

Abstract
High spatial resolution Raman maps of fixed cells in an aqueous environment are reported. These maps were obtained by collecting individual Raman spectra via a Raman microspectrometer in a raster pattern on a 0.5-μm grid and assembling pseudocolor maps from the spectral hypercubes by multivariate methods. The Raman maps show the nucleus and the nucleoli of cells as well as subcellular organization in the cytoplasm. In particular, the distribution of mitochondria in the perinuclear region could be demonstrated by correlating distinct areas of the Raman maps with corresponding areas of fluorescence maps of the same cells after staining with mitochondria-specific labels. To the best of our knowledge, this is the first report of label-free detection of mitochondria inside a somatic mammalian cell using Raman microspectroscopy.
INTRODUCTION
The study of distribution, dynamics, and physiological activity of mitochondria in living cells is an increasingly important field of research in cellular biology. In addition to being the site of oxidative phosphorylation and, therewith, being a major component in the energy conversion in a cell, mitochondria have been found to contribute to many other cellular functions, such as apoptosis. Furthermore, aberrant mitochondria are associated with certain diseases, such as cardiomyopathy and cancer. Mitochondria also play a vital role in the production of energy-containing compounds needed in great abundance for developing embryos or fertilized eggs (1,2).
Mitochondria are subcellular organelles of ∼1 μm in length and 0.5 μm in width and height. However, mitochondria are dynamic structures and they may fuse to form large, tubular networks (3,4). The structure of the mitochondrion consists of a smooth outer membrane, a highly folded inner membrane on which the proteins responsible for oxidative phosphorylation are located, the intermembrane space between the two membranes, and the matrix (the space enveloped by the inner membrane). Electrons generated from glycolysis are carried through the electron transport chain in the inner membrane. This process creates a pH gradient across the inner membrane by pumping protons from the matrix to the intermembrane space. As protons are forced back into the matrix, the proton-motive force drives the synthesis of ATP, thus providing the energy for the cell.
Visualization of mitochondria or other subcellular organelles can be carried out via electron microscopy (EM) or more recently by cryoelectron microscopy, in which cells are embedded in vitreous ice (5). EM methods are rather invasive and involve extensive sample preparation and are, therefore, unsuitable for studies on living cells. Another possibility is to stain for mitochondria using fluorescent dyes. Fluorescence microscopy, using mitochondria-specific stains such as rhodamine derivatives, can be used to visualize regions of high mitochondrial concentration. The method requires the incubation of cells with a fluorescent dye, which presumably does not alter mitochondrial or cellular activity and viability.
In this article, we report a comparison of standard fluorescence microscopic methods with a label-free, confocal Raman microspectroscopic method to visualize mitochondrial distribution in cells. In confocal Raman microspectroscopy, the inherent vibrational spectroscopic signatures of biochemical constituents of cells are observed. This method permits the detection of Raman scattering from small volume elements of ∼0.3 × 0.3 × 1.3 μm3 in size, depending on the exciting wavelength used in the experiment. This spatial resolution is comparable to that obtainable in standard optical microscopy.
Here, we report Raman images of cells constructed via multivariate methods of analysis from Raman hyperspectral data sets, and we demonstrate the enormous advantages of these image reconstruction algorithms over simple univariate methods. We further demonstrate the feasibility of using confocal Raman microspectroscopy coupled with multivariate methods of analysis to visualize mitochondrial distribution by comparing Raman spectral images with those obtained using fluorescence microscopy. This method sets the stage for observing biological processes in living cells, such as the migration of mitochondria in oocytes without the use of any dyes or other contrasting agents.
MATERIALS AND METHODS
Cell culture
Human HeLa cells (cell line CCL-2, ATCC, Manassas, VA) were grown in 75-cm3 culture flasks (Fisher Scientific, Loughborough, Leicestershire, UK) with 7 mL of Dulbecco's modified Eagle's medium (ATCC) and 10% fetal bovine serum (ATCC) at 37°C and 5% CO2. Cells were seeded onto and allowed to attach to polished CaF2 substrates (Wilmad LabGlass, Buena, NJ), which were chosen to avoid background scattering that is observed from regular glass windows. The CaF2 substrates were removed from the culture medium after 12–24 h, and the cells were fixed in a 10% phosphate buffered formalin solution (Sigma-Aldrich, St. Louis, MO) and washed in phosphate buffered saline. For Raman and fluorescence measurements, the CaF2 windows with the attached and fixed HeLa cells were submerged in buffer solution during the measurement. The fixed cells are completely immobilized on the substrates.
