

Abstract
The key event in the switch from lysogenic to lytic growth of phage lambda is the self-cleavage of lambda repressor, which is induced by the formation of a RecA-ssDNA-ATP filament at a site of DNA damage. Lambda repressor cleaves itself at the peptide bond between Ala111 and Gly112, but only when bound as a monomer to the RecA-ssDNA-ATP filament. Here we have designed a hyper-cleavable fragment of lambda repressor containing the hinge and C-terminal domain (residues 101-229), in which the monomer-monomer interface is disrupted by two point mutations and a deletion of seven residues at the C terminus. This fragment crystallizes as a monomer and its structure has been determined to 1.8 Å resolution. The hinge region, which bears the cleavage site, is folded over the active site of the C-terminal oligomerization domain (CTD) but with the cleavage site flipped out and exposed to solvent. Thus, the structure represents a non-cleavable conformation of the repressor, but one that is poised for cleavage after modest rearrangements that are presumably stabilized by binding to RecA. The structure provides a unique snapshot of lambda repressor in a conformation that sheds light on how its self-cleavage is tempered in the absence of RecA, as well as a framework for interpreting previous genetic and biochemical data concerning the RecA-mediated cleavage reaction.
Keywords: lambda repressor, RecA protein, DNA repair, LexA, bacteriophage lambda
Introduction
The genetic switch of bacteriophage lambda has for many years been a model system for understanding gene regulation.1 In the lysogenic state, a repressor protein, cI, binds to multiple operator sites on the DNA, blocking transcription of genes necessary for assembly of phage particles. In the lytic state, the repressor is proteolytically inactivated, the phage genes are expressed, and the host cell is ultimately lysed. Proteolytic cleavage of cI is mediated by its binding to a helical filament of the host RecA protein formed at a site of DNA damage.2 Though initially RecA was thought to be a protease that cleaves cI, it eventually became clear that the repressor actually undergoes an intra-molecular self-cleavage,3 but only when bound as a monomer to a RecA-ssDNA-ATP filament.4-6 Similar RecA-mediated cleavages occur for the host LexA repressor and UmuD proteins,7-8 which activate the SOS response to DNA damage and translesion mutagenesis, respectively. Thus, to control its optimal mode of replication, the phage probes the cellular environment by tapping into the host’s own mechanism for sensing DNA damage.
cI protein (Mr 26 kDa; 236 amino acid residues) contains an N-terminal DNA-binding domain and a C-terminal oligomerization domain (CTD) that are connected by a flexible “hinge” (Figure 1).9 The CTD forms dimers and higher-ordered oligomers (tetramers and octamers) that mediate cooperative binding of cI to multiple operator sites. The CTD also forms the proteolytic active site, including Ser149 and Lys192,10 and its fold is conserved in LexA and UmuD.11-13 Cleavage occurs within the hinge region at the peptide bond between Ala111 and Gly112.14 While at neutral pH self-cleavage of cI is entirely dependent on its binding as a monomer to RecA filaments, at elevated pH (~10) it can cleave itself in the absence of RecA in a reaction termed “autodigestion”.3 RecA is thought to stimulate cleavage not by participating directly in catalysis, but rather by stabilizing a conformation in which the cleavage site is properly oriented within the active site on the CTD.12,15
Figure 1.
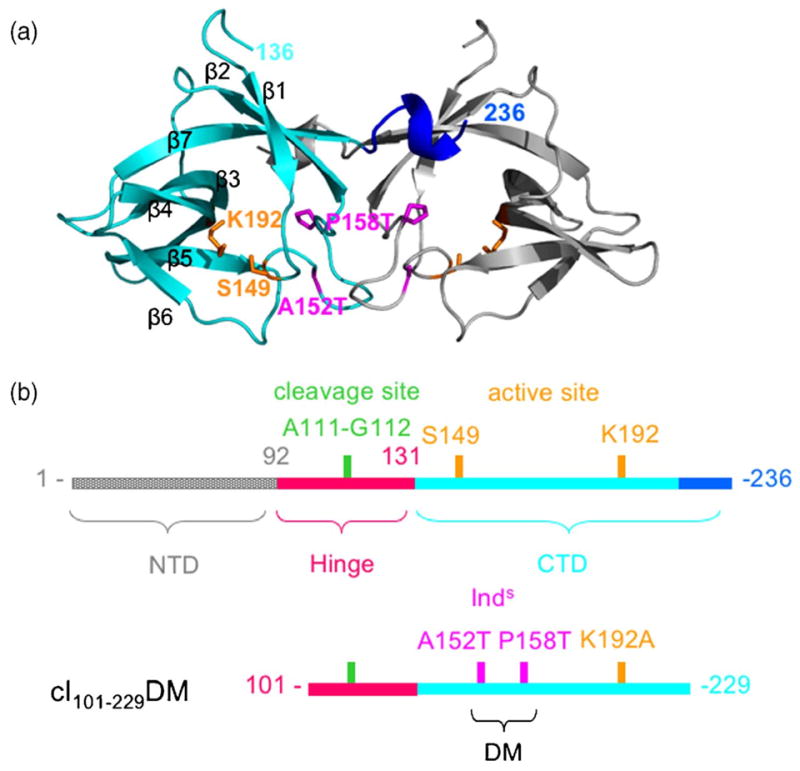
Design of hypercleavable fragments of cI. (a) Crystal structure of the CTD dimer (PDB code 1F39)13 is shown with one monomer in cyan. Side-chains of active site residues are in orange, residues affected by Inds mutations are in magenta, and the C-terminal 310 helix, residues 230-236, in blue, (b) Cartoons representing full length cI (top) and the hyper-cleavable fragments used in this study (bottom) are shown with the residues of interest color coded as in (a) and Figure 4.
A model for the RecA-mediated cleavage of LexA has been put forward in which LexA exists in two conformations that are in equilibrium: a non-cleavable form in which the cleavage site is removed from the active site, and a cleavable form in which the cleavage site is correctly positioned within the active site. 12, 15 In the absence of RecA, this conformational equilibrium strongly favors the non-cleavable form, such that cleavage is extremely slow. According to the model, RecA activates cleavage by selectively binding and stabilizing the cleavable form. Both the cleavable and non-cleavable forms of LexA have been observed in crystal structures12 where the latter was captured by mutating the active site lysine (K156A) and introducing three “super-inducible” (Inds) mutations. In the cleavable form of LexA, the Ala-Gly cleavage site is bound within the active site in a manner appropriate for hydrolysis. In the non-cleavable form the cleavage site is bound to a different surface of LexA, about 20 Å away from the active site, which is solvent exposed. From these structures it is apparent that a significant energetic barrier to forming the cleavable conformation is the burial of the active site lysine, which would require it to be deprotonated. This provides the basis for the observed pH dependence of autodigestion, which occurs at pH ~10 in the absence of RecA.
