of each lobe rotate in a rigid manner around the hinge. The difference in the hinge regions of the two lobes of hTF, both in size and secondary structure, suggests a role for this region in the differing iron affinities between the lobes which has thus far not been explained. The structure also suggests that only the N2- and C2- subdomains undergo significant movement in transitioning between the open and closed conformations, with interesting ramifications for interactions with the TFR. While it is clear that a structure of apo-hTF is crucial to a complete understanding of the interaction of TF with the TFR, this structure per se does not explain discrepancies in the current model of the TF/TFR complex. However, the structure of apo-hTF suggests that conformational changes in either the TFR and/or hTF are required to form the complex.
Supplementary Material
The abbreviations used are
- hTF
human serum transferrin
- hTF-NG
recombinant non-glycosylated human serum transferrin
- hTF-Gly
commercially available glycosylated human serum transferrin
- TF
transferrin
- apo-TF
transferrin lacking iron
- oTF
ovotransferrin
- LTF
lactoferrin
- TFR
transferrin receptor
- cryo-EM
cryo-electron microscopy
- BHK
baby hamster kidney
- SeMet
selenomethionine
- NCS
non-crystallographic symmetry
Footnotes
Coordinates- The atomic coordinates and structure-factor amplitudes of hTF have been deposited in the Protein Data Bank (accession codes 2HAV & 2HAU).
We thank Robert Sweet and the RapiData 2004 program for access to synchrotron radiation facilities. We also thank Lothar Esser (NCI, NIH), Gali Prag (NIDDK, NIH), and Sangho Lee (NIDDK, NIH) for advice and discussions, and Tim Fritz (NIDDK, NIH) for critically reading the manuscript. Data were also collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. This work is financially supported by U.S. Public Health Service Grant R01 DK 21739 (ABM). The work of JLW and SKB is supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. We want to thank Igor Kaltashov for providing the mass spectrometry data on the selenomethionine samples.
References
- 1.Harris DC, Aisen P. Physical Biochemistry of the Transferrins. In: Loehr TM, editor. Iron Carriers and Iron Proteins. VCH Publishers, Inc; New York: 1989. [Google Scholar]
- 2.Aisen P, Leibman A, Zweier J. JBiolChem. 1978;253:1930–1937. [PubMed] [Google Scholar]
- 3.Klausner RD, Ashwell G, van Renswoude J, Harford JB, Bridges KR. ProcNatlAcadSciUSA. 1983;80:2263–2266. doi: 10.1073/pnas.80.8.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker HM, Anderson BF, Baker EN. ProcNatlAcadSciUSA. 2003;100:3579–3583. doi: 10.1073/pnas.0637295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambert LA, Perri H, Meehan TJ. CompBiochemPhysiol[B] 2005;140:11–25. doi: 10.1016/j.cbpc.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 6.Lambert LA, Perri H, Halbrooks PJ, Mason AB. CompBiochemPhysiol[B] 2005;142:129–141. doi: 10.1016/j.cbpb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Kurokawa H, Mikami B, Hirose M. JMolBiol. 1995;254:196–207. doi: 10.1006/jmbi.1995.0611. [DOI] [PubMed] [Google Scholar]
- 8.Kurokawa H, Dewan JC, Mikami B, Sacchettini JC, Hirose M. JBiolChem. 1999;274:28445–28452. doi: 10.1074/jbc.274.40.28445. [DOI] [PubMed] [Google Scholar]
- 9.Princiotto JV, Zapolski EJ. JBiolChem. 1975;253:1930–1937. [Google Scholar]
- 10.Baldwin DA, De Sousa DMR, Von Wandruszka RMA. BiochimBiophysActa. 1982;719:140–146. doi: 10.1016/0304-4165(82)90317-8. [DOI] [PubMed] [Google Scholar]
