

Abstract
Previous studies have shown that acyl-coenzyme A:cholesterol acyl transferase (ACAT), an enzyme that controls cellular equilibrium between free cholesterol and cholesteryl esters, modulates proteolytic processing of APP in cell-based and animal models of Alzheimer’s disease. Here we report that ACAT-1 RNAi reduced cellular ACAT-1 protein by ~50% and cholesteryl ester levels by 22%. This correlated with reduced proteolytic processing of APP and 40% decrease in Aβ secretion. Similarly to pharmacological inhibition, ACAT-1 RNAi increased free cholesterol content of ER membranes. These data show that even a modest decrease in ACAT activity can have robust suppressive effects on Aβ generation.
Keywords: RNAi, cholesterol, cholesteryl esters, amyloid-β, Alzheimer’s disease, Amyloid Precursor Protein, ACAT
1. INTRODUCTION
Progressive accumulation of amyloid β-peptide (A β) in senile plaques in brain regions responsible for memory and cognitive functions is a major pathological hallmark of Alzheimer’s disease [1]. Aβ is a 39- to 43-amino acid peptide generated from β-amyloid precursor protein (APP) by sequential proteolytic cleavages mediated by β- and γ-secretases. Alternatively, APP can be processed by the nonamyloidogenic α-secretase pathway, which cuts APP in the middle of the Aβ region [1]. Genetic, epidemiological and biochemical studies have suggested that cholesterol is an important risk factor for AD [2,3]. Statins, a highly successful class of drugs that inhibit HMG CoA reductase, have been shown to attenuate Aβ production in cell-based and animal models of AD and in humans [4]. Although the beneficial effects of statins for AD may be at least partially due to their pleiotropic actions, other cholesterol-modifying strategies for suppression of Aβ production in Alzheimer’s disease have recently gained considerable interest.
ACAT is an endoplasmic reticulum (ER)-resident enzyme responsible for conversion of excess free cholesterol to cholesteryl esters [5–7]. Of the two human ACAT isoforms (two different genes in most mammals), ACAT-1 is ubiquitously expressed whereas ACAT-2 expression is restricted to the liver and the intestine [6]. Inhibition of ACAT function in cells by pharmacological means has been shown to efficiently suppress Aβ generation in vitro [8]. Importantly, a two-month treatment with ACAT inhibitor CP-113,818 remarkably reduced amyloid pathology and correlated with improved spatial learning in transgenic mice expressing human APP751 containing the London (V717I) and Swedish (K670M/N671L) mutations [9]. To provide an important cell biological proof of principle for ACAT-1 as a therapeutic target and as a modulator of APP metabolism, we set up and characterized a cell-based RNAi model for ACAT-1. Here we show that reduction of cellular ACAT-1 protein level to half by a single transfection of ACAT-1 siRNA oligonucleotides reduces cellular cholesteryl ester levels while significantly suppressing amyloidogenic processing of APP and Aβ production.
2. MATERIALS AND METHODS
Cell culture and RNA interference
Human H4 neuroglioma cells (ATCC) were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% (v/v) FBS (Atlanta Biologicals), 1% (v/v) L-Glutamine-Penicillin-Streptomycin solution (Sigma) at 37°C in a water-saturated air/5% CO2 atmosphere. H4 cells were transfected with pcDNA3.1-APP751, selected and maintained with G418 sulphate (Calbiochem). A clone (H4APP751) with ~5-fold overexpression of APP was used in this study. For silencing ACAT-1 expression, cells were transfected with 1 or 3 μg of a manufacturer-optimized mixture of human-specific ACAT-1 siRNA (Santa Cruz Biotechnology) using Nucleofector™ technology according to the manufacturer’s instructions (Amaxa). Control cells were transfected with 3 μg of a mixture of mouse-specific ACAT-1 siRNA (Santa Cruz Biotechnology). Culture media was changed once at 72 h post-transfection, and 24-h conditioned media was collected when the cells were harvested 96 h post-transfection.