Cells from adherent cultures (i.e., most cell lines originating from epithelial tissues, but not from lymphocytes or leukocytes) will attach to most substrates (glass, quartz, CaF2, plastics). Thus, any of these materials can readily be used for Raman studies, as long as they do not exhibit fluorescence signals. Nonadherent cells can be spin deposited onto suitable slides using any methodology commonly used in cytological preparations. These cells adhere similarly strongly and are completely immobilized on the substrates.
Fluorescence staining for mitochondrial distribution was carried out by adding a rhodamine derivative, green fluorescent Mitotracker Green FM (Invitrogen, Carlsbad, CA) to a final concentration of 200 nM. Fluorescence images were collected ∼20 min after the stain was added.
Raman data acquisition
Raman spectra were acquired using a WITec (Ulm, Germany) Model CRM 2000 confocal Raman microscope. Excitation (∼30 mW at 488 nm) was provided by an air-cooled Ar ion laser (Melles Griot, Carlsbad, CA; Model 532). The exciting laser radiation was coupled into a Zeiss (Thornwood, NY) microscope through a wavelength-specific single-mode optical fiber. The incident laser beam was collimated via an achromatic lens and passed through a holographic band-pass filter before it was focused onto the sample through the microscope objective. A Nikon (Tokyo, Japan) Fluor (60×/1.00 numerical aperture, working distance = 2.0 mm) water immersion objective was used in the studies reported here.
The sample was located on a piezoelectrically driven microscope scanning stage with an x,y resolution of ∼3 nm and a repeatability of ±5 nm, and z resolution of ∼0.3 nm and ±2 nm repeatability. The sample was scanned through the laser focus in a raster pattern at a constant stage speed of fractions of a micrometer per second. The continuous motion prevented sample degradation in the focal point of the laser beam. Spectra were collected at a 0.5-μm grid with a dwell time of 0.5 s.
Raman backscattered radiation was collected through the microscope objective and passed through a holographic edge filter to block Rayleigh scattering and reflected laser light before being focused into a multimode optical fiber. The 50-μm-diameter single-mode input fiber and the 50-μm-diameter multimode output fiber provided the optical apertures for the confocal measurement. The light emerging from the output optical fiber was dispersed by a 30-cm focal length, f/4 Czerny-Turner monochromator, incorporating interchangeable gratings (1800/mm, blazed at 500 nm and 600/mm, blazed at 500 nm). The light was finally detected by a back-illuminated deep-depletion, 1024 × 128 pixel charge-coupled device camera operating at −82°C.
Spatial resolution of the acquired confocal sampling area was determined by the diffraction limit and was between 250 and 350 nm (depending on the laser wavelength utilized). Depth resolution was about twice the lateral resolution. Spectral resolution depends on the excitation wavelength and grating groove density: for 488 nm excitation and the 600/mm grating, the optical resolution varied from ∼5.5 cm−1/data point at ∼200 cm−1 to ∼3.3 cm−1/data point at 4500 cm−1. Laser power at the sample is estimated to be ∼10 mW.
Fluorescence measurements
The green fluorescence of the Mitotracker stain was detected either using a Nikon Diaphot fluorescence microscope or the confocal Raman setup described above, using 488-nm excitation. For a comparison of Raman and fluorescence images to observe mitochondrial distribution, the cells were first scanned at 488-nm excitation to collect the Raman spectra at a dwell time of 0.5 s per data point. Subsequent to the Raman measurement, Mitotracker stain was carefully added and the fluorescence emission of the cell was rescanned at significantly lower laser power and a dwell time of 0.2 s per data point. The laser power was reduced to ∼1 mW for fluorescence measurements.