- 11.Baker EN, Baker HM, Kidd RD. BiochemCell Biol. 2002;80:27–34. doi: 10.1139/o01-153. [DOI] [PubMed] [Google Scholar]
- 12.Lestas AN. BrJHaematol. 1976;32:341–350. [Google Scholar]
- 13.Mason AB, He QY, Halbrooks PJ, Everse SJ, Gumerov DR, Kaltashov IA, Smith VC, Hewitt J, MacGillivray RTA. Biochemistry. 2002;41:9448–9454. doi: 10.1021/bi025927l. [DOI] [PubMed] [Google Scholar]
- 14.Halbrooks PJ, He QY, Briggs SK, Everse SJ, Smith VC, MacGillivray RTA, Mason AB. Biochemistry. 2003;42:3701–3707. doi: 10.1021/bi027071q. [DOI] [PubMed] [Google Scholar]
- 15.Mason AB, Halbrooks PJ, James NG, Connolly SA, Larouche JR, Smith VC, MacGillivray RTA, Chasteen ND. Biochemistry. 2005;44:8013–8021. doi: 10.1021/bi050015f. [DOI] [PubMed] [Google Scholar]
- 16.Dewan JC, Mikami B, Hirose M, Sacchettini JC. Biochemistry. 1993;32:11963–11968. doi: 10.1021/bi00096a004. [DOI] [PubMed] [Google Scholar]
- 17.He QY, Mason AB, Tam BM, MacGillivray RTA, Woodworth RC. Biochemistry. 1999;38:9704–9711. doi: 10.1021/bi990134t. [DOI] [PubMed] [Google Scholar]
- 18.Halbrooks PJ, Giannetti AM, Klein JS, Bjorkman PJ, Larouche JR, Smith VC, MacGillivray RTA, Everse SJ, Mason AB. Biochemistry. 2005;44:15451–15460. doi: 10.1021/bi0518693. [DOI] [PubMed] [Google Scholar]
- 19.He Q-Y, Mason AB. Molecular aspects of release of iron from transferrins. In: Templeton DM, editor. Molecular and Cellular Iron Transport. Marcel Dekker, Inc; New York: 2002. [Google Scholar]
- 20.Baker EN. AdvInorgChem. 1994;41:389–463. [Google Scholar]
- 21.Zak O, Tam B, MacGillivray RTA, Aisen P. Biochemistry. 1997;36:11036–11043. doi: 10.1021/bi970628v. [DOI] [PubMed] [Google Scholar]
- 22.Hamilton DH, Turcot I, Stintzi A, Raymond KN. JBiolInorgChem. 2004;9:936–944. doi: 10.1007/s00775-004-0592-6. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Cell. 2004;116:565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- 24.Young SP, Bomford A, Williams R. BiochemJ. 1984;219:505–510. doi: 10.1042/bj2190505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mason AB, He QY, Tam BM, MacGillivray RTA, Woodworth RC. BiochemJ. 1998;330:35–40. [Google Scholar]
- 26.Evans RW, Crawley JB, Garratt RC, Grossmann JG, Neu M, Aitken A, Patel KJ, Meilak A, Wong C, Singh J, Bomford A, Hasnain SS. Biochemistry. 1994;33:12512–12520. doi: 10.1021/bi00207a019. [DOI] [PubMed] [Google Scholar]
- 27.Hall DR, Hadden JM, Leonard GA, Bailey S, Neu M, Winn M, Lindley PF. Acta CrystallogrDBiolCrystallogr. 2002;58:70–80. doi: 10.1107/s0907444901017309. [DOI] [PubMed] [Google Scholar]
- 28.Zuccola HJ. The crystal structure of monoferric human serum transferrin. Georgia Institute of Technology; Atlanta, GA: 1993. [Google Scholar]
- 29.Mason AB, He QY, Adams TE, Gumerov DR, Kaltashov IA, Nguyen V, MacGillivray RTA. Protein ExpPurif. 2001;23:142–150. doi: 10.1006/prep.2001.1480. [DOI] [PubMed] [Google Scholar]
- 30.Mason AB, Halbrooks PJ, Larouche JR, Briggs SK, Moffett ML, Ramsey JE, Connolly SA, Smith VC, MacGillivray RTA. Protein ExprPurif. 2004;36:318–326. doi: 10.1016/j.pep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods in Enzymology, Part A. Academic Press; San Diego: 1997. [DOI] [PubMed] [Google Scholar]
- 32.Schneider TR, Sheldrick GM. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 33.La Fortelle E, Bricogne G. Methods Enzym. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 34.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. In: Doublie S, editor. Crystallographic Methods. Humana Press; Totowa, NJ: 2006. [DOI] [PubMed] [Google Scholar]
- 35.Collaborative Computational Project, N. 4. Acta Crystallogr D Biol Crystallogr. 1994;D50:760–763. [Google Scholar]
