

Abstract
To understand the complex nature of the atherogenic response initiated by oxidative stress in vascular smooth muscle cells (vSMCs), computational prediction methodology was employed to define putative gene-gene and gene-environment interactions in vSMCs subjected to oxidative chemical stress. Computational relationships were derived from the global gene expression profiles of murine cells challenged with a chemical pro-oxidant to cause oxidative stress or cells treated with anti-oxidant prior to oxidative injury. Target clones were chosen based on their biological relevance within the context of the atherogenic response and included lysyl oxidase, matrix metalloproteinase 2, insulin like growth factor binding protein 5, and lymphocyte antigen 6c. Established biological relationships were derived computationally confirming the usefulness of the algorithm in uncovering novel biological relationships worthy of future investigation. Thus, the predictive algorithm can be a useful tool to advance the frontiers of biological discovery.
Keywords: gene networks, vascular cells, oxidative stress, redox
Background
The formation of atherosclerotic lesions involves the activation of vSMCs leading to modulation from a normally quiescent, contractile phenotype to a lesser differentiated and proliferative (i.e. atherogenic) phenotype. [1] This phenotypic modulation process is manifested by migration of vSMCs from the tunica media to the vessel lumen where they proliferate uncontrollably and give rise to occluding lesions that accumulate large amounts fat, undergo cellular necrosis and recruit clotting factors. To date, the interactive gene networks responsible for induction of atherogenic vSMC phenotypes have not been identified with certainty. We have previously established that oxidative chemical injury of vSMCs in vivo or in vitro mediate the phenotypic modulation of vSMCs to atherogenic phenotypes. [1]
To understand the complex nature of the atherogenic process, a computational approach was used to examine global patterns of gene expression and to define putative gene-gene interactions predictive of critical biological relationships during the course of atherogenesis. Several genes were chosen as targets for prediction using a method first described by Kim et al. [2] The target genes selected for analysis were lysyl oxidase, matrix metalloproteinase 2, insulin like growth factor binding protein 5, and lymphocyte antigen 6c. These genes encode for proteins known to be involved in the regulation of cellular growth and differentiation.
The experimental system employed involved acute challenge of vSMCs with benzo(a)pyrene (BaP), an aromatic hydrocarbon that causes oxidative stress in vSMCs [3] and initiates a cascade of genomic changes that culminates in induction of atherogenic phenotypes. [4] Our goal was to identify small sets of genes whose transcriptional states were predictive of the chosen targets, whether lying upstream or downstream within the gene interaction network, or based on chains of interaction among various intermediates. There was no assumption of causality in the prediction method and its sole focus was to identify sets of genes that may be associated with the target gene, and that could constitute the basis of hypothesis-driven biological investigations.
Methodology
To define genomic profiles during the early phase of the atherogenic response, G0 synchronized cultures of vSMCs from C57BL/6J (6 wk old) mice at passage 12 and 75% confluence were released into growth by addition of fetal bovine serum (10%) in the presence of benzo (a)pyrene (BaP) (3 μM; Sigma-Aldrich) or dimethyl sulfoxide (DMSO, 0.0075%; Sigma-Aldrich) for 24 h. A separate set of cultures was pretreated for 1 h with 0.5 mM N-acetylcysteine (NAC) (Sigma-Aldrich), a water-soluble antioxidant and precursor of cellular glutathione, dissolved in culture medium prior to BaP treatment to enhance antioxidant activity. Cultures were allowed to recover for 1 wk before mRNA isolation. Mouse cDNA arrays developed at National Institute of Environmental Health Sciences (NIEHS) were used for gene expression profiling. A complete listing of the 8,976 transcripts represented on the chip is available at http://dir.niehs.nih.gov/microarray/chips.htm. Comparisons between three treatment groups and one control were duplicated four times for a total of 12 independent hybridizations. Poly(A)RNA samples (2–4μg) were labeled with cyanine-3 (Cy3) or cyanine-5 (Cy5)- conjugated dUTP (Amersham) by reverse transcription using SuperScript (Invitrogen) and oligo-dT (Amersham). A subset of 200 differentially expressed genes was selected based on ANOVA p values that had been derived from gene expression profiles in response to pro-oxidant and anti-oxidant treatment as defined by cDNA microarrays. [5]
Using a heuristic method to discretize the data into ternary states that describe their behavior, the algorithm started by categorizing transcript levels into ternary expression data: -1 for down-regulated, 0 for invariant, and +1 for up-regulated genes. Invariant genes were defined as genes whose expression was not changed by the treatment relative to control. The data were then divided into training and test sets. Based on the training data, the conditional probability that the target gene takes on one of the three transcriptional states was calculated for all possible patterns of the predictor genes, and the predicted target value defined as the state with the largest conditional probability. In considering a predictor set with two genes, the relationships can be defined as: t1,..., t9 equal –1, 0, or +1. The analysis then reverted to the test data to examine the performance of these predictors. The error for each of the predictor function is given by |Tobs – T*| , where Tobs is the observed and T* is the predicted transcriptional state, which could be the optimal predictor state Tψ obtained by the designed filter or the reference predictor state, Tμ obtained by the reference filter.
