

Abstract
The anticonvulsant effect of felbamate (FBM) is ascribable to inhibition of N-methyl-d-aspartate (NMDA) currents. Using electrophysiological studies in rat hippocampal neurons to examine the kinetics of FBM binding to and unbinding from the NMDA channel, we show that FBM modifies NMDA channel gating via a one-to-one binding stoichiometry and has quantitatively the same enhancement effect on NMDA and glycine binding to the NMDA channel. Moreover, the binding rates of FBM to the closed and the open/desensitized NMDA channels are 187.5 and 4.6 × 104 M−1 s−1, respectively. The unbinding rates of FBM from the closed and the open/desensitized NMDA channels are ∼6.2 × 10−2 and ∼3.1 s−1, respectively. From the binding and unbinding rate constants, apparent dissociation constants of ∼300 and ∼70 μM could be calculated for FBM binding to the closed and the open/desensitized NMDA channels, respectively. The slight (approximately fourfold) difference in FBM binding affinity to the closed and to the open/desensitized NMDA channels thus is composed of much larger differences in the binding and unbinding kinetics (∼250- and ∼60-fold difference, respectively). These findings suggest that the effects of NMDA and glycine binding coalesce or are interrelated before or at the actual activation gate, and FBM binding seems to modulate NMDA channel gating at or after this coalescing point. Moreover, the entrance zone of the FBM binding site very likely undergoes a much larger conformational change along the gating process than that in the binding region(s) of the binding site. In other words, the FBM binding site becomes much more accessible to FBM with NMDA channel activation, although the spatial configurations of the binding ligand(s) for FBM themselves are not altered so much along the gating process.
INTRODUCTION
It has been found that N-methyl-d-aspartate (NMDA) channel antagonists possess a broad-spectrum antiepileptic effect on experimental seizures (1). In clinical trials, however, most NMDA channel antagonists have demonstrated serious neurobehavioral complications that have limited further pharmaceutical development (2). Felbamate (FBM; 2-phenyl-1,3-propanediol dicarbamate) is the only marketed anticonvulsant that shows evident NMDA channel inhibitory effect at therapeutic concentrations (3–6). Although some side effects (e.g., idiosyncratic hematologic and hepatic toxicities) have precluded its use as the first-line antiepileptic therapy, FBM has remained as an important therapeutic option for different types of seizures that are refractory to the other anticonvulsants in both children and adults (7–9).
FBM has been shown to inhibit [3H]5,7-dichlorokynurenic acid (DCKA, a high-affinity competitive glycine site antagonist) binding in the rat brain (10,11). Glycine was also reported to compensate the antiepileptic action of FBM in the NMDA-induced seizures (12), and the inhibitory effect of FBM on Ca2+ influxes induced by NMDA/glycine exposure in cultured cerebellar granular cells (13). These studies suggested that FBM competed with glycine binding to the NMDA channel. However, Subramanian et al. (6) showed that FBM competitively inhibited [3H]MK-801 binding but not [3H]5,7-DCKA binding. Also, exogenous addition of glycine failed to modulate the excitoprotective effect of FBM on cultured cortical neurons exposed to glutamate or NMDA (14), and there were no competitive interactions between FBM and glycine in studies of NMDA currents (5). Furthermore, it has been reported that FBM produced an increase rather than a decrease in [3H]glycine binding to the NMDA channel (15). These reports are seemingly complicated or even conflicting. FBM thus probably has an effect on glycine binding, but the details remain to be defined.