Protein extraction and Western blotting
Cells were washed twice, scraped in ice-cold PBS and extracted on ice for 30 minutes in a buffer containing 10 mM Tris-HCl, pH 6.8, 1 mM EDTA, 150 mM NaCl, 0.25% Nonidet P-40, 1% Triton X-100 and a protease inhibitor mixture (Roche Molecular Biochemicals). Cell debris was removed by a spin at 16,000 × g. The protein concentrations were determined using the BCA protein assay kit (Pierce). For Western blot analysis, 30 μg of total protein per lane was resolved in a 4-12% gradient Bis-Tris gels (Novex) under reducing conditions. The filters were probed with a C-terminal APP antibody (A8717; Sigma), ACAT-1 (Santa Cruz) and GAPDH (Chemicon) antibodies. Calreticulin (Calbiochem) and GM130 (BD Biosciences) antibodies were used to identify ER and Golgi fractions, respectively. After incubation with horseradish-conjugated secondary antibodies the signal was developed using ECL Western Blotting detection reagent (Amersham). Western blot images were quantitated using Quantity One software package (Bio-Rad).
Lipid extraction and cholesterol assay
For the determination of cellular pools of free cholesterol (FC) and cholesteryl esters (CE), cells were extracted in chloroform/methanol/ddH2O (4:2:1; v/v/v). Chloroform phase was separated, mixed with 1:100 volume of polyoxyethylene 9-lauryl ether (‘polidocanol’; Sigma), dried and resuspended in assay reaction buffer (100 mM potassium phosphate, pH 7.4, 50 mM NaCl, 5 mM cholic acid, 0.1% Triton X-100). Free cholesterol was measured enzymatically using Amplex Red Cholesterol Assay kit (Molecular Probes/Invitrogen). To directly measure cholesteryl esters in samples, free cholesterol was first converted to cholest-4-ene-3-one by cholesterol oxidase and the resulting hydrogen peroxide decomposed by catalase after which the enzymatic cholesterol assay was performed in the presence of cholesterol esterase [10].
Lipid droplet staining
Four days after transfection with 3.0 μg of ACAT1 siRNA and 0.5 μg of pEGFP plasmid (Amaxa), cells were washed once with PBS and fixed with 3% paraformaldehyde in PBS for 20 minutes. Cells were stained with HCS LipidTOX™ red neutral lipid stain and Hoechst 33342 (Molecular Probes/Invitrogen) for 20 minutes according to manufacturers instructions. Images were taken with Olympus DSU/IX70 spinning disc confocal microscope. Control cells were treated for 4 days with 10 μM CP-113,818 or vehicle (DMSO).
Aβ ELISA
For Aβ determination, the conditioned media was cleared from debris and secreted Aβ40 and Aβ42 were quantitated by standard sandwich ELISA (Aβ ELISA Core Facility, Center for Neurological Diseases, Harvard Institutes of Medicine, Harvard Medical School).
Subcellular fractionation
ER membranes from transfected H4APP751 cells were prepared as described previously [11].
3. RESULTS
Knockdown of ACAT-1 reduces cholesteryl ester levels in cells
Human H4 neuroglioma cells overexpressing human APP751 (H4APP751) were transfected with an increasing dose of chemically synthesized ACAT-1 siRNA oligonucleotides (specific for human ACAT-1). As a control, the cells were transfected with siRNA oligonucleotides specific for mouse ACAT-1 which had no detectable effect on endogenous ACAT-1 protein levels in H4APP751 cells. In previous studies, we have noted that for ACAT inhibitors the maximal efficacy in reducing cholesteryl ester levels in cultured cells requires prolonged (up to 4 days) incubation times [8]. Thus, cells were harvested 96 hours after transfection with ACAT-1 siRNA oligonucleotides for analysis of ACAT-1 expression and cholesterol levels. At this point, ACAT-1 protein levels were down by 42.7±7.6% (p=0.0052) for 1.0 μg siRNA dose and 54.4±11.0% (p=0.0067) for 3.0 μg of siRNA dose as compared to the control siRNA-transfected cells (Fig. 1A–B).
Figure 1. Knockdown of ACAT-1 Reduces Cholesteryl Ester Levels.