The above procedure was repeated by randomly splitting the data into training and test sets, in a fixed proportion. The test error was estimated by averaging the prediction error across all iterations, and this error was computed for all possible predictor combinations. The performance of a set of predictors was determined by a statistic known as the coefficient of determination (COD. [6] This coefficient measured the degree to which the transcriptional levels of a set of genes can be used to improve the prediction of the transcriptional state of a target gene relative to the best possible prediction in the absence of predictors. In this case, the mean of the target gene was used as the reference metric, (its transcriptional state represented by Tμ ). The COD (θ) is defined as
Where ε. is the average error for the best predictor in absence of observation and εψ is the average error due to the optimal predictor designed. The errors with respect to n observations is given by,
The higher the COD θ (close to 1), the more accurate the prediction of the target's transcriptional state, i.e., the higher the degree of relationship between the target and predictor genes. All possible combinations of 1, 2 and 3 gene predictors for the chosen targets were studied with possible predictors runs in the order of millions for multiple gene combinations for each target. Predictors were ordered with respect to their errors and the COD's, and the analysis focused on COD's greater than 0.9 and a test error less than 0.05. Information obtained was suggestive of biological commonality between predictor genes and their specified targets.
Results and Discussion
The present study was undertaken to understand the complex nature of the atherogenic process initiated by chemical atherogens present in tobacco smoke using a novel computational approach. Based on ANOVA p-values ≤ 0.01 several clones were selected for further analysis using the computational target clone-predictor approach. This strategy selected for genes within the dataset that displayed a high probability to behave as superior singleton predictors. Target clones included lysyl oxidase, matrix metalloproteinase 2, insulin like growth factor binding protein 5, and lymphocyte antigen 6c. Multiple clone predictor combinations were ranked based on prediction error. Predictor combinations with CODs greater than 0.9 and errors less than 0.05 were selected for further analysis. A large number of threeclone combinations met these criteria for most targets, with one or two clones identified as predominant predictors within the sample pool.
The development and validation of analytical tools that detect multivariate influences on cellular decision-making within complex genetic networks is essential. COD methodology provides an advantage over linear correlations because gene associations are measured based on categorization of discrete variables into a finite numbers of subgroups that enhance the accuracy of prediction. This is in contrast to Pearson's correlation where a pair of continuous variables is examined in the absence of criteria that examine putative interactions among multiple genes. CoD can in fact be used for nonlinear filtering of small datasets such as those often encountered in DNA microarray experiments as CoD is based on error estimation of patterns of gene expression. The determination coefficient permits biologists to focus on particular connections in the genome and coefficient estimates are useful even if they are biased and not overly precise, because at least the estimated coefficients provide a practical means of discrimination among potential predictor sets.