- 36.Vagin AA, Isupov MN. Acta Crystallogr D Biol Crystallogr. 2001;57:1451–1456. doi: 10.1107/s0907444901012409. [DOI] [PubMed] [Google Scholar]
- 37.Jones TA. JApplCrystallogr. 1978;11:268–272. [Google Scholar]
- 38.Abrahams JP, Leslie AG. Acta Crystallogr D Biol Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 39.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 40.Winn MD, Isupov MN, Murshudov GN. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 41.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 42.Mason AB, Miller MK, Funk WD, Banfield DK, Savage KJ, Oliver RWA, Green BN, MacGillivray RTA, Woodworth RC. Biochemistry. 1993;32:5472–5479. doi: 10.1021/bi00071a025. [DOI] [PubMed] [Google Scholar]
- 43.Gumerov DR, Mason AB, Kaltashov IA. Biochemistry. 2003;42:5421–5428. doi: 10.1021/bi020660b. [DOI] [PubMed] [Google Scholar]
- 44.Baker EN, Hubbard RE. ProgBiophysMolBiol. 1984;44:97–179. doi: 10.1016/0079-6107(84)90007-5. [DOI] [PubMed] [Google Scholar]
- 45.MacGillivray RTA, Moore SA, Chen J, Anderson BF, Baker H, Luo YG, Bewley M, Smith CA, Murphy ME, Wang Y, Mason AB, Woodworth RC, Brayer GD, Baker EN. Biochemistry. 1998;37:7919–7928. doi: 10.1021/bi980355j. [DOI] [PubMed] [Google Scholar]
- 46.Jeffrey PD, Bewley MC, MacGillivray RTA, Mason AB, Woodworth RC, Baker EN. Biochemistry. 1998;37:13978–13986. doi: 10.1021/bi9812064. [DOI] [PubMed] [Google Scholar]
- 47.Anderson BF, Baker HM, Norris GE, Rumball SV, Baker EN. Nature. 1990;344:784–787. doi: 10.1038/344784a0. [DOI] [PubMed] [Google Scholar]
- 48.Khan JA, Kumar P, Paramasivam M, Yadav RS, Sahani MS, Sharma S, Srinivasan A, Singh TP. JMolBiol. 2001;309:751–761. doi: 10.1006/jmbi.2001.4692. [DOI] [PubMed] [Google Scholar]
- 49.Rawas A, Muirhead H, Williams J. Acta Crystallogr D Biol Crystallogr. 1997;53:464–468. doi: 10.1107/S0907444997000838. [DOI] [PubMed] [Google Scholar]
- 50.He QY, Mason AB, Nguyen V, MacGillivray RTA, Woodworth RC. BiochemJ. 2000;350:909–915. [PMC free article] [PubMed] [Google Scholar]
- 51.Adams TE, Mason AB, He Q-Y, Halbrooks PJ, Briggs SK, Smith VC, MacGillivray RTA, Everse SJ. JBiolChem. 2003;278:6027–6033. doi: 10.1074/jbc.M210349200. [DOI] [PubMed] [Google Scholar]
- 52.Zak O, Ikuta K, Aisen P. Biochemistry. 2002;41:7416–7423. doi: 10.1021/bi0160258. [DOI] [PubMed] [Google Scholar]
- 53.Bates GW, Billups C, Saltman P. JBiolChem. 1967;242:2810–2815. [PubMed] [Google Scholar]
- 54.Marques HM, Watson DL, Egan TJ. InorgChem. 1991;30:3758–3762. [Google Scholar]
- 55.Harris WR, Wang Z, Brook C, Yang B, Islam A. InorgChem. 2003;42:5880–5889. doi: 10.1021/ic034009o. [DOI] [PubMed] [Google Scholar]
- 56.Harris WR, Wang Z, Hamada YZ. InorgChem. 2003;42:3262–3273. doi: 10.1021/ic026027w. [DOI] [PubMed] [Google Scholar]
- 57.Giannetti AM, Halbrooks PJ, Mason AB, Vogt TM, Enns CA, Bjorkman PJ. Structure. 2005;13:1613–1623. doi: 10.1016/j.str.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 58.Beatty EJ, Cox MC, Frenkiel TA, Tam BM, Mason AB, MacGillivray RTA, Sadler PJ, Woodworth RC. Biochemistry. 1996;35:7635–7642. doi: 10.1021/bi960684g. [DOI] [PubMed] [Google Scholar]
- 59.Kurokawa H, Mikami B, Hirose M. JBiolChem. 1994;269:6671–6676. [PubMed] [Google Scholar]
- 60.Day CL, Stowell KM, Baker EN, Tweedie JW. JBiolChem. 1992;267:13857–13862. [PubMed] [Google Scholar]
- 61.Lin L, Mason AB, Woodworth RC, Brandts JF. Biochemistry. 1991;30:11660–11669. doi: 10.1021/bi00114a008. [DOI] [PubMed] [Google Scholar]
- 62.Lin LN, Mason AB, Woodworth RC, Brandts JF. Biochemistry. 1994;33:1881–1888. doi: 10.1021/bi00173a035. [DOI] [PubMed] [Google Scholar]