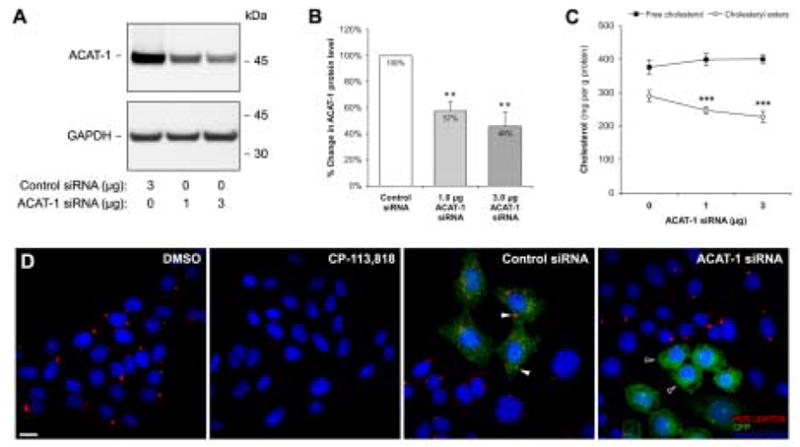
H4APP751 cells transfected with either mouse ACAT-1-specific (control siRNA) or an increasing dose of human ACAT-1-specific siRNA oligonucleotides were analyzed 96 hours after transfection for ACAT-1 expression and cholesterol levels. ACAT-1 expression determined by Western blot analysis (A) was quantitated and normalized to GAPDH levels (B). (C) Cellular free cholesterol and cholesteryl ester levels are shown as the means ± standard deviation of three independent experiments. (D) H4APP751 cells were treated with DMSO or 10 μM CP-113,818 or transfected with 0.5 μg GFP plasmid together with 3.0 μg of either mouse ACAT-1 (control) or human ACAT-1 siRNA oligonucleotides. After 96 h, cells were stained with HCS LipidTOX™ red neutral lipid stain (lipid droplets) and Hoechst 33342 (nuclei). ** p < 0.01, *** p < 0.001. Bar = 10 μm.
To determine how this reduction in ACAT-1 protein levels affect cellular cholesterol levels, both free cholesterol and cholesteryl esters were determined from chloroform:methanol-extracted total lipids by enzymatic assay. We noted a moderate, statistically insignificant rise [5.9% for 1.0 μg siRNA (from 376.4±18.5 to 398.4±14.2 mg/g protein) and 6.2% for 3.0 μg siRNA (from 376.4±18.5 to 399.7±12.2 mg/g protein)] in free cholesterol level and highly significant 14.6±2.9% (from 289.8±16.9 to 247.2±10.8 mg/g protein; p=0.00013) and 21.6±4.4% (from 289.8±16.9 to 227.2±17.0 mg/g protein; p=0.00010) decreases in cholesteryl ester levels in cells transfected with 1.0 μg and 3.0 μg ACAT-1 siRNA, respectively (Fig. 1C).
To verify that ACAT-1 RNAi affects ACAT activity in the transfected cells, we cotransfected GFP with ACAT-1 siRNA oligonucletides and stained the cells with HCS LipidTOX™ red neutral lipid dye and Hoechst 33342 nuclei dye to visualize lipid droplets. As shown in Fig. 1D, treatment of cells with ACAT inhibitor CP-113,818 abolished lipid droplet staining from cells as compared to vehicle-treated (DMSO) cells. In control cells transfected with mouse ACAT-1 siRNA and GFP, lipid droplets are also clearly detectable (white arrowheads in Fig. 1D). Remarkably, in cells transfected with human ACAT-1 siRNA and GFP, the number and size of lipid droplets is clearly reduced (black arrowheads) whereas the nontransfected cells in the same culture display many lipid droplets suggesting that in the cells transfected with ACAT-1 siRNA ACAT-1 activity is almost completely suppressed. Altogether, these results show that in this RNAi model, a maximum of 54% reduction of ACAT-1 protein levels can be achieved resulting in 22% decrease in cellular cholesteryl esters and that transfection efficiency is the limiting factor for the efficacy of ACAT-1 RNAi in this cell-based model.