In the case of cI repressor, crystal structures of the isolated N-terminal DNA-binding and C-terminal oligomerization domains have been determined,13,16-18 but there is currently no structure reported for intact cI, and residues 92-131 of the hinge, which contain the cleavage site, are absent from the structures of the isolated N and C-terminal domains. In order to gain further insight into the mechanism of RecA-mediated cleavage and how it is controlled for cI, we have designed a hyper-cleavable fragment of cI consisting of residues 101-229, characterized its biochemical properties, and determined its structure at pH 6.5 to 1.8 Å resolution. Although this fragment of cI is hyper-cleavable in the RecA-mediated reaction, it does not undergo autodigestion at all at neutral pH in the absence of RecA. In the structure, the hinge region folds over the active site of the CTD, but with the cleavage site flipped out and exposed to solvent, in what is likely to be a non-cleavable form of the repressor.
Results
Design and characterization of hyper-cleavable fragments of cI
As part of an effort to design cI molecules suitable for structural studies in complex with RecA, we have constructed hyper-cleavable truncated versions of cI consisting of residues 93-229 or 101-229 and containing two mutations, P158T and A152T. We refer to these constructs as cI93-229DM and cI101-229DM, respectively, where the DM stands for double mutant (Figure 1). The two mutations, which are termed Inds for super-inducible, were identified in a genetic screen for hyper-cleavable repressor molecules.19 The mutations disrupt dimer formation, increasing the pool of cI monomer that can bind to RecA and undergo self-cleavage. The residues affected by these mutations are located at the dimer interface in the crystal structure of the CTD (Figure 1(a)). Removing the N-terminal domain also enhances cleavage by destabilizing dimer formation.14,19 Removal of the C-terminal residues 230-236 was intended to further disrupt the dimer, since these residues form a 310 helix that is swapped in the dimer and is the site of several other mutations that affect dimerization.4,13,20
To evaluate the effects of these alterations, the RecA-mediated cleavage of four different cI fragments was compared (Figure 2(a)). As seen with full-length cI,19 incorporation of the two Inds mutations into the C-terminal domain fragments leads to significantly enhanced RecA-mediated cleavage, since cI93-236DM is cleaved considerably faster than the same fragment without the mutations (cI93-236). Somewhat surprisingly, the fragment consisting of residues 93-229 (cI93-229DM) is cleaved significantly faster than the fragment containing the further N-terminal truncation (cI101-229DM), indicating a potential role (either direct or indirect) for residues 93-100 in RecA-mediated cleavage. cI93-229DM is cleaved at about the same rate as cI93-236DM, indicating that deletion of the C-terminal helix (residues 230-236) does not disrupt dimerization to a significantly greater extent than the two Inds mutations. Despite having enhanced cleavage for the RecA-mediated reaction, all of the hyper-cleavable fragments have the same or even lower rates of autodigestion at pH 10 as cI93-236 (Figure 2(b)). Importantly, none of the fragments autodigest to detectable levels at neutral pH (pH 7.4) in the absence of RecA (data not shown). Thus, incorporation of the Inds mutations in cI specifically increases the rate of RecA-mediated cleavage, as was seen for the full-length protein.19
Figure 2.
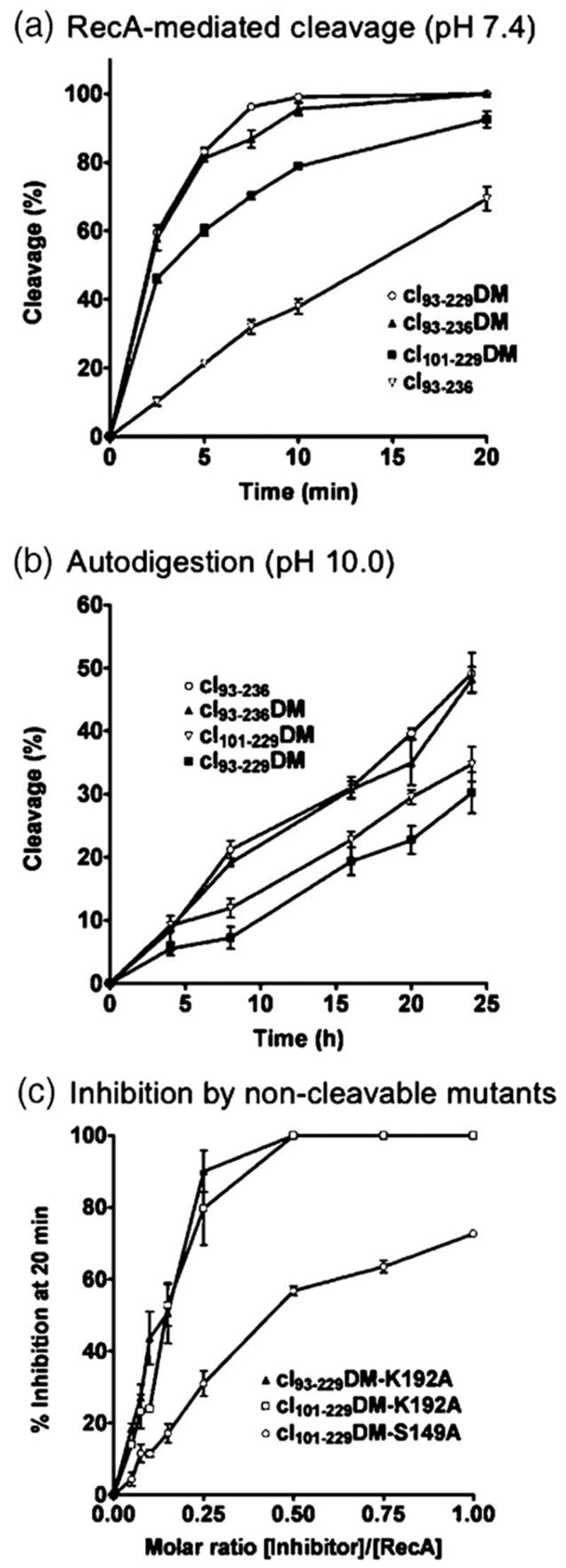
Biochemical characterization of hyper-cleavable cI fragments. (a) RecA-mediated cleavage of cI fragments in the presence of ADP-AlF4 and a TGG-repeating 15-mer oligonucleotide. Notice that incorporation of the two Inds mutations (DM) significantly increases the cleavage rate, removal of residues 230-236 only slightly increases the cleavage rate, and removal of residues 93-100 decreases the cleavage rate, (b) Autodigestion of cI fragments at pH 10 in the absence of RecA. The hyper-cleavable cI fragments autodigest at the same rate or slower than cI93-236. (c) Inhibition of RecA-mediated cleavage of cI93-236 by cI constructs rendered non-cleavable by mutation of the active site Ser149 or Lys192 residues. Notice that the K192A mutants bind to RecA significantly more tightly than the S149A mutant, and give complete inhibition at a 0.5 molar equivalent relative to RecA.