- 63.Okamoto I, Mizutani K, Hirose M. Biochemistry. 2004;43:11118–11125. doi: 10.1021/bi049147j. [DOI] [PubMed] [Google Scholar]
- 64.Ward PP, Zhou X, Conneely OM. JBiolChem. 1996;271:12790–12794. doi: 10.1074/jbc.271.22.12790. [DOI] [PubMed] [Google Scholar]
- 65.Abdallah FB, El Hage Chahine JM. EurJBiochem. 1999;263:912–920. doi: 10.1046/j.1432-1327.1999.00596.x. [DOI] [PubMed] [Google Scholar]
- 66.Abdallah FB, El Hage Chahine JM. JMolBiol. 2000;303:255–266. doi: 10.1006/jmbi.2000.4101. [DOI] [PubMed] [Google Scholar]
- 67.El Hage Chahine JM, Fain D. EurJBiochem. 1994;223:581–587. doi: 10.1111/j.1432-1033.1994.tb19028.x. [DOI] [PubMed] [Google Scholar]
- 68.Williams J, Evans RW, Moreton K. BiochemJ. 1978;173:535–542. [Google Scholar]
- 69.Bali PK, Aisen P. Biochemistry. 1991;30:9947–9952. doi: 10.1021/bi00105a019. [DOI] [PubMed] [Google Scholar]
- 70.Bali PK, Aisen P. Biochemistry. 1992;31:3963–3967. doi: 10.1021/bi00131a011. [DOI] [PubMed] [Google Scholar]
- 71.Guha TP, Choudhury D, Dasgupta R, Dattagupta JK. Acta Crystallogr D Biol Crystallogr. 2003;59:1773–1781. doi: 10.1107/s0907444903016652. [DOI] [PubMed] [Google Scholar]
- 72.Rawas A, Muirhead H, Williams J. Acta Crystallogr D Biol Crystallogr. 1996;52:631. doi: 10.1107/S0907444996000212. [DOI] [PubMed] [Google Scholar]
- 73.Anderson BF, Baker HM, Norris GE, Rice DW, Baker EN. JMolBiol. 1989;209:711–734. doi: 10.1016/0022-2836(89)90602-5. [DOI] [PubMed] [Google Scholar]
- 74.Zak O, Aisen P. Biochemistry. 2003;42:12330–12334. doi: 10.1021/bi034991f. [DOI] [PubMed] [Google Scholar]
- 75.Bali PK, Zak O, Aisen P. Biochemistry. 1991;30:324–328. doi: 10.1021/bi00216a003. [DOI] [PubMed] [Google Scholar]
- 76.Giannetti AM, Snow PM, Zak O, Bjorkman PJ. PLoSBiol. 2003;1:341–350. [Google Scholar]
- 77.Penhallow RC, Brown-Mason A, Woodworth RC. JCell Physiol. 1986;128:251–260. doi: 10.1002/jcp.1041280217. [DOI] [PubMed] [Google Scholar]
- 78.Young SP, Garner C. BiochemJ. 1990;265:587–591. doi: 10.1042/bj2650587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu G, Liu R, Zak O, Aisen P, Chance MR. MolCell Proteomics. 2005;4:1959–1967. doi: 10.1074/mcp.M500095-MCP200. [DOI] [PubMed] [Google Scholar]
- 80.Mason AB, Tam BM, Woodworth RC, Oliver RWA, Green BN, Lin LN, Brandts JF, Savage KJ, Lineback JA, MacGillivray RTA. BiochemJ. 1997;326:77–85. doi: 10.1042/bj3260077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Turkewitz AP, Schwartz AL, Harrison SC. JBiolChem. 1988;263:16309–16315. [PubMed] [Google Scholar]
- 82.Bennett MJ, Lebrón JA, Bjorkman PJ. Nature. 2000;403:46–53. doi: 10.1038/47417. [DOI] [PubMed] [Google Scholar]
- 83.Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. JStructBiol. 2005;152:204–210. doi: 10.1016/j.jsb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 84.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. JApplCrystallogr. 1993;26:283–291. [Google Scholar]
- 85.Kleywegt GJ. Acta Crystallogr D Biol Crystallogr. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 86.Vriend G. JMolGraph. 1990;8:52–6. 29. [Google Scholar]
- 87.Liu RT, Guan JQ, Zak O, Aisen P, Chance MR. Biochemistry. 2003;42:12447–12454. doi: 10.1021/bi0352973. [DOI] [PubMed] [Google Scholar]
- 88.Teh M, Hewitt J, Ung C, Griffiths TA, Nguyen V, Briggs SK, Mason AB, MacGillivray RT. FEBS J. 2005;272:6344–6353. doi: 10.1111/j.1742-4658.2005.05028.x. [DOI] [PubMed] [Google Scholar]
- 89.Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 90.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.