Multivariate Raman data analysis
A more detailed description of hierarchical cluster analysis (HCA) has been published before (6,7). For HCA, the pair-wise similarity of all spectra is expressed as a matrix of correlation coefficients 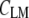 :
:
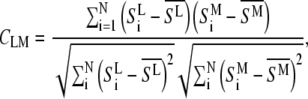 |
where 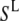 and
and 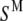 are two individual spectra of the data set, and
are two individual spectra of the data set, and 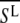 and
and 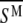 are the mean values of the spectral vectors. The correlation matrix
are the mean values of the spectral vectors. The correlation matrix 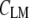 contains N2 entries, where N is the total number of spectra. The correlation coefficients can also be seen as normalized vector dot products ranging from zero to one, with
contains N2 entries, where N is the total number of spectra. The correlation coefficients can also be seen as normalized vector dot products ranging from zero to one, with 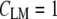 for identical spectra and
for identical spectra and 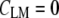 for spectra with no overlap. The correlation matrix was then searched for the most similar spectra, which were merged into a new object employing an algorithm introduced by Ward (8). The correlation matrix was recalculated and the merging process repeated until all spectra had been combined into a small number of clusters. Each cluster was assigned a color, and these colors were displayed at the coordinates at which the spectra were collected, creating a pseudocolor map based on the spectral similarity. Mean cluster spectra, obtained by averaging all spectra in a cluster, provide insight into the spectral differences between clusters and offer a measure of the compositional variations of the cellular regions associated with the clusters. The mean cluster spectra exhibit vastly improved signal/noise (S/N) ratios over individual spectra. HCA, using Ward's algorithm as a merging method, was found to produce the best clusters as judged by the homogeneity of the spectra in each cluster (9).
for spectra with no overlap. The correlation matrix was then searched for the most similar spectra, which were merged into a new object employing an algorithm introduced by Ward (8). The correlation matrix was recalculated and the merging process repeated until all spectra had been combined into a small number of clusters. Each cluster was assigned a color, and these colors were displayed at the coordinates at which the spectra were collected, creating a pseudocolor map based on the spectral similarity. Mean cluster spectra, obtained by averaging all spectra in a cluster, provide insight into the spectral differences between clusters and offer a measure of the compositional variations of the cellular regions associated with the clusters. The mean cluster spectra exhibit vastly improved signal/noise (S/N) ratios over individual spectra. HCA, using Ward's algorithm as a merging method, was found to produce the best clusters as judged by the homogeneity of the spectra in each cluster (9).
RESULTS
Fig. 1 shows results for a fixed epithelial cancer (HeLa) cell grown onto a CaF2 substrate. The cells assume a characteristic shape and size, typically between 40 and 60 μm in the x,y dimension and ∼5-μm thick. Panel A is a bright-field image of a cell in aqueous medium, taken through 20× objective, and Panel B depicts the total integrated Raman intensity of the CH-stretching region. The quality of the bright-field image is low since the aqueous medium, in which the cell is immersed, tends to reduce the visual contrast. However, when the same cell was viewed in the dried state, the nucleoli in the nucleus could be identified unambiguously.
FIGURE 1.

(A) Bright-field microscopic image of a HeLa cell in buffer solution, 20× objective. (B) Integrated Raman intensities in the 2800–3000 cm−1 region of the cell shown in A, collected at a dwell time of 0.5 s/point and a point spacing of 0.5 μm. The spectra were excited by ∼10 mW at 488-nm laser radiation. Bright yellow hues indicate highest, and orange hues low integrated C-H stretching intensities. (C) Nine cluster pseudocolor Raman map, based on the Raman data shown in B. Data were vector normalized and correlated in the 1200–1800 cm−1 region.
The univariate x,y map (Fig. 1 B) was collected ∼2 μm above the interface between the cell and the CaF2 window. Easy to recognize is the nucleus with distinct nucleoli, surrounded by the cytoplasm with varying density. Panel C shows the results of the HCA of the data set shown in Fig. 1 B for nine clusters. Hierarchical clustering was performed in the spectral range between 1200 and 1800 cm−1. This region exhibits the most predominant protein peaks and was found to contain sufficient spectral information to give the best clustering results. Alternatively, HCA can be performed on the C-H stretching region between 2800 cm−1 and 3020 cm−1, which resulted in almost identical images, but failed to contrast the nucleoli against the nucleus, using <10 clusters. Clustering over both regions did not change the images significantly. To avoid clustering by sample density, the spectra were vector normalized over the spectral region employed. The number of clusters, represented by the number of colors in the pseudocolor map, was chosen according to the number of clusters needed to contrast the nucleoli inside the nucleus. The clustering reproduces the position of nucleus and nucleoli, as well as several regions within the cytoplasm, reflecting different composition of the cytoplasm with varying subcellular organelles. Spectral differences will be discussed below.