To assess the binding of the hyper-cleavable cI constructs to RecA, the active site residues Lys192 and Ser149 of cI were separately mutated to alanine, and the ability of the resulting non-cleavable cI molecules to inhibit RecA-mediated cleavage of cI93-236 was measured (Figure 2(c)). cI101-229DM with a K192A substitution inhibits RecA-mediated cleavage of cI93-236 significantly better than cI101-229DM with a S149A substitution, as was observed for the equivalent active site mutants of LexA.21 In fact, a substoichiometric amount of cI101-229DM K192A relative to RecA (approximately a 0.5 molar equivalent) completely inhibits cleavage of cI93-236. At lower concentrations, slightly enhanced inhibition was observed with cI93-229DM K192A as compared to cI101-229DM K192A, although this difference is not significant enough to conclude that residues 93-100 of cI are involved in or important for binding to RecA.
Structure of a hyper-cleavable cI fragment containing the cleavage site region
To gain further insight into the mechanism of RecA-mediated cleavage, crystal structures of the cI101-229DM and cI101-229DM K192A constructs were determined to 2.5 Å and 1.8 Å resolution, respectively (Table 1 and Figures 3 and 4). Aside from the K192A mutation in cI101-229DM K192A, the two structures are essentially indistinguishable, superimposing to an rms deviation of 0.33 Å for all atoms. We were not able to crystallize the slightly extended fragments cI93-229DM and cI93-236DM, presumably because the additional residues are flexible and would disrupt potential crystal contacts, and this is indeed indicated by the crystal packing interactions in the crystal form observed for the fragment consisting of residues 101-229.
Table 1.
Crystallographic data and refinement statistics
| A. Crystal data | ||
| Crystal | CI101-229DM K192A | CI101-229DM |
| Space group | P6122 | P6122 |
| Cell dimensions (Å) | a = b=59.6, c = 148.5 | a=b=59.6, c=148.4 |
| Resolution (Å) | 30-1.8 | 30-2.5 |
| Observed reflections | 119,808 | 31,534 |
| Unique reflections | 15,045 | 5891 |
| Completeness (%) | 97.6 (83.2)a | 99.6 (99.1) |
| Redundancy | 7.9 (2.12) | 5.3 (5.6) |
| Rmerge (%)b | 6.6 (31.0) | 10.9 (33.6) |
| I/σ | 18.6 (3.2) | 10.7 (4.9) |
| B. Refinement statistics | ||
| Number protein atoms | 1,007 | 998 |
| Number water molecules | 120 | 80 |
| Number of reflections (working/free set) | 13,101/1944 | 5066/825 |
| R-factor (%)c | 25.2 (43.6) | 25.3 (31.1) |
| Free R-factor (%)c | 28.2 (46.9) | 32.1 (39.1) |
| Estimated coordinate error (Å)d | 0.32 | 0.49 |
| Mean B-factor (Å2) | 32.4 | 33.4 |
| RMS deviation from ideality: | ||
| Bonds (Å) | 0.005 | 0.006 |
| Angles (°) | 1.3 | 1.2 |
Numbers in parentheses refer to the highest resolution shell only
Rmerge=Σ|Ih − <I>h|/Σ Ih, where <I>h is average intensity over symmetry equivalents, and h is reflection index. I/σ is the mean of the intensity/sigma of the unique averaged reflections.
R-factor=Σ|Fobs − Fcalc|/ΣFobs. R free is calculated from 10% of the reflections that are omitted from the refinement.
The estimated coordinate error is the value from the cross-validated sigma plot.
Figure 3.
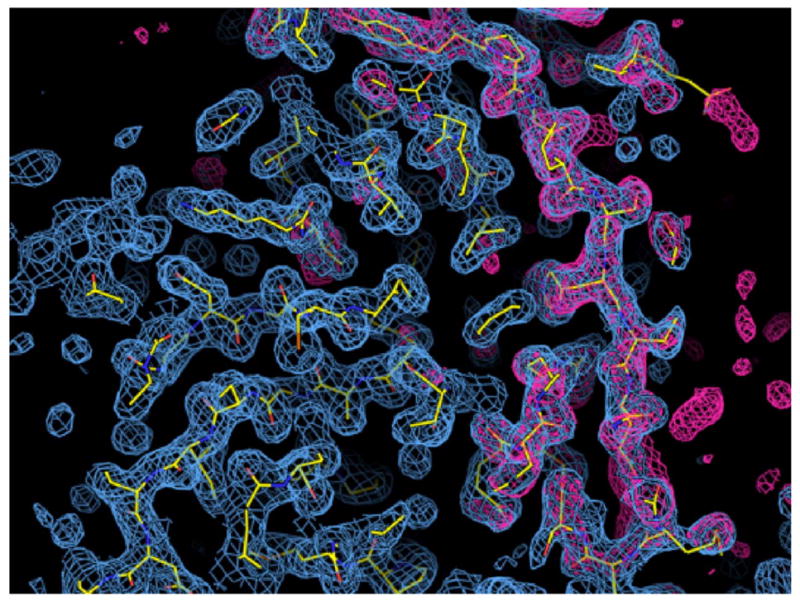
Electron density for the crystal structure of a hyper-cleavable monomeric fragment of cI containing the hinge region. The structure of the cI101-229DM K192A construct is shown superimposed on the 2Fo-Fc electron density map (blue cage) contoured at 1 sigma and calculated at 1.8 Å resolution. The red cage shows the Fo-Fc map contoured at +3 sigma after simulated annealing refinement with the atoms of the hinge region (residues 101-132) omitted. The atoms in the model are colored yellow for carbon, blue for nitrogen, and red for oxygen.