A complete listing of target-predictor clones is presented as Appendix 1. Biologically relevant three gene combinations for each selected target are presented in Figure 1 and 2. The combination of lysyl hydroxylase, syk tyrosine kinase, and osteopontin was shown to predict the behavior of lysyl oxidase (COD 0.91). Lysyl oxidase functions in the maturation of collagen and elastin and is a putative tumor suppressor through a Ras related mechanism. [7] The two matrix related targets, lysyl oxidase (LO) and matrix metalloproteinase-2 (mmp-2) shared two common predictors syk tyrosine kinase (Syk) and osteopontin (OPN). The substitution of stat1 for lysyl hydroxylase and the combination of syk tyrosine kinase, and osteopontin were shown to predict the behavior of matrix metalloproteinase-2 (COD 0.95). This is significant given the role of these two targets in matrix remodeling during atherogenesis. The prediction of genes related to insulin like growth factor binding protein 5 included squalene monooxygenase, osteopontin, and connective tissue growth factor (fisp12) (COD 0.935). Lastly, the best predictors of lymphocyte antigen 6c included MSSP, pip92 and CD6 antigen (COD 0.945).
Figure 1.
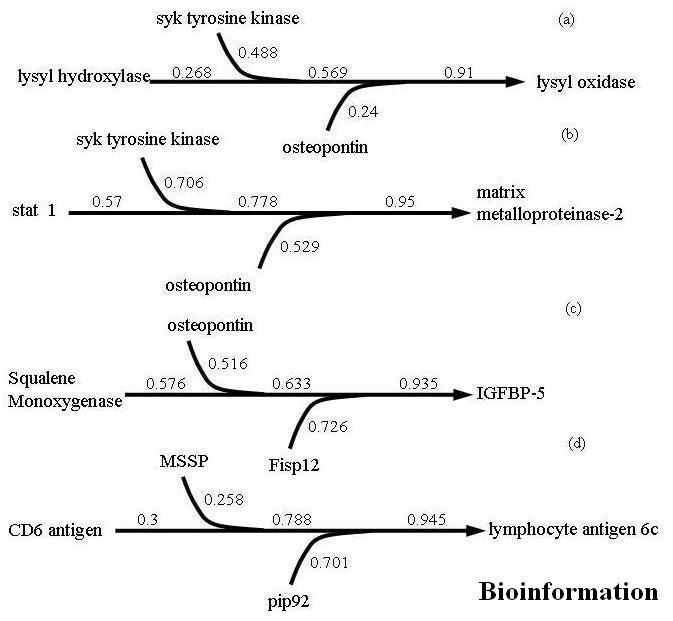
Three gene combinations to predict the behavior of selected target genes. The values are the COD values for each predictor and the effect that addition of each predictor has on overall model prediction potential
Figure 2.

Network inference based on overlapping edges in the predictor-target relationships resolved using CoD values
MMP-2 functions in the digestion collagens of and elastin to promote cell migration and vascular remodeling. Syk plays a critical role in signaling of various receptors of the adaptive immune system and functions as an inhibitor of breast cancer cell growth and metastasis. [8] Syk participates in the integrin signaling pathway in monocytic cells leading to activation of NF-kB and increased levels of cytokine mRNAs. [9] Studies have shown that OPN induces activation of mmp-2 through the IkB alpha/IKK signaling pathways [10], and that mmp-2 plays a direct role in OPN-induced cell migration, invasion, and tumor growth. Both LO and OPN are activated by platelet-derived growth factor in vSMC. [11]
The difference between LO and mmp-2 in our predictor model is the addition of lysyl hydroxylase in the case of LO and stat1 in the case of mmp-2. Lysyl hydroxylase catalyzes hydroxylation of lysyl residues in collagens and other proteins with collagenous domains. [12] Both LO and lysyl hydroxylase are involved in post-translational modifications of collagen as part of the cross-linking pathway and would thus be expected to behave is a similar manner. The regulation of mmp-2 by stat1 has been demonstrated in tumor cells. [13] Stat1 is a signal transducer and activator of early transcription factor by adherence, and a modulator of ICAM gene expression. Studies have shown the involvement of Stat1 in IFN and growth factor-dependent signaling and in positive, negative and constitutive regulation of gene expression. [14]
Insulin like growth factor binding protein 5 (IGFBP-5) was predicted by OPN, squalene monooxygenase (SMO) and connective tissue growth factor (Fisp12). IGFBP-5 is the most conserved IGFBP across species and as an essential regulator in bone, kidney and mammary gland. In addition, IGFBP-5 plays a decisive role in the control of proliferation of specific tumor cell types. [15] In vSMCs, IGFBP-5 and OPN promote IGF-I effects and OPN binds to IGFBP-5 with high affinity. [16] These interactions are important for concentrating intact IGFBP-5 in the extracellular matrix and modulation of the cooperative interaction between the IGF-I receptor and integrin αvβ3 signaling pathways in atherosclerotic lesion. [17] Fisp12 mediates cell adhesion and migration through integrin αvβ3, and promotes cell survival, and induces angiogenesis in vivo. [18] Fisp12 is also known as insulin-like growth factor binding protein related proteins (IGFBP-rPs). [19] Therefore, it is likely that Fisp12 is regulated in a like fashion to IGBP-5.