Knockdown of ACAT-1 expression reduces proteolytic processing of APP and generation of Aβ
Pharmacological ACAT inhibition as well as a selective disruption of ACAT-1 gene in AC29 cells has previously been shown to affect all three secretases (α, β and γ) responsible for proteolytic processing of APP [8]. To characterize the effects of ACAT-1 RNAi on APP metabolism, we first analyzed the levels of APP holoprotein as well as α-secretase (APP-C83) and β-secretase (APP-C99) cleavage products of APP on Western blots. As compared to the cells transfected with control siRNA, the levels of amyloidogenic APP-C99 were down by 11.7±2.7% (p=0.0085) and 48.4±5.2% (p=0.0019) in cells transfected with 1.0 μg and 3.0 μg of ACAT-1 siRNA, respectively (Fig. 2A–B). The nonamyloidogenic APP-C83 level was reduced by 27.4±4.1% (p=0.0036) in cells transfected with 3.0 μg of ACAT-1 siRNA but was unaffected in cells transfected with 1.0 μg of ACAT-1 siRNA (Fig. 2A–B). APP holoprotein levels remained unaffected in all the samples.
Figure 2. Decreased ACAT-1 Expression Levels Correlate with Reduced Proteolytic Processing of APP.
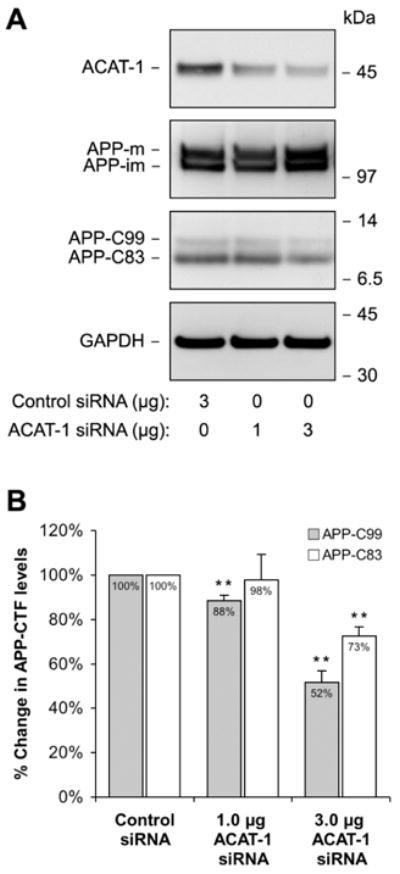
H4APP751 cells were transfected with either control siRNA (mouse ACAT-1) or siRNA specific for human ACAT-1 and analyzed 96 h later for ACAT-1 expression and APP proteolytic fragements (A). The levels of the APP C-terminal fragments (APP-C99 generated by β-secretase and APP-C83 produced by α-secretase) were quantitated in four independent experiments (B). The values were normalized to GAPDH and the immature form of APP holoprotein (APP-im). * p < 0.05, ** p < 0.01.
Soluble Aβ levels in the conditioned media from the siRNA-transfected cells were analyzed by sandwich ELISA. As compared to the cells transfected with control siRNA, conditioned media from cells transfected with 1.0 μg of ACAT-1 siRNA contained 18.5% (p=0.0194) less total Aβ and 21.8% (p=0.0316) less Aβ42 (from 28.7±3.7 to 23.4±6.6 pmol/l/mg protein and from 5.5±1.4 to 4.3±2.2 pmol/l/mg protein; Fig. 3). Cells transfected with 3.0 μg of ACAT-1 siRNA produced 39.2% (p=0.0027) less total Aβ and 27.8% (p=0.0237) less Aβ42 as compared to the control siRNA-transfected cells (from 28.7±3.7 to 17.5±4.6 pmol/l/mg protein and from 5.5±1.4 to 4.0±1.1 pmol/l/mg protein; Fig. 3). These results show that by reducing cellular ACAT-1 protein levels by ~50%, a significant suppression of amyloidogenic APP processing can be achieved.
Figure 3. Knockdown of ACAT-1 Inhibits Aβ Generation.