We have demonstrated that FBM has a higher affinity to the open and especially the desensitized NMDA channels (dissociation constant ∼55–110 μM) than to the closed channels (dissociation constant ∼200 μM) (4). Also, FBM slows the recovery from desensitization in the NMDA channel. The selective binding of FBM to the open/desensitized channels well explains the use-dependent inhibition of NMDA currents and consequently the nonsedative anticonvulsant effect of FBM. FBM thus is an effective gating modifier of the NMDA channel at its therapeutic concentrations (50–300 μM). The overall gating conformational changes in the FBM binding site, however, do not seem to be dramatic, considering the small (approximately two-to-fourfold) difference in FBM affinity to the closed and to the open/desensitized NMDA channels (4). To characterize the molecular action of FBM in more detail, we explored the kinetics of FBM binding to and unbinding from the NMDA channel, as well as the effect of FBM on the affinity of NMDA or glycine to the channel. We find that FBM has quantitatively the same effect on both the NMDA and glycine binding to the NMDA channel, indicating that the effects of NMDA and glycine binding coalesce or are interrelated before or at the actual activation gate, and FBM binding seems to modulate NMDA channel gating at or after this coalescing point with a one-to-one binding stoichiometry (one FBM per NMDA channel). Most interestingly, FBM has much faster binding and unbinding rates (∼250-fold and ∼60-fold, respectively) to the open/desensitized than to the closed NMDA channels. The latter findings strongly suggest that the FBM binding site in the NMDA channel becomes much more accessible to FBM after channel activation, signaling a much larger gating conformational change at the entrance zone than at the binding region(s) of the FBM binding site.
MATERIALS AND METHODS
Dissociated neuron preparation
Neonatal (7- to 14-day-old) Wistar rats were used for the preparation of coronal slices of the whole brain. The CA1 region of hippocampus dissected from the slices was cut into small chunks. Tissue chunks were treated with protease XXIII (1 mg/ml; Sigma, St. Louis, MO) in dissociation medium (82 mM Na2SO4, 30 mM K2SO4, 3 mM MgCl2, and 5 mM HEPES, pH 7.4) for 3–5 min at 35°C, and then were moved into dissociation medium containing no protease but bovine serum albumin (1 mg/ml; Sigma). Each time two to three chunks were picked and triturated to release single neurons for whole-cell recordings.
Electrophysiological recordings
Acute dissociated neurons were placed in a recording chamber filled with Tyrode's solution (150 mM NaCl, 4 mM KCl, 2 mM MgCl2, 2 mM CaCl2, and 10 mM HEPES, pH 7.4). The fire-polished pipettes pulled from borosilicate capillaries (1.55–1.60 mm outer diameter; Hilgenberg, Malsfeld, Germany) were used for whole-cell recordings. When filled with the internal solution (75 mM CsCl, 75 mM CsF, 10 mM HEPES, and 5 mM EGTA, pH 7.4), the pipettes had resistances of 1–2 MΩ. After the whole-cell configuration was obtained in Tyrode's solution, the cell was lifted and moved in front of a set of square-glass three-barrel tubes (0.6 mm internal diameter) or theta-glass tubes (2.0 mm outer diameter pulled to an opening of ∼300 μm in width; Warner Instruments, Hamden, CT) emitting different external recording solutions. The standard external solution was Mg2+-free Tyrode's solution (pH 7.4) containing 0.5 μM tetrodotoxin. The holder of glass tubes was connected to a stepper control (SF-77B perfusion system, Warner Instruments) to carry out fast switches between different glass tubes and thus rapid solution exchange. The rate of solution exchange was quantified by the method described previously (4). In short, the rate of change in current amplitude between two different external solutions containing different cations, namely Tyrode's solution and a solution of the same constituents, except that the Na+ ion was replaced with the impermeable N-methyl-d-glucamine ion. The 50% current change time is ∼6 ms and ∼40 ms for the theta-glass and the square-glass, respectively (4). Theta-glass tubes were thus used in the experiments studying the kinetics of FBM binding to and unbinding from the channel in Figs. 5 and 8, which require a better resolution in the time domain. In the other experiments, square-glass tubes were used to facilitate the switch between a larger number of different external solutions. NMDA and glycine (Sigma) were dissolved in water and FBM (Tocris Cookson, Bristol, UK) was dissolved in dimethyl sulfoxide to make 100, 10, and 100 mM stock solutions, respectively. The stock solutions were diluted into Mg2+-free Tyrode's solution to make 0.1 μM to 1 mM NMDA, 10 nM to 30 μM glycine, and 10 μM to 1 mM FBM right before the experiment. Three-hundred micromolar was the most commonly used concentration of FBM for the characterization of the gating-modification effects because it is within the therapeutic concentration range (50–300 μM) (16,17), and because submillimolar FBM chiefly behaves as a gating modifier (4). The final concentration of dimethyl sulfoxide (≦∼1%) was found to have no detectable effect on NMDA currents. The NMDA currents were recorded at a membrane potential of −70 mV and at room temperature (∼25°C) with an Axoclamp 200A amplifier, filtered at 1 kHz with a four-pole Bessel filter, digitized at 200–500 μs intervals, and stored using a Digidata-1322A analog/digital interface as well as the pCLAMP software (all from Axon Instruments, Union City, CA). All data are expressed as mean ± SE. For comparisons between experimental groups, the Student's t-test was used and p < 0.05 was considered as statistically significant.