It is unclear how squalene monooxygenase (squalene epoxidase) is related to IGBP-5, but squalene monooxygenase catalyzes the second committed step in cholesterol biosynthesis from farnesyl pyrophosphate to squalene. [20] Studies have shown that squalene monooxygenase is bound to the endoplasmic reticulum of cells in association with NADPH-cytochrome P450 reductase, its electron transfer partner. [21] Squalene monooxygenase is regulated at the transcriptional level in response to sterol levels and may compete with HMG-CoA reductase as the regulated step in cholesterol synthesis. Studies have identified a link between farnesyl pyrophosphate and post-translational processing of Ras and Rasrelated proteins. [20] The Ramos laboratory has demonstrated that Ras is a key factor in atherogenesis [22], and others have reviled that OPN is also critical for Ras expression. [23] Thus, LO, MMP-2 and IGBP-5 share OPN and integrin signaling as common factors, a relationship identified by computational methodology.
The last target examined was lymphocyte antigen 6c, which plays a role in the T cell activation cascade and is modified by atherogenic challenge in vSMC. [5] This was predicted by the combination of CD6 antigen, pip92 and MSSP. CD6 belongs to the scavenger receptor cysteine-rich protein super family that triggers co-activating signaling of T cells. Its regulation during T cell ontogeny and activation has been extensively investigated. [24] MSSP promotes ras/myc cooperative cell transforming activity by binding to c-Myc [25], while Pip92 is an early response gene, activated by growth factors. The activation of pip92 is mediated by JNK and p38 kinase, but not ERK. [26] Type I interferon is the primary regulator of inducible Ly-6C expression on T cells [27], and studies have shown that interferon-alpha down regulates c-myc. [28] c-myc single-strand binding protein (MSSP) may function in a similar fashion, a pattern that fits well with our current understanding of chemical-induced atherosclerosis. [29]
Conclusion
The greatest challenge in computational prediction is biological validation. Clearly, biological relevance is required to establish the true significance of findings derived computationally. Based on established biological relationships, we demonstrate that predictor gene combinations derived computationally using the COD algorithm can be related at the biological level and consistent with established biological contexts. We have applied a similar strategy to define gene network structure and function of LINE-1 and found that unknown predicted relationships could be validated using functional genomics approaches. [30] Thus, the fidelity of computational relationships lends confidence that COD methodology can in fact predict undiscovered relationships. These findings may yield new areas of exploration for those interested in atherogenesis and computational biology.