Figure 4.
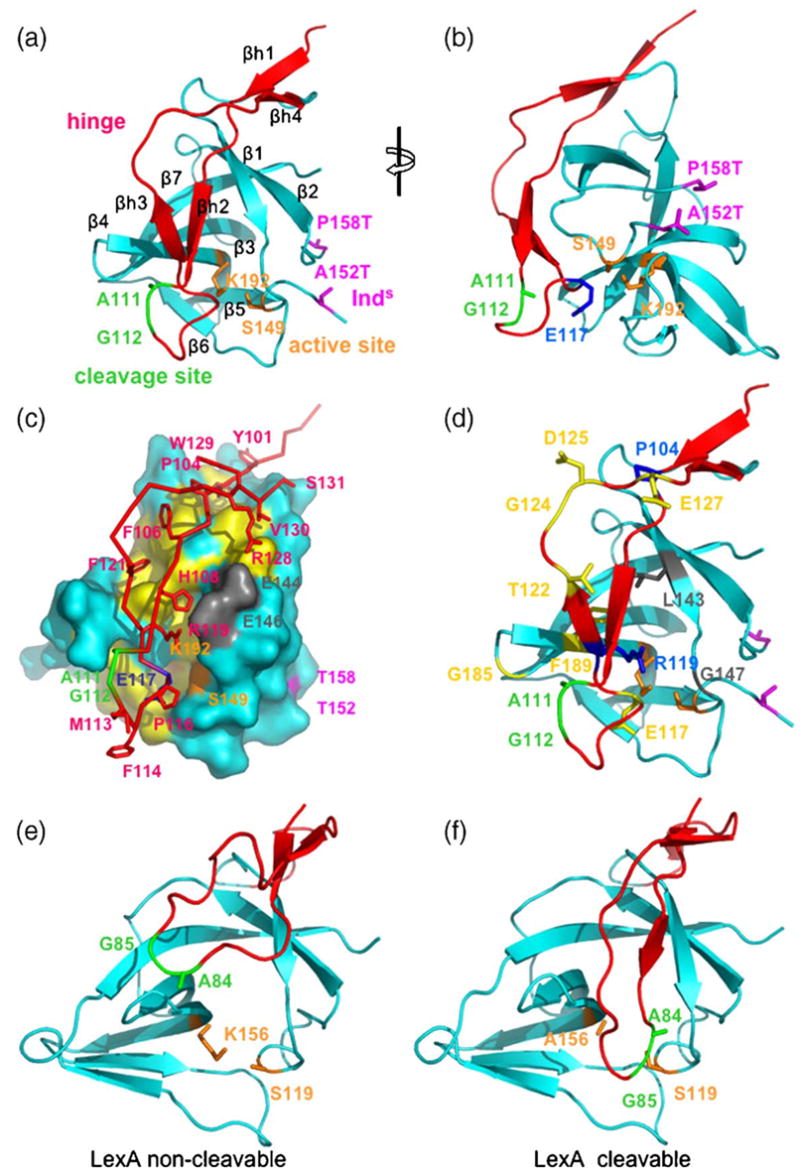
Crystal structure of a hyper-cleavable monomeric fragment of cI containing the hinge region. (a) and (b) Ribbon diagrams showing the folding of the hinge region (red) onto the CTD (cyan). All Figures were drawn using coordinates of the cI101-229 DM structure, which has lysine at position 192. The monomer in (a) is in a similar orientation as the left subunit of the dimer in Figure 1(a). (b) View from the right of (a), as indicated by the rotation symbol. The cleavage site (green) is flipped out of the active site (orange), which is instead occupied by Glu117 (blue in (b)). The loop connecting the residues bearing the super-inducible Inds mutations (magenta) is disordered. β-Strands of the hinge and CTD are labeled βh1-βh4 and β1-β7, respectively. (c) Interactions between the hinge and the CTD. The hinge region is shown as a red trace, with side-chains of interest in sticks. The CTD is represented as a molecular surface in cyan, with apolar residues underneath the hinge in yellow, active site residues in orange, and two glutamate residues that form ion pairs with arginine residues of the hinge in grey. The orientation is the same as in (a). (d) Locations of inducible-deficient (Ind−) mutations. Residues affected by mutations that disrupt RecA-mediated cleavage but not autodigestion (termed RecA-specific) are in yellow (E117K, T122I, G124D/D125V, E127K, G185E, G185R, and F189L). Residues affected by mutations that disrupt both RecA-mediated cleavage and autodigestion are in grey or green (A111T, G112E, L143F and G147D). Residues affected by mutations that lead to instability and rapid proteolysis of the hinge are in blue or green (P104L, G112R and R119K). (e) and (f) Structures of a C-terminal fragment of LexA in the non-cleavable and cleavable conformations, respectively. The structures are drawn using PDB codes 1JHC (e) and 1JHE. (f) Notice that in the non-cleavable form, the region containing the cleavage site is removed from the active site, while in the cleavable form the cleavage site lies directly in the active site.12
As expected, cI101-229DM (and cI101-229DM K192A) crystallizes as a monomer. The threonine residues substituted for Ala152 and Pro158 are resolved in the structure, but density for the intervening loop (residues 153-157) is not observed, presumably because without dimer formation the loop is disordered. Aside from the disordering of this loop, the monomer does not show any large conformational changes relative to its structure in the dimer (PDB code 1F39)13 as the two superimpose to an rmsd value of 0.64 Å. The hinge region, residues 101-131, is fully resolved in the electron density maps (Figure 3), and forms a long, twisted hairpin that folds down over the active site of the CTD (Figure 4(a) and (b)), with essentially the same overall topology as the corresponding regions of LexA and phage 186 repressor.12,22 The hinge folds into two pairs of β-strands that pack onto the CTD, one at the top of the structure (βh1,βh4) and a second just above the active site (βh1-βh3). Addition of strands βh2 and βh3 onto the β-sheet of the CTD results in the formation of a seven-stranded closed β-barrel.