As pointed out before, the Raman maps were collected immediately before staining with Mitotracker. After addition of the Mitotracker stain (see Materials and Methods), the 488-nm laser was used for fluorescence measurements without disturbing any of the optical settings. Fig. 2, A–C, shows fluorescence images of three individual HeLa cells stained with Mitotracker for mitochondrial distribution. Bright green areas correspond to high mitochondria content. The highest Mitotracker fluorescence intensity was observed in the perinuclear region. Fig. 2, D–F, shows images of the corresponding Raman maps constructed as previously described for five (Fig. 2 D) or six (Fig. 2, E and F) clusters. A comparison of the images in the two columns of Fig. 2 reveals that the regions, from which significant fluorescence due to the Mitotracker stain is observed, correspond very well with the Raman cluster shown in light green. Consequently, we assign the light green cluster in the Raman images to correspond to the regions of the highest abundance of mitochondria.
FIGURE 2.

(A–C) Fluorescence images of three HeLa cells after staining with green fluorescent Mitotracker stain. Excitation wavelength: 488 nm, integrated fluorescence intensity between 510 and 540 nm. Bright green hues indicate highest fluorescence intensities. (D–F) Five cluster Raman maps of the same cells, taken before addition of Mitotracker stain and fluorescence measurement. Notice the congruence of the light green areas in the Raman and the fluorescence maps.
The number of spectra contributing to the mean cluster spectra is on the order of several hundred. Consequently, the observed S/N ratio for the mean cluster spectra is very good, allowing interpretation of these spectra and the spectral differences corresponding to different regions in the cell. For example, a comparison of the mean cluster spectra observed in the nuclei of the cells in Fig. 2 is shown in Fig. 3. Spectra were vector normalized between 1200 and 1800 cm−1 to remove dependence on laser power, sampling volume, and number of spectra averaged into the mean cluster spectra. The three cells show excellent agreement between the spectral traces, and even small details in the spectra are reproduced in all of them. Small spectral differences are observed in the extended amide III region (1270–1350 cm−1), which exhibits slight intensity variations. A vibrational assignment of bands observed in these spectra is summarized in Table 1.
FIGURE 3.

Raman spectra of the nuclei of cells 1, 2, and 3 shown in Fig. 2 (D–F), extracted as the mean cluster spectra of all clusters shown in gray in Fig. 2.
TABLE 1.
Assignment of predominant bands in the Raman spectra of cellular components
| Mode | Frequency (cm−1) | Description |
|---|---|---|
| CH stretching | 2800–3020 | C-H stretching modes (CH3, CH2, CH) |
| Amide I | 1655 | C=O stretching mode, peptide linkage |
| CH2 and CH3 deformations | 1425–1475 | Antisymmetric methyl and methylene deformations, peptide side chains, phospholipids |
| Extended amide III | 1250–1350 | Coupled C-H, N-H deformation modes, peptide backbone |
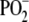 |
1095 | Symmetric stretching mode of phosphate esters, DNA, RNA, phospholipids |
| Phenylalanine | 1002 | Symmetric stretching (ring breathing) mode of phenyl group |
| Pyrimidine bases | 785 | Ring breathing modes, C, T, and U |
The spectra of the mitochondrial regions in comparison with the mean spectra of the nuclei and the remaining cytoplasm are shown in Fig. 4. The three spectra show subtle, but reproducible differences from both the nuclear spectra as well as the spectra from the cytoplasm. All three spectra exhibit distinct protein bands (the amide I vibration at 1655 cm−1, the extended amide III region between 1270 to 1350 cm−1, and the sharp phenylalanine ring-stretching vibration at 1002 cm−1), which is not surprising, given that the major component in all three regions—i.e., nuclei, cytoplasm, and mitochondrial areas—is protein. Spectral differences are also observed in the C-H-stretching region. In particular the shoulder between 2850 and 2900 cm−1 is more pronounced in the spectra of the cytoplasm and the mitochondrial regions compared to the spectrum of the nucleus. Apart from spectral features from proteins, lipid contributions may be observed in the spectra from within the cytoplasm (10). Various organelles, for instance the endoplasmatic reticulum, the Golgi apparatus, lysosomes, mitochondria, or intracellular vesicles, are encased in often highly folded membranes, which mainly consist of phospholipids. The Raman spectra of phospholipids are distinctly different from the spectra of proteins.