FIGURE 5.

Kinetics of FBM binding to and unbinding from the activated NMDA channel. (A) Inhibition (left panel) and relaxation (recovery from inhibition, right panel) of the steady-state inward NMDA (1 mM) currents by fast application of FBM with theta-glass tubes. (Left) The decay phase of the current after application of FBM is well fitted with single-exponential functions to give binding time constants of 105 and 23 ms for 100 and 1000 μM FBM, respectively. The dashed line indicates zero current level. (Right) The increment phase of the current after wash-off of FBM can be well fitted with single-exponential functions and gives unbinding time constants of 340 and 325 ms for 100 and 1000 μM FBM, respectively. To avoid the effect from incomplete solution change, the start of the fitting curve was carefully selected after a 10-ms delay of the decay or increment phase of the current trace. The dashed line indicates zero current level. (B) Cumulative results are obtained from six cells with the experiments described in panel A. (Left) The binding time constants are 115 ± 12, 58 ± 3, and 20 ± 1 ms in the presence of 100, 300, and 1000 μM FBM, respectively. (Right) The unbinding time constants are 330 ± 25, 319 ± 17, and 321 ± 23 ms after the wash-off of 100, 300, and 1000 μM FBM, respectively. (C) The inverses of the binding and unbinding time constants in panel B are plotted against the FBM concentration. The lines are linear regression fits to the two sets of mean values. For the binding rates, the slope and intercept are 4.6 × 104 M−1 s−1 and 3.5 s−1, respectively. For the unbinding rates, the intercept is 3.1 s−1 and the slope is zero (i.e., an essentially horizontal line indicating that the unbinding rates are unrelated to the FBM concentration).
FIGURE 8.

Measurement of the unbinding rate of FBM from the resting NMDA channel with a different experimental approach. (A) NMDA currents were elicited by 1 mM NMDA. The neuron was then rapidly shifted to an external solution containing no NMDA to study the decay kinetics of the currents with theta-glass tubes. The experiment was repeated both in the absence (control) and presence of 300 μM FBM. The decay phase of the current is fitted with a single-exponential function, which gives time constants of 40 and 52 ms in the absence and presence of 300 μM FBM, respectively. (B) Cumulative results are obtained from four cells with the experimental protocol described in panel A. The inverses of time constants of NMDA current decay are 26.3 ± 1.3, and 20.7 ± 1.2 s−1 in the absence (control) and presence of 300 μM FBM, respectively. *p < 0.05, compared with the control data. (C) The unbinding rate of FBM from the resting NMDA channel is obtained with an experimental approach different from that in Fig. 7. An 8-s NMDA (1 mM) pulse was given to elicit the NMDA current. For the next 3 s, 300 μM FBM (along with the 1 mM NMDA) was applied to the cell. The cell was then washed with NMDA- and FBM-free solution for different periods of time (the wash time) before a final pulse with 1 mM NMDA was given for 6 s. The numbers above the raw sweeps represent the wash time (4–32 s). It is evident that the peak current in the second NMDA pulse gets larger (and reaches a similar size to the control peak current in the first NMDA pulse) as the wash time gets longer. (D) Cumulative results are obtained from six cells with the experimental protocol described in panel C. The peak NMDA currents in the final NMDA pulses are normalized to those in the first control NMDA pulses, and are plotted against the duration of the wash time. The line is a single-exponential fit of the form: relative current = 1–0.37 × exp(−t/17.4) (t denotes length of wash time in seconds, the horizontal axis).