The cleavage site is located within the loop at the bottom of the hinge, which we call the cleavage-site loop. Although this loop is folded over the active site of the CTD, the residues of the cleavage site (Ala111 and Gly112) do not insert into the active site, as would be expected for a cleavable conformation, but instead are flipped out to the surface of the structure (Figure 4(b)). The closest approach of the hinge to the active site is made by the side-chain of Glu117 of the cleavage-site loop, which comes within 4.5 Å of Lys192. Lys192 is almost fully buried in the structure, but presumably partially neutralized by Glu117, although the two side-chains are not oriented properly to form a strong ion pair. Interestingly, Glu117 is a residue that is unique to cI, as alanine is found at this position in most other LexA family members, including LexA and UmuD. Moreover, genetic and biochemical studies implicate Glu117 as having a key role in stabilizing the non-cleavable form of cI, since the E117K mutation increases the rate of autodigestion (see below).24,25
The hinge region of cI forms an integral part of the structure of the CTD, consistent with the observation that in the related P22 repressor, fragments corresponding to the hinge and CTD remain associated after protease digestion.23 A total of 1922 Å2 of solvent accessible surface area is buried between the hinge and the CTD, and water molecules are excluded from much of the interface. The β-strand regions of the hinge interact intimately with the CTD, while the cleavage-site loop folds up on itself but does not form close contacts with the CTD. The contacts between the hinge and CTD (Figure 4(c)) include a combination of hydrophobic, ion-pair, and hydrogen bonding interactions. Several apolar residues, including Tyr101, Tyr103, Pro104, Phe106, Phe121, and Val130 of the hinge, and Phe141, Trp142, Leu143, Leu165, Ile167, and Phe189 of the CTD, are buried at the interface. The binding of the hinge to the CTD is also stabilized by ten hydrogen bonds, six between side-chain and main-chain atoms, and four between main-chain and main-chain atoms. In addition, two close ion-pairs are formed between Arg119 and Arg128 of the hinge, and Glu146 and Glu144 of the CTD, respectively. Arg119 also forms a hydrogen bond to the backbone carbonyl group of Pro116 of the cleavage-site loop, and thus appears to play a dual role in stabilizing the binding of the hinge to the CTD. Accordingly, the conservative R119K mutation destabilizes the folding of the hinge, as determined by the susceptibility of the hinge region of this mutant to rapid proteolysis in cell lysates.24
Discussion
Folding of the hinge in intact cI protein
The structure of the hinge region of cI within a monomeric form of the CTD has been captured using a construct that is quite different from native cI: 100 residues from the N terminus and seven residues from the C terminus have been deleted, and two mutations were introduced to disrupt the dimer interface. Thus, it is prudent to consider to what extent the observed structure is biologically meaningful. Importantly, the fragment used for crystallization maintains the expected cleavage properties; the effects of the mutations on the fragment recapitulate their effects on full-length cI, as seen in previous genetic and biochemical studies.19,24 The deletions in the crystallized construct apparently facilitate crystallization because slightly extended fragments could not be crystallized. Based on the structural information, it is likely that the additional residues of the extended fragments (93-100 and 230-236) are highly flexible in the monomer such that they prevent formation of crystal contacts.
One must also consider the possible influence of crystal contacts on the observed conformation of the hinge region. The hinge region of cI does make two contacts with symmetry related molecules in the crystal. The more extensive of these contacts involves several residues at the base of the hinge, located at the top of the structure as viewed in Figure 4, about 25 Å away from the cleavage site. This contact buries a total of 1534 Å2 of solvent-accessible surface area. The second of these crystal contacts involves Met113 and Phe114 of the cleavage site loop, but buries a total of only 521 Å2 of surface area. Given that the crystal contact involving the cleavage-site loop is considerably less extensive than the interactions between the hinge and the C-terminal domain (1922 Å2), it is likely that crystal contacts do not significantly distort the conformation of the hinge that is observed in the crystal structure. Moreover, the structure of the hinge is consistent with the available genetic data (as described below), and has a similar overall topology as the corresponding regions of LexA and phage 186 repressors.
Superposition of this structure with a subunit from the CTD octamer shows that the folding of the hinge would not appear to block dimer formation, consistent with the observation that a disulfide-cross-linked dimer of cI undergoes autodigestion at a similar rate as wild-type. However, the hinge would appear to interfere with the dimer-dimer interaction that builds up the octamer and is responsible for cooperative binding of repressor dimers to adjacent operator sites. Thus, we would predict that for intact dimers, the hinge is likely folded as observed in this structure, but upon formation of higher-ordered oligomers that bind to multiple operator sites, the hinge is displaced and possibly unfolded. This makes sense mechanistically, since release of the hinge upon oligomerization would give the N-terminal domains more freedom to adopt their optimal positions on the different operator sites, which are themselves restrained by virtue of being connected via the DNA duplex.
Mapping of cI induction mutants onto the structure
A genetic screen identified 16 Ind− mutations in cI that maintain a functional repressor but prevent RecA-mediated cleavage, thereby conferring an induction-deficient phenotype.25 All of these mutations affect residues within the hinge region or the underlying surface of the CTD, and thus the new structure that we have determined containing the hinge region provides a three-dimensional context for interpreting them. It must be noted, however, that there may be significant conformational changes that occur within cI upon RecA binding. The mutations are mapped onto the crystal structure of cI101-229DM in Figure 4(d). Biochemical characterization of the purified proteins identified four mutants defective in both RecA-mediated cleavage and autodigestion, nine RecA-specific mutants that diminish RecA-mediated cleavage but not autodigestion, and three that could not be purified due to rapid proteolysis of the hinge region.24 Two of the mutants that are defective in both RecA-mediated cleavage and autodigestion directly affect the cleavage site (A111T and G112E), and one is very near the active site Ser149 residue (G147D). The fourth (L143P) would likely disrupt the folding of the hinge over the active site, since Leu143 is tightly packed among residues of the hinge (Phe121, His108) and CTD (Phe141, Phe189).