FIGURE 4.

Mean cluster spectra of the nuclei (black), mitochondrial areas (medium gray), and cytoplasm (light gray) of the cells shown in Fig. 2. Between 600 and 1800 cm−1, the intensities were expanded by a factor of 2.5 to better demonstrate the spectral differences between the three regions.
The relatively long alkane chains give rise to high C-H-stretching intensities between 2850 and 2935 cm−1. In addition to the carbonyl stretches and the CH2 deformations, the ester linkage C=O stretching is observed around 1735 cm−1. Furthermore, distinct peaks, which can be related to deformations of saturated and unsaturated fatty acid side chains, appear between 1200 and 1350 cm−1. Phospholipids often have pronounced Raman bands below 800 cm−1, which can be assigned to different residues at the phosphate-ester headgroup (11,12). For the observed spectra from within the cytoplasm, contributions that can be assigned to lipids or phospholipids are rather small. As mentioned before, the C-H stretching bands of the spectra corresponding to mitochondria-rich regions and other regions in the cytoplasm exhibit enhanced intensities between 2850 and 2900 cm−1, which arise from the alkane chains of lipids. In addition, we found a reproducible peak at 715 cm−1 in these spectra that is not present in the spectra from the nuclei. It is possible that this band originates from the symmetric stretching vibration of the N+(CH3)3 choline group of phosphocholine, which is vastly abundant in membranes. Previously, there has been a report that associated the spectra of mitochondria in yeast cells with spectral features from phospholipids (12). Although we have frequently observed strong phospholipid spectra from small regions in the Raman maps of other cell types, we were not able to colocalize these with the position of mitochondria and to reproduce these features for the HeLa cell model utilized here.
The only nucleic acid feature that is reproducible with relative certainty is the symmetric uracil or thymine ring breathing at 785 cm−1, which is observed quite strongly for the nuclear and the cytoplasmic regions but very small for the mitochondria. We attribute the nucleic acid features in the cytoplasm to be due to RNA in ribosomes. We have shown previously via high resolution, synchrotron-based Fourier transform infrared microspectroscopy that actively dividing cells show significant nucleic acid contributions in the cytoplasm. These contributions disappear completely after treatment of the cell with RNase and are, therefore, attributed to ribosomal RNA (13).
DISCUSSION
Univariate and multivariate Raman spectral maps of individual cells have been reported previously by several research groups (10,14,15). The main advantages of Raman imaging over other techniques include the high spatial resolution achievable, which is on the same order as visible microscopy, and the compositional sensitivity of vibrational spectroscopic methods. Thus, cellular images can be constructed without the use of molecular labels or dyes. The differences in Raman spectra from different regions of a eukaryotic cell are generally quite small and not necessarily amenable to interpretation by standard methods, such as visual inspection and peak assignments. Therefore, Raman images based on univariate methods, such as displaying band intensity or intensity ratios, reveal some information on the distribution of biochemical constituents but cannot rival multivariate methods in the analysis of Raman hyperspectral data sets. Multivariate analyses use the entire spectrum collected at each data point and produce pseudocolor images of the cell under investigation by a correlation approach of all spectra collected. As pointed out in the Results section, the clusters produced by multivariate methods correlate well with bright-field images (see Fig. 1) and with regions indicating high mitochondrial distribution using fluorescence microscopy. Thus, this report demonstrates the excellent sensitivity of Raman microspectroscopy coupled to multivariate methods of data analysis.