RESULTS
FBM binding increases the affinity of NMDA to the NMDA channel
We have shown that FBM has a higher affinity to the open/desensitized state than to the closed state of NMDA channels (4). Because NMDA channel activation and desensitization are conformational changes triggered by NMDA binding, it is very likely that FBM binding also increases the affinity of NMDA to the NMDA channel. To quantify NMDA affinity to the NMDA channel, we chose the steady-state data (late-sustained currents) to estimate the dissociation constant of NMDA binding to the resting channel. The NMDA concentration-response curves are constructed by application of different concentrations of NMDA with 0.3 μM glycine in the absence and presence of 300 μM FBM (Fig. 1 A). Fig. 1 B shows two simple gating schemes of the NMDA channel. Two molecules of NMDA are presumed to bind to two binding sites on the NMDA receptor before the channel could open. As a first approximation, these two sites are assumed to have the equal binding affinity to NMDA, and the binding of one NMDA molecule will not affect the binding of the other. The relative steady-state occupancy of states C, CN, CN2, ON2, and DN2 thus are 1, 2[N]/Kn, ([N]/Kn)2, p × ([N]/Kn)2, and m × p × ([N]/Kn)2, respectively, in which [N] is the concentration of NMDA, Kn is the dissociation constant of NMDA binding to either of the two sites in the closed state, p is the relative probability that the channel would be open (versus remaining closed) when both sites are bound with NMDA molecules, and m is the ratio between the steady-state distribution of the desensitized and the open states.
FIGURE 1.

FBM modification of the NMDA concentration-response curves. (A) NMDA currents were elicited by application of 0.3 μM glycine and different concentrations of NMDA in the absence (control) and presence of 300 μM FBM. The relative current is defined as the ratio between the sustained current amplitude in different concentrations of NMDA and that in 1 mM NMDA (approximating the Imax in Eq. 1), and is plotted against the NMDA concentration (n = 6–9). The same experiments were repeated in the absence (control) and presence of 300 μM FBM with each set of data separately normalized to their respective Imax, which is smaller in FBM than in control (data not shown; see also (4)). Note that FBM shifts the NMDA concentration-response curve to the left, indicating an enhancement effect on the NMDA affinity. Quantitatively, the solid lines are best fits to the data points using Eq. 1 with p- and m-values fixed at 2.6 and 4, respectively (see also (4)). The Kn values given by the best fits are 54 and 27 μM in the control condition and 300 μM FBM, respectively. One may also fix Kn at 54 μM and fit for the p-value in 300 μM FBM. In this case, the best fit gives p = 8.1 and a fitting curve (dotted line) very similar to that with p = 2.6 and Kn = 27 μM. (B) Two simple gating schemes describe that two molecules of NMDA bind to the channel and thus result in opening and desensitization. The closed, open, and desensitized states of the channel are denoted by C, O, and D, respectively. The NMDA molecule is represented by N. The closed channels with one and two bound molecules of NMDA are indicated by CN and CN2, respectively. Although the NMDA channel may not necessarily open before being desensitized ((34); but see (35)), we chose the basic C-O-D linear scheme (in the upper panel) for the sake of simplicity, and also for the possible coupling between channel opening and desensitization. Because the data analysis here is based on steady-state considerations, the major conclusions should remain similar if one chooses more complicated schemes like the one in the lower panel.
The amplitude of the steady-state NMDA current (I) is proportional to the ratio between the number of open and total channels, or
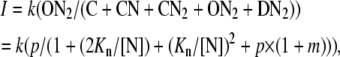 |
where k is a constant for conversion and is in the unit of current (μA).