Since the RecA-specific Ind− mutations diminish RecA-mediated cleavage but not autodigestion, the residues at which they occur have been implicated as being possible sites of interaction with RecA protein.24 Interestingly, RecA-specific Ind− mutations are not observed for LexA.21 Six of the RecA-specific mutations occur at four residues, Thr122, Gly124, Asp125, and Glu127, which are located on a flexible surface loop following strand βh3. This loop would appear to be suitably positioned for a possible interaction with RecA (Figure 4(d)). One of the RecA-specific mutations occurs at Glu117 (E117K), which projects into the active site in the current structure, but would need to be displaced (and likely exposed) when the hinge adopts the cleavable conformation. Interestingly, the E117K mutation actually increases the rate of autodigesion by about fourfold, while completely abolishing RecA-mediated cleavage. Based on the structure, the E117K mutation could enhance autocleavage by destabilizing the non-cleavable conformation via unfavorable electrostatic interactions with the active site lysine residue. RecA-specific mutations also occur at Gly185, located on the β3-β4 hairpin turn at the surface of cI, as well as at Phe189, which is buried at the interface of the CTD and the hinge. A role in RecA binding for Phe189 is thus not likely, and the F189L Ind− mutation likely disrupts the proper RecA-cI interaction more indirectly, possibly by disrupting the folding of the hinge region.
The structure represents the non-cleavable form of cI
Assuming a two-state conformational equilibrium model for cI similar to that proposed for LexA in which the protein exists in cleavable and non-cleavable conformations,12,15 the structure presented here most likely corresponds to the non-cleavable form, since in the structure the cleavage site is not correctly positioned within the active site. Interestingly, however, the overall conformation of the hinge region of cI, which folds down over the active site, more closely resembles that of LexA in its cleavable form. In the non-cleavable form of LexA (Figure 4(e)), the entire portion of the hinge containing the cleavage site packs against a different surface of the CTD, such that the active site is completely exposed to solvent. Thus, our structure suggests that the non-cleavable conformations of LexA and cI are quite different from one another, and we would predict that the conformational changes necessary to inter-convert between the non-cleavable and cleavable forms of cI are significantly less extensive than is seen for LexA.
Given the large difference in conformation between the non-cleavable form of LexA and the structure we have determined for cI, one could argue that our structure does not represent the actual non-cleavable conformation for cI, but is instead much closer to the cleavable form, but with the cleavage site loop distorted by crystal contacts. However, this is very unlikely because the cI101-229DM construct we have crystallized, which contains an intact active site (lysine at position 192), does not cleave itself at neutral pH in the absence of RecA, and thus presumably does not adopt a cleavable conformation to any significant extent in solution. In addition, the cI101-229DM construct does not autodigest to a significant degree under the crystallization conditions (pH=6.5), since the intact, non-cleaved molecule is observed in the crystal structure. Therefore, the most reasonable conclusion is that our structure of cI represents the actual non-cleavable form cI, which happens to be quite different from the non-cleavable form of LexA. While it is conceivable that the structure we have determined is only one of many non-cleavable conformations that cI could adopt, the observed structure nicely accounts for the biochemical properties of several of the cI mutant proteins, most notably R119K and E117K (see above).
What are the conformational changes that would need to occur for the hinge to assume a cleavable conformation? Starting from the structure we have determined, a fairly modest rearrangement of the cleavage-site loop could reorient the cleavage site to project into the active site. Such a conformational change would seem quite feasible, since the hinge is anchored to the CTD mostly via contacts involving residues of the β-strands, while the cleavage-site loop itself does not make intimate contacts with the CTD. Significant movements of Arg119 and Glu117, which are positioned close to the active site, would also need to occur, and accordingly both of these residues are identified by Ind− mutations as having key roles in RecA-mediated cleavage. Three apolar residues at the tip of the cleavage-site loop, Met113, Phe114, and Pro116, are exposed at the surface in this structure, but might help to stabilize the cleavable form of cI by binding to a hydrophobic patch on RecA or on the CTD of cI itself. An exposed apolar patch on the CTD, including Leu184 and Ile213, is in fact located underneath the cleavage-site loop (Figure 4(c)).
It is notable that for LexA, mutating the active site lysine and introducing three Inds mutations allowed for the cleavable conformation to be captured crystallographically, whereas for cI a seemingly similar set of mutations (two Inds and a K192A mutation) results in the non-cleavable conformation. In fact, this mirrors the biochemical properties, since Inds mutants of LexA (without the K192A mutation) rapidly autodigest at neutral pH,15 and Inds mutants of cI do not.19 For cI the Inds mutations increase the rate of RecA-mediated cleavage but have very little effect on autodigestion, whereas for LexA exactly the opposite is true. The Inds mutations in cI act by destabilizing the dimer interface, whereas the mutations in LexA more directly affect the interactions between the hinge and the CTD. All of the structures of LexA are dimers, including the cleavable form with three Inds mutations, whereas the structure of cI with two Inds mutations reported here is a monomer. The different nature of the Inds mutants in LexA and cI is intriguing, and may reflect a conformational equilibrium for cI that more strongly favors the non-cleavable form, since cI undergoes both RecA-mediated cleavage and autodigestion much more slowly than LexA.
Insights into binding of cI to RecA
The structure and biochemical data offer some new insights into the binding of cI to the RecA-DNA filament. First, as described above, mapping of the RecA-specific mutants onto the structure suggests regions on the surface of cI that are possible binding sites for RecA. Although these mutants do not provide a definitive view of the complete RecA binding site or sites on cI, the new structural context for the mutants provides an improved picture of the general regions on the surface of cI that are likely to be involved in RecA binding. Second, the biochemical data implicate residues 93-100 as having a role in RecA-mediated cleavage: removing them significantly decreases RecA-mediated cleavage but not autodigestion. Although a possible role for these residues would be direct involvement in RecA binding, based on the inhibition studies with the active site lysine mutants, removing them does not significantly decrease the binding of cI to RecA. It is conceivable that these residues are involved in an interaction with RecA that promotes cleavage but does not significantly increase the stability of the binding that is measured in the inhibition experiments. Residues 93-100 are not present in the crystallized fragment, but the position of residue 101 indicates they would be at the top of the structure (as viewed in Figure 4), fairly distant from the active site. Third, the observation that sub-stoichiometric amounts of cI101-229DM K192A relative to RecA (a ~0.5 molar ratio) completely inhibit RecA-mediated cleavage of cI93-236 suggests that binding of cI to a given site on the RecA filament prevents binding of a second molecule of cI to a neighboring site, either by physical occlusion or an allosteric effect. The 0.5:1 binding stoichiometry for cI estimated from the inhibition experiment is remarkably consistent with earlier electron microscopy studies of LexA in complex with RecA-DNA, which also gave a stoichiometry of about 0.5:1.26
Finally, the structure of the monomeric, hyper-cleavable form of cI that we have captured provides some further clues into why only the monomer of cI is capable of undergoing RecA-mediated cleavage. Possible explanations for the selective cleavage of the monomer, which are not exclusive of one another, include (1) conformational changes in the monomer relative to the dimer, (2) the hinge could fold appropriately only in the monomer, (3) the RecA binding site on cI overlaps with the dimerization surface, and (4) the dimer is sterically occluded from the cI binding site within the RecA-ssDNA filament. The current structural information is most consistent with the latter possibility, since there is no evidence of dramatic conformational changes in the monomer relative to the dimer, the folding of the hinge region appears to be compatible with dimer formation, and the RecA binding site implicated by the RecA-specific Ind− mutants does not overlap with the surface of cI used for dimerization. It must be pointed out, however, that there could be significant conformational changes that occur within a cI monomer upon RecA binding that could be restricted from occurring within the dimer. It is also possible that the RecA binding site on the surface of cI covers a significantly larger area than is implicated by the known RecA-specific Ind− mutations, and thus possibly extend into the region involved in dimerization. Clearly, further structural information on the complex between cI and the RecA-ssDNA filament will provide more definitive insight into this issue.