Although the cells reported in this study were fixed, the use of a water immersion objective, in principle, allows live cells to be studied (12). Raman microspectroscopy, therefore, is a novel and potentially powerful method to study unlabeled cells noninvasively in their native environment. The water immersion objective also permits the dissipation of the laser energy without destroying the cell. When using an air objective for the observation of Raman microspectra of cells, we frequently observed destruction of cellular material, even at low laser power.
Although the spectral images reported in this article were collected using the 488-nm line of an Argon ion laser, we observed no fluorescent interference in the Raman spectra. At this point, it is not clear why there was no fluorescence for the HeLa cells reported here, since the generally accepted wisdom is that visible excitation cannot be used for work on cells and tissue. For cultured cells, we mostly observed fluorescence-free spectra for both fixed and unfixed cells, both in aqueous medium and as dried cells. We have observed fluorescence in other cell types, usually originating from very small regions within the cells.
In the absence of fluorescence, the 488-nm Argon ion line offers enormous advantages over red or near infrared laser excitation wavelengths at, for example, 633 or 785 nm. These include a higher scattering cross section (by about a factor of 6), significantly higher detector sensitivity, and better spatial resolution due to the shorter wavelength.
Inspection of the cluster images of the same cell (Figs. 1 C and 2 E) reveals that increasing the number of clusters first causes better differentiation in the cytoplasm before more details can be observed in the nucleus. This can be understood in terms of the compositional variations in the nucleus and the cytoplasm. The spectral differences between the nucleus (composed predominantly of protein and DNA) and the nucleoli (composed mainly of protein, DNA, and ribosomal RNA) are expected to be small; thus, finer discrimination is required for their distinction. In the cytoplasm, on the other hand, there are regions with quite different composition (e.g., membrane-based structures such as the ER or Golgi apparatus, which show larger spectral differences). In some cell types, distinct clusters due to pure phospholipids have been observed and assigned to be due to liposomes (16). With sufficiently high signal quality, it will most likely be possible to monitor the presence of other components, such as glycogen, which can be seen easily in infrared microspectroscopy (17).
We also collected Raman spectral maps of the same cells at a different excitation wavelength. Since the cytochromes in mitochondria have pronounced and well-understood ultraviolet absorption and resonance Raman spectra (18), we anticipated that excitation with 514.5-nm laser radiation would allow the detection of cytochrome resonance Raman features in the mitochondrial cluster and would add another level of evidence for the assignment of the observed spectra. In isolated mitochondria, excitation with 514.5-nm radiation produces a strong resonance Raman spectrum of the cytochrome complexes. However, at the S/N ratio of the present spectra, only minute changes were evident for the mitochondrial cluster spectra excited with 488 and 514.5 nm. These changes may or may not have been due to resonance Raman bands of cytochrome. Excitation at 413 nm (krypton ion laser) or any other source closer to the strong cytochrome absorptions (19) could reveal the cytochrome resonance spectra more clearly and would provide further confirmation of our assignment of the mitochondrial spectra.
CONCLUSIONS
We have demonstrated that Raman microspectroscopy, coupled to multivariate methods of analysis such as HCA, can distinguish the nucleus, nucleolus, cytoplasm, and mitochondria of intact fixed cells by their biochemical composition. This opens the door for noninvasive, in vitro studies of cell biological aspects such as the dynamics of mitochondrial movement, drug uptake, apoptosis, and other effects. The spatial resolution of confocal Raman microspectroscopy is comparable to that achieved in other confocal optical microscopic methods. Yet the biggest advantage of Raman over other microspectroscopic techniques is the fact that no external labels or fluorophores are required, making future applications to living cells highly likely.
Acknowledgments
Partial support of this research from grants CA 090346 from the National Institutes of Health (to M.D.) and program EEC-9986821 from the National Science Foundation—Engineering Research Centers (to C.M.W.) is gratefully acknowledged.
Editor: David W. Piston.