If [N] is very large or [N] ≫ Kn, then I approaches its maximum (Imax) and the equation above is simplified to
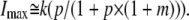 |
The relative steady-state current therefore could be expressed as
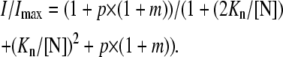 |
(1) |
The best fit with Eq. 1 to the data points in the control condition gives Kn = 54 μM (with p and m values fixed at 2.6 and 4, respectively; see also (4)). With the same fixed parameters, the data in 300 μM FBM can be best fitted with Kn = 27 μM, indicating a twofold increase in the affinity of NMDA to the NMDA channel in the presence of 300 μM FBM. Alternatively, one may fix Kn at 54 μM, and fit for p. The best fit to the data in 300 μM FBM then gives p = 8.1, indicating that NMDA channels bound with two NMDA molecules have roughly a threefold higher chance of opening in 300 μM FBM. These results provide a quantitative description of the modification effect of FBM on the gating process of the NMDA channel, and are very much consistent with the small but significant (two-to-fourfold) differences in the binding affinity of FBM toward different gating states of the NMDA channel (4).
FBM cannot elicit NMDA currents by itself but enhances the currents elicited by very low concentrations of NMDA
If FBM binding effectively alters the conformation of the NMDA channel, moving the resting (closed) channel toward the open state and thus increasing the affinity of NMDA, it would be interesting to see whether FBM itself can elicit NMDA currents in the complete absence of NMDA. After repeated trials we cannot demonstrate discernible currents with FBM and glycine in the absence of NMDA (Fig. 2 A), although FBM indeed enhances rather than inhibits the current elicited by a very low concentration (4 μM) of NMDA (Fig. 2 B). These findings imply that FBM is a partial but not a full allosteric agonist of the NMDA binding site, because FBM binding would increase the efficacy of NMDA binding to the NMDA (or glutamate) site, but FBM binding by itself cannot replace NMDA and move the NMDA channel to the open conformation.
FIGURE 2.

FBM enhancement of the NMDA currents elicited by very low concentrations of NMDA. (A) No discernible currents were elicited by application of 300 μM FBM and 0.3 μM glycine in the absence of NMDA. (B) 300 μM FBM enhances rather than inhibits the current elicited by a very low concentration (4 μM) of NMDA.
FBM binding also allosterically interacts with the glycine site
We have argued that FBM is a partial allosteric agonist of the NMDA channel and increases the affinity of NMDA to the NMDA channel. Glycine is essential for NMDA channel activation and is described as a coagonist of glutamate (18,19). Since an allosteric interaction between the NMDA and glycine binding sites of the NMDA channel has been well established (20,21), we also investigated the interaction between FBM and glycine binding on the NMDA channel (Fig. 3). NMDA currents were elicited by application of saturating concentrations of NMDA (1 mM) and different concentrations of glycine in the absence and presence of 300 μM FBM. It is interesting that FBM significantly inhibits the current in the presence of 30 μM glycine, but has only negligible inhibitory effect on the current with 0.1 μM glycine (Fig. 3 A). Fig. 3, B and C, shows that FBM decreases the maximum response elicited by 1 mM NMDA with a decrease in the EC50 of glycine, indicating an enhancement effect on glycine binding by FBM. To have a more quantitative analysis of the glycine affinity to the NMDA channel, we took an approach similar to that in Fig. 1. With saturating concentrations of NMDA, the relative steady-state current in different concentrations of glycine could be given by
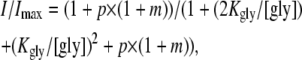 |
(2) |
where [gly] is the glycine concentration, Kgly is the dissociation constant of glycine binding to either of the two glycine binding sites in the closed channel, p is the relative chance that the channel is open (versus staying closed) when both sites are occupied by glycine, and m is the ratio between the steady-state occupancy of the desensitized state and the open state.
FIGURE 3.