Experimental Procedures
Protein expression and purification
Expression and purification of Escherichia coli RecA protein were performed as described.27 DNA for the cI constructs was PCR-amplified from phage γ genomic DNA (New England Biolabs) and ligated into the Ndel and BamHI sites of pET-14b, which expresses the proteins with an N-terminal 6His tag and a thrombin proteolytic site. Site-directed mutagenesis to incorporate the A152T, P158T, K192A, and S149A mutations was performed using the QuickChange procedure (Stratagene). The resulting plasmids were transformed into the arabinose-inducible BL21-AI strain of E. coli (Invitrogen), and expressed in one liter cultures at 37 °C. The cells were harvested by centrifugation, resuspended in 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, and 10 mM imidazole, and lysed by sonication. After centrifugation, the supernatant was loaded onto a Ni-NTA column (Qiagen) at 4 °C. After washing with buffer containing 30 mM imidazole, proteins were eluted with a linear gradient of 30 mM-500 mM imidazole. Collected fractions were dialyzed into 20 mM NaH2PO4 (pH 7.4), 150 mM NaCl, and digested at 22 °C with 50 units of thrombin for 24 h. After a second nickel affinity step, the cleaved cI protein was further purified by anion exchange chromatography on HiTrap Q HP (GE Healthcare), dialyzed into 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM DTT, concentrated to 90-120 mg/ml, and frozen in small aliquots at −80 °C. Protein concentrations were determined by optical density at 280 nm using extinction coefficients calculated from the amino acid sequences. All of the proteins used in this study contain the extra N-terminal GSHM sequence from the expression vector.
RecA-mediated cleavage and autodigestion reactions
RecA-mediated cleavage reactions (50 μl) contained 20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 30 μM (nucleotides) of 15-mer TGG-repeating oligonucleotide, 1 mM ADP, 2 mM aluminum nitrate, 10 mM NaF, 2 mM MgCl2, and 10 μM E. coli RecA. This mixture was incubated at 25 °C for 30 min, at which point 10 μM of a cI construct was added and incubated at 25 °C. At the indicated time points aliquots were removed and quenched by adding 0.25 volumes of 5× SDS-PAGE loading buffer and immediately heating to 95 °C for 5 min. The samples were electrophoresed on SDS −15% (w/v) polyacrylamide gels and visualized by Coomassie brilliant blue staining. Gels were digitally scanned and the intensity of each band was quantified with Kodak Digital Science™ 1D image analysis software. The percent cleavage was calculated from the ratio of the net intensities of the bands corresponding to cleaved product and uncleaved substrate, where a correction factor was applied to the intensity for each cleaved product to account for its smaller size. Each reaction was performed in triplicate. Cleavage-inhibition experiments were performed as describe above, except that the indicated concentration (0-10 mM) of the cleavage-inactivated cI construct (cI101-229DM K192A, cI93-229DM K192A, or cI101-229DM S149A) was included in the RecA pre-incubation mixture prior to the addition of the cleavable cI93-236 construct. Autodigestion reactions in the absence of RecA-DNA were performed at 37 °C in 20 mM glycine-NaOH (pH 10), 10 mM CaCl2.
Crystallization and X-ray structure determination
cI101-229DM and cI101-229DM K192A were crystallized at 10 mg/ml by hanging drop vapor diffusion at 22 °C where the reservoir solution consisted of 36% (w/v) PEG 550, 0.1 M sodium cacodylate (pH 6.5), 0.2 M calcium acetate. For X-ray data collection, crystals were transferred to the solution above with PEG 550 increased to 40%, and frozen in liquid nitrogen. X-ray diffraction data were collected at −180 °C using a Rigaku RU300 rotating anode generator and a RAXIS-IV++ image plate detector. Images were processed with CrystalClear software (Molecular Structure Corporation). Crystals belong to space group P6122 with one monomer per asymmetric unit (see Table 1). The structure of cI101-229DM was solved by molecular replacement using a search model consisting of a monomer of the CTD (PDB accession code 1F39) with residues 230-236 removed. After initial refinement using CNS,28 the R-factor was high (40%) but the resulting electron density map allowed for the hinge region (residues 101-135) to be traced using O.29 Continued refinement at 1.8 Å used the conjugate gradient minimization and individual temperature factor protocols of CNS. Residues 153-157 were not included in the model due to lack of electron density. The cI101-229DM K192A structure was solved from the cI101-229DM structure by difference Fourier methods. Figures were prepared using Pymol (Delano Scientific).
Protein Data Bank accession numbers
The coordinates and structure factors for the crystal structures of cI101-229DM and cI101-229DM K192A have been deposited with the RCSB Protein Data Bank under the accession numbers 2HO0 and 2HNF, respectively.
Acknowledgments
This work was supported by NIH grant GM067947 to C.E.B.