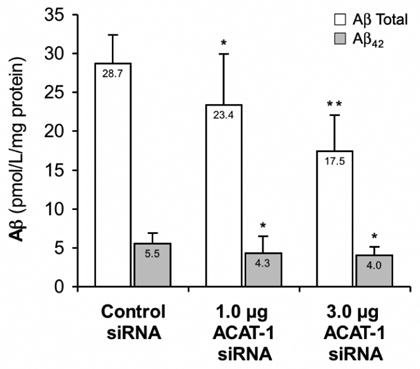
Conditioned media collected from the cells examined in Figure 2 was analyzed for Aβtotal and Aβ42 by sandwich ELISA. The results shown represent the means ± standard deviation of four independent experiments. * p < 0.05, ** p < 0.01.
ACAT-1 RNAi increases unesterified cholesterol content in ER membranes
Our previous studies have indicated that lack of ACAT-1 activity in cells results in transient increases in free cholesterol levels in the ER [8]. As it is likely that this is mechanistically relevant, we tested whether ACAT-1 RNAi has similar effect on ER free cholesterol as treatment with a well-characterized ACAT inhibitor CP-113,818. Cells were transfected with 3.0 μg of control or ACAT-1 siRNA oligonucleotides or treated with 10 μM CP-113,818 for 4 days. Microsomes were harvested and subjected to further fractionation in Optiprep gradients. ACAT-1 and calreticulin-containing ER fractions (as indicated in Fig. 4A) were assayed for free cholesterol. Treatment with CP-113,818 increased ER free cholesterol content by 47.6% (from 14.0±3.5 to 20.7±3.8 mg/g protein; p=0.0013) whereas ACAT-1 RNAi caused a 16.3% increase (from 13.1±1.9 to 15.3±2.1 mg/g protein; p=0.0248) (Fig. 4B).
Figure 4. ACAT-1 inhibitor and RNAi increases cholesterol content of ER membranes.
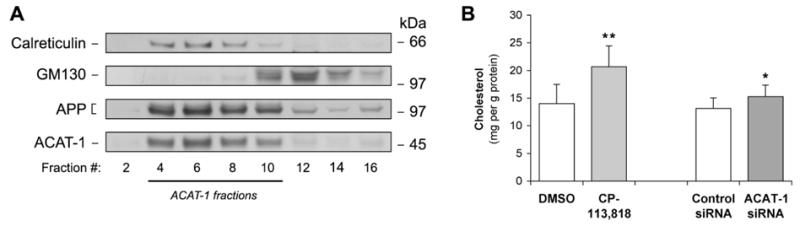
Microsomes from H4APP751 cells were isolated by centrifugation at 100,000 g, rehomogenized and separated in Optiprep gradients. (A) Fractions were analyzed for ER (calreticulin) and Golgi markers (GM130), and for ACAT-1 and APP. (B) Cells were treated with 10 μM CP-113,818 or transfected with 3 μg of ACAT-1 or control siRNA. After 96 h, ER fractions were prepared and membrane cholesterol content in fractions containing ACAT-1 and calreticulin was assayed and normalized to protein levels. * p < 0.05, ** p < 0.01.
4. DISCUSSION
Cholesterol homeostasis has proven to be intimately linked to various aspects of Aβ biology [2,3]. In addition to statins, ACAT inhibitors are promising therapeutic strategies for Alzheimer’s disease since both reduce amyloid plaque density in animal models of the disease. It appears that the balance between amyloidogenic and nonamyloidogenic APP processing is especially sensitive to changes in intracellular cholesterol distribution [8,9]. As ACAT is a key regulator of the equilibrium between free cholesterol and cholesteryl ester pools, it is not surprising that inhibition of ACAT suppresses proteolytic processing of APP. In this study, we investigated whether a knockdown of ACAT-1 expression by RNAi in cells affects proteolytic processing of APP and production of Aβ. To detect secreted Aβ species in the culture medium we used a model overexpressing human APP751. It should be noted that we have detected similar suppressive effects of ACAT-1 RNAi on the proteolytic processing of endogenous APP in H4 naïve cells (data not shown). To control the specificity of the RNAi we used a mixture of three siRNA oligonucleotides specific for mouse ACAT-1. Importantly, even in the high 3 μg dose the control siRNA did not display any effects on the key parameters of this study, ACAT-1 expression, FC and CE levels and APP/Aβ levels. As we routinely perform 4-day treatments in cell-based ACAT-inhibitor studies, this study was designed to be a 4-day study for comparability. Importantly, RNAi-induced decrease in ACAT-1 expression to roughly half of the control cell levels resulted in a significant suppression of Aβ production (~40%) despite the rather modest ~22% decrease in cholesteryl ester levels. Both Aβ40 and Aβ42 were affected more or less equally. These results are comparable to the previously published results that used ACAT-1 inhibitors [8]. Interestingly, ACAT-1 RNAi caused a similar although smaller increase in the free cholesterol content of ER membranes as pharmacological ACAT inhibition. This may have important mechanistic implications and require further studies.