Footnotes
Citation:Johnson et al., Bioinformation 1(10): 379-383 (2007)
References
- 1.Ramos KS. Ann Rev Pharmacol Toxicol. 1999;29:243. doi: 10.1146/annurev.pharmtox.39.1.243. [DOI] [PubMed] [Google Scholar]
- 2.Kim S, et al. Genomics. 2000;67:201. doi: 10.1006/geno.2000.6241. [DOI] [PubMed] [Google Scholar]
- 3.Kerzee KK, Ramos KS. Mol Pharmacol. 2000;58:152. doi: 10.1124/mol.58.1.152. [DOI] [PubMed] [Google Scholar]
- 4.Lu KP, et al. Atherosclerosis. 2002;160:273. doi: 10.1016/s0021-9150(01)00581-0. [DOI] [PubMed] [Google Scholar]
- 5.Johnson CD, et al. Physiol Genomics. 2003;13:263. doi: 10.1152/physiolgenomics.00006.2003. [DOI] [PubMed] [Google Scholar]
- 6.Dougherty ER, et al. Signal Processing. 2000;80:2219. [Google Scholar]
- 7.Contente S, et al. Science. 1990;249:796. doi: 10.1126/science.1697103. [DOI] [PubMed] [Google Scholar]
- 8.Coopman PJ, et al. Nature. 2000;406:742. doi: 10.1038/35021086. [DOI] [PubMed] [Google Scholar]
- 9.Lin TH, et al. J Biol Chem. 1995;270:16189. doi: 10.1074/jbc.270.27.16189. [DOI] [PubMed] [Google Scholar]
- 10.Philip S, et al. J Biol Chem. 2001;276:44926. doi: 10.1074/jbc.M103334200. [DOI] [PubMed] [Google Scholar]
- 11.Green RS, et al. Lab Invest. 1995;73:476. [PubMed] [Google Scholar]
- 12.Kielty CM, et al. J Cell Sci. 1992;103:445. doi: 10.1242/jcs.103.2.445. [DOI] [PubMed] [Google Scholar]
- 13.Huang S, et al. Oncogene. 2002;21:2504. doi: 10.1038/sj.onc.1205341. [DOI] [PubMed] [Google Scholar]
- 14.Malmgaard L. J Virol. 2002;76:4520. doi: 10.1128/JVI.76.9.4520-4525.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider MR, et al. J Endo. 2002;172:423. doi: 10.1677/joe.0.1720423. [DOI] [PubMed] [Google Scholar]
- 16.Nam TJ, et al. Endocrinology. 2000;141:1100. doi: 10.1210/endo.141.3.7386. [DOI] [PubMed] [Google Scholar]
- 17.Nichols TC, et al. Circ Res. 1999;85:1040. doi: 10.1161/01.res.85.11.1040. [DOI] [PubMed] [Google Scholar]
- 18.Babic M, et al. Mol Cell Biol. 1999;19:2958. doi: 10.1128/mcb.19.4.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horn V, et al. Int J Mol Med. 2002;9:645. [PubMed]
- 20.Holstein SA, et al. Biochemistry. 2002;41:13698. doi: 10.1021/bi026251x. [DOI] [PubMed] [Google Scholar]
- 21.Ono T, Bloch K. J Biol Chem. 1975;250:1571. [PubMed] [Google Scholar]
- 22.Ramos KS, et al. Arch Biochem Biophys. 1996;332:213. doi: 10.1006/abbi.1996.0335. [DOI] [PubMed]
- 23.Wu Y, et al. Br J Cancer. 2000;83:156. doi: 10.1054/bjoc.2000.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skonier JE, et al. Protein Eng. 1997;10:943. doi: 10.1093/protein/10.8.943. [DOI] [PubMed] [Google Scholar]
- 25.Niki T, et al. Genes Cells. 2000;5:127. doi: 10.1046/j.1365-2443.2000.00311.x. [DOI] [PubMed] [Google Scholar]
- 26.Chung KC, et al. Eur J Biochem. 2000;267:4676. doi: 10.1046/j.1432-1327.2000.01517.x. [DOI] [PubMed] [Google Scholar]
- 27.Schlueter AJ, et al. J Interferon Cytokine Res. 2001;21:621. doi: 10.1089/10799900152547885. [DOI] [PubMed] [Google Scholar]
- 28.Radaeva S, et al. Gastroenterology. 2002;122:1020. doi: 10.1053/gast.2002.32388. [DOI] [PubMed] [Google Scholar]
- 29.Fujimoto M, et al. Int J Oncol. 2000;16:245. doi: 10.3892/ijo.16.2.245. [DOI] [PubMed] [Google Scholar]
- 30.Ramos KS, et al. Genomics. 2007 [Google Scholar]