Abbreviations
- CTD
C-terminal oligomerization domain
- ss
single-stranded
Footnotes
Edited by K. Morikawa
References
- 1.Ptashne M. A Genetic Switch. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2004. [Google Scholar]
- 2.Roberts JW, Roberts CW. Proteolytic cleavage of bacteriophage lambda repressor in induction. Proc Natl Acad Sci USA. 1975;72:147–151. doi: 10.1073/pnas.72.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Little JW. Autodigestion of lexA and phage lambda repressors. Proc Natl Acad Sci USA. 1984;81:1375–1379. doi: 10.1073/pnas.81.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen S, Knoll BJ, Little JW, Mount DW. Preferential cleavage of phage lambda repressor monomers by recA protease. Nature. 1981;294:182–184. doi: 10.1038/294182a0. [DOI] [PubMed] [Google Scholar]
- 5.Craig NL, Roberts JW. E. coli recA protein-directed cleavage of phage lambda repressor requires polynucleotide. Nature. 1980;283:26–30. doi: 10.1038/283026a0. [DOI] [PubMed] [Google Scholar]
- 6.Phizicky EM, Roberts JW. Kinetics of RecA protein-directed inactivation of repressors of phage lambda and phage P22. J Mol Biol. 1980;139:319–328. doi: 10.1016/0022-2836(80)90133-3. [DOI] [PubMed] [Google Scholar]
- 7.Shinagawa H, Iwasaki H, Kate T, Nakata A. RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc Natl Acad Sci USA. 1988;85:1806–1810. doi: 10.1073/pnas.85.6.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horii T, Ogawa T, Nakatani T, Hase T, Matsubara H, Ogawa H. Regulation of SOS functions: purification of E. coli LexA protein and determination of its specific site cleaved by the RecA protein. Cell. 1981;27:515–522. doi: 10.1016/0092-8674(81)90393-7. [DOI] [PubMed] [Google Scholar]
- 9.Pabo CO, Sauer RT, Sturtevant JM, Ptashne M. The lambda repressor contains two domains. Proc Natl Acad Sci USA. 1979;76:1608–1612. doi: 10.1073/pnas.76.4.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slilaty SN, Little JW. Lysine-156 and serine-119 are required for LexA repressor cleavage: a possible mechanism. Proc Natl Acad Sci USA. 1987;84:3987–3991. doi: 10.1073/pnas.84.12.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peat TS, Frank EG, McDonald JP, Levine AS, Woodgate R, Hendrickson WA. Structure of the UmuD’ protein and its regulation in response to DNA damage. Nature. 1996;380:727–730. doi: 10.1038/380727a0. [DOI] [PubMed] [Google Scholar]
- 12.Luo Y, Pfuetzner RA, Mosimann S, Paetzel M, Frey EA, Cherney M, et al. Crystal structure of LexA: a conformational switch for regulation of self-cleavage. Cell. 2001;106:585–594. doi: 10.1016/s0092-8674(01)00479-2. [DOI] [PubMed] [Google Scholar]
- 13.Bell CE, Frescura P, Hochschild A, Lewis M. Crystal structure of the lambda repressor C-terminal domain provides a model for cooperative operator binding. Cell. 2000;101:801–811. doi: 10.1016/s0092-8674(00)80891-0. [DOI] [PubMed] [Google Scholar]
- 14.Sauer RT, Ross MJ, Ptashne M. Cleavage of the lambda and P22 repressors by recA protein. J Biol Chem. 1982;257:4458–4462. [PubMed] [Google Scholar]
- 15.Roland KL, Smith MH, Rupley JA, Little JW. In vitro analysis of mutant LexA proteins with an increased rate of specific cleavage. J Mol Biol. 1992;228:395–408. doi: 10.1016/0022-2836(92)90829-9. [DOI] [PubMed] [Google Scholar]
- 16.Pabo CO, Lewis M. The operator-binding domain of lambda repressor: structure and DNA recognition. Nature. 1982;298:443–447. doi: 10.1038/298443a0. [DOI] [PubMed] [Google Scholar]
- 17.Jordan SR, Pabo CO. Structure of the lambda complex at 2.5 A resolution: details of the repressor-operator interactions. Science. 1988;242:893–899. doi: 10.1126/science.3187530. [DOI] [PubMed] [Google Scholar]
- 18.Bell CE, Lewis M. Crystal structure of the lambda repressor C-terminal domain octamer. J Mol Biol. 2001;314:1127–1136. doi: 10.1006/jmbi.2000.5196. [DOI] [PubMed] [Google Scholar]
- 19.Gimble FS, Sauer RT. Lambda repressor mutants that are better substrates for RecA-mediated cleavage. J Mol Biol. 1989;206:29–39. doi: 10.1016/0022-2836(89)90521-4. [DOI] [PubMed] [Google Scholar]
- 20.Whipple FW, Kuldell NH, Cheatham LA, Hochschild A. Specificity determinants for the interaction of lambda repressor and P22 repressor dimers. Genes Dev. 1994;8:1212–1223. doi: 10.1101/gad.8.10.1212. [DOI] [PubMed] [Google Scholar]
- 21.Lin LL, Little JW. Autodigestion and RecA-dependent cleavage of Ind- mutant LexA proteins. J Mol Biol. 1989;210:439–452. doi: 10.1016/0022-2836(89)90121-6. [DOI] [PubMed] [Google Scholar]
- 22.Pinkett HW, Shearwin KE, Stayrook S, Dodd IB, Burr T, Hochschild A, et al. The structural basis of cooperative regulation at an alternate genetic switch. Mol Cell. 2006;21:605–615. doi: 10.1016/j.molcel.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 23.De J, Sauer RT. P22 c2 repressor. Domain structure and function. J Biol Chem. 1983;258:10536–10542. [PubMed] [Google Scholar]
- 24.Gimble FS, Sauer RT. Lambda repressor inactivation: properties of purified ind- proteins in the autodigestion and RecA-mediated cleavage reactions. J Mol Biol. 1986;192:39–47. doi: 10.1016/0022-2836(86)90462-6. [DOI] [PubMed] [Google Scholar]
- 25.Gimble FS, Sauer RT. Mutations in bacteriophage lambda repressor that prevent RecA-mediated cleavage. J Bacteriol. 1985;162:147–154. doi: 10.1128/jb.162.1.147-154.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu X, Egelman EH. The LexA repressor binds within the deep helical groove of the activated RecA filament. J Mol Biol. 1993;231:29–40. doi: 10.1006/jmbi.1993.1254. [DOI] [PubMed] [Google Scholar]
- 27.Xing X, Bell CE. Crystal structures of Escherichia coli RecA in a compressed helical filament. J Mol Biol. 2004;342:1471–1485. doi: 10.1016/j.jmb.2004.07.091. [DOI] [PubMed] [Google Scholar]
- 28.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallog, sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]