As has been previously shown for pharmacological ACAT inhibition [8], the results presented here suggest that ACAT-1 RNAi affects all of the three α-, β- and γ-secretases as not only Aβ and APP-C99 but also APP-C83 levels were decreased by ACAT-1 RNAi. Although the mechanistic details on how ACAT inhibition modulates APP processing require further studies, it seems that pharmacological ACAT inhibition and ACAT-1 RNAi affect the same or similar pathways in the early secretory pathway that are important for APP metabolism. Recently, the specificity of certain ACAT inhibitors has been questioned [12]. Thus, this study provides an important, nonpharmacological proof-of-principle confirming that reduction of cellular ACAT activity is a viable and specific approach for modulating Aβ generation.
Acknowledgments
This work was supported by grants from the NIH/NINDS (D.M.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–9. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat Neurosci. 2003;6:345–51. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 3.Wolozin B. Cholesterol and the biology of Alzheimer’s disease. Neuron. 2004;41:7–10. doi: 10.1016/s0896-6273(03)00840-7. [DOI] [PubMed] [Google Scholar]
- 4.Shobab LA, Hsiung GY, Feldman HH. Cholesterol in Alzheimer’s disease. Lancet Neurol. 2005;4:841–52. doi: 10.1016/S1474-4422(05)70248-9. [DOI] [PubMed] [Google Scholar]
- 5.Chang TY, Chang CC, Cheng D. Acyl-coenzyme A:cholesterol acyltransferase. Annu Rev Biochem. 1997;66:613–38. doi: 10.1146/annurev.biochem.66.1.613. [DOI] [PubMed] [Google Scholar]
- 6.Buhman KF, Accad M, Farese RV. Mammalian acyl-CoA:cholesterol acyltransferases. Biochim Biophys Acta. 2000;1529:142–54. doi: 10.1016/s1388-1981(00)00144-x. [DOI] [PubMed] [Google Scholar]
- 7.Chang TY, Chang CC, Lin S, Yu C, Li BL, Miyazaki A. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and -2. Curr Opin Lipidol. 2001;12:289–96. doi: 10.1097/00041433-200106000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Puglielli L, et al. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat Cell Biol. 2001;3:905–12. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- 9.Hutter-Paier B, et al. The ACAT Inhibitor CP-113,818 Markedly Reduces Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Neuron. 2004;44:227–38. doi: 10.1016/j.neuron.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 10.Mizoguchi T, Edano T, Koshi T. A method of direct measurement for the enzymatic determination of cholesteryl esters. J Lipid Res. 2004;45:396–401. doi: 10.1194/jlr.D300024-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Plonne D, Cartwright I, Linss W, Dargel R, Graham JM, Higgins JA. Separation of the intracellular secretory compartment of rat liver and isolated rat hepatocytes in a single step using self-generating gradients of iodixanol. Anal Biochem. 1999;276:88–96. doi: 10.1006/abio.1999.4311. [DOI] [PubMed] [Google Scholar]
- 12.Chambers K, Brown WJ. Characterization of a novel CI-976-sensitive lysophospholipid acyltransferase that is associated with the Golgi complex. Biochem Biophys Res Commun. 2004;313:681–6. doi: 10.1016/j.bbrc.2003.12.016. [DOI] [PubMed] [Google Scholar]