FBM enhancement of glycine affinity to the NMDA channel. (A) NMDA currents were elicited by application of saturating concentrations of NMDA (1 mM) and different concentrations of glycine in the absence (control) and presence of 300 μM FBM. FBM clearly inhibits the current in the presence of 30 μM glycine but has only negligible inhibitory effect on the current with 0.1 μM glycine. (B) The relative current is defined as the ratio between the sustained current amplitude in different concentrations of glycine (and 0 or 300 μM FBM) and that in 30 μM glycine (and 0 FBM), and is plotted against the glycine concentration (n = 5–8). (C) The same data as in panel B, but the currents in 300 μM FBM are now normalized to the current in 30 μM glycine (and 300 μM FBM). The two very similar solid lines are the best fits to the mean data in the control condition using Eq. 2 with a p-value of 1 and an m-value of 5 or 3, respectively. The best fits give relatively constant Kgly values of 308 and 243 nM, respectively. With the same fixed parameters, the best fits (dotted lines) to the data in 300 μM FBM give Kgly = 165 (m = 5), and 129 nM (m = 3), respectively. We also fixed Kgly at 308 or 243 nM, and fit for the p-value in 300 μM FBM (dashed lines, p = 2.6). Note the close proximity between the dotted and dashed lines.
It has been suggested that glycine could decrease desensitization of the NMDA channel (22,23). Here the m-value also becomes smaller with increasing concentrations of glycine in our study. For example, the peak current in Fig. 3 A is approximately six times and approximately four times the size of the sustained current in 0.1 and 30 μM glycine, respectively. We therefore set the p-value at 1 and the m-value at 5 (0.1 μM glycine) or 3 (30 μM glycine) as a first approximation in the analysis of the data points in the control condition in Fig. 3 C. One may obtain very similar fits with Kgly values of 308 and 243 nM with m-values set at 5 and 3, respectively. It is interesting that the Kgly values remain relatively constant (308 and 243 nM) with different m-values, and are consistent with the previously reported EC50 (e.g., 185 nM in (23); 290 nM in (22); and 430 nM in (3)) of glycine to the NMDA channel despite the different experimental approaches in different studies. On the other hand, with the same fixed parameters, the data in 300 μM FBM can be best fitted with Kgly = 165 (m set at 5) and 129 nM (m set at 3), indicating a twofold increase in the affinity of glycine to the NMDA channel in the presence of 300 μM FBM. Alternatively, one may also fix Kgly at 308 (m set at 5) or 243 nM (m set at 3), and fit for p. The best fit to the data in 300 μM FBM then gives p = 2.6, suggesting that glycine-bound channels have a 2.6-fold higher chance of opening in 300 μM FBM than in the control condition. It is intriguing that 300 μM FBM has quantitatively the same effects on the NMDA and glycine binding to the NMDA channel (i.e., approximately twofold increase in the NMDA or glycine binding affinity, or alternatively, an approximately threefold increase in the opening probability of the NMDA- and glycine-bound channel). This quantitative similarity suggests that the effects of NMDA and glycine binding coalesce or are interrelated at or before the actual activation gate, and FBM binding seems to modulate channel gating at or after this coalescing point. In other words, the FBM binding site is probably close to or right at the actual activation gate, which should be controlled by an integrative effect, rather than a simple summation of each individual effect, of NMDA and glycine binding to the different subunits of the channel. FBM may thus affect the NMDA or glycine binding in each different subunit to a similar extent via a similar allosteric mechanism (see below and Discussion).
FBM potentiates the currents elicited by very low concentrations of glycine
We have seen that FBM enhances rather than inhibits the NMDA currents elicited by very low concentrations of NMDA in Fig. 2. We therefore also investigated the effect of FBM on the NMDA currents elicited in the presence of a very low concentration (0.01 μM) or in the complete absence of glycine (Fig. 4). One-millimolar NMDA could elicit very small currents in the absence of glycine. We found that FBM does not definitely enhance this very small current elicited by 1 mM NMDA