References
- 1.Warner, C. M., J. A. Newmark, M. Comisey, S. R. De Fazio, D. M. O'Malley, M. Rajadhyaksha, D. J. Townsend, S. McKnight, B. Roysam, P. J. Dweyer, and C. A. DiMarzio. 2004. Genetics and imaging to assess oocyte and preimplantation embryo health. Reprod. Fertil. Dev. 16:729–741. [DOI] [PubMed] [Google Scholar]
- 2.Van Blerkom, J. 2004. Mitochondria in human oogenisis and preimplantation embryogenisis: engines of metabolism, ionic regulation and developmental competence. Reproduction. 128:269–280. [DOI] [PubMed] [Google Scholar]
- 3.Collins, T. J., and M. D. Bootman. 2003. Mitochondria are morphologically heterogeneous within cells. J. Exp. Biol. 206:1993–2000. [DOI] [PubMed] [Google Scholar]
- 4.Karbowski, M., and R. J. Youle. 2003. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 10:870–880. [DOI] [PubMed] [Google Scholar]
- 5.Grimm, R., M. Bärmann, W. Häckl, D. Typke, E. Sackmann, and W. Baumeister. 1997. Energy filtered electron tomography of ice-embedded actin and vesicles. Biophys. J. 72:482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lasch, P., W. Haensch, D. Naumann, and M. Diem. 2004. Imaging of colorectal adenocarcinoma using FT-IR microspectroscopy and cluster analysis. Biochim. Biophys. Acta. 1688:176–186. [DOI] [PubMed] [Google Scholar]
- 7.Diem, M., M. Romeo, S. Boydston-White, M. Miljković, and C. Matthäus. 2004. A decade of vibrational micro-spectroscopy of human cells and tissue (1994–2004). Analyst. 129:880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward, J. H. 1963. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58:236–244. [Google Scholar]
- 9.Helm, D., H. Labischinski, G. Schallen, and D. Naumann. 1991. Classification and identification of bacteria by FT-IR spectroscopy. J. Gen. Microbiol. 137:69–79. [DOI] [PubMed] [Google Scholar]
- 10.Krafft, C., T. Knetschke, R. H. W. Funk, and R. Salzer. 2006. Studies on stress-induced changes at the subcellular level by Raman microspectroscopic mapping. Anal. Chem. 78:4424–4429. [DOI] [PubMed] [Google Scholar]
- 11.Krafft, C., L. Neubert, T. Simat, and R. Salzer. 2005. Near infrared Raman spectra of human brain lipids. Spectrochim. Acta A Mol. Biomol. Spectrosc. 61:1529–1535. [DOI] [PubMed] [Google Scholar]
- 12.Huang, Y. S., T. Karashima, M. Yamamoto, and H. Hamaguchi. 2005. Molecular-level investigation of the structure, transformation, and bioactivity of single living fission yeast cells by time- and space-resolved Raman spectroscopy. Biochemistry. 44:10009–10019. [DOI] [PubMed] [Google Scholar]
- 13.Lasch, P., A. Pacifico, and M. Diem. 2002. Spatially resolved IR microspectroscopy of single cells. Biopolymers. 67:335–338. [DOI] [PubMed] [Google Scholar]
- 14.Uzunbajakava, N., A. Lenferic, Y. Kraan, E. Volokhina, G. Vrensen, J. Greve, and C. Otto. 2003. Nonresonant confocal Raman imaging of DNA and protein distribution in apoptotic cells. Biophys. J. 84:3968–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthäus, C., S. Boydston-White, M. Miljković, M. Romeo, and M. Diem. 2006. Raman and infrared microspectral imaging of mitotic cells. Appl. Spectrosc. 60:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Manen, H. J., Y. M. Kraan, D. Roos, and C. Otto. 2005. Single-cell Raman and fluorescence microscopy reveal the association of lipid bodies with phagosomes in leukocytes. Proc. Natl. Acad. Sci. USA. 102:10159–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiriboga, L., P. Xie, H. Yee, V. Vigorita, D. Zarou, D. Zakim, and M. Diem. 1998. Infrared spectroscopy of human tissue I. Biospectroscopy. 4:47–53. [DOI] [PubMed] [Google Scholar]
- 18.Adar, F., and M. Erecińska. 1978. Resonance Raman spectra of whole mitochondria. Biochemistry. 17:5484–5488. [DOI] [PubMed] [Google Scholar]
- 19.Berezhna, S., H. Wohlrab, and P. M. Champion. 2003. Resonance Raman investigations of cytochome c conformational change upon interaction with the membranes of intact and Ca2+-exposed mitochondria. Biochemistry. 42:6149–6158. [DOI] [PubMed] [Google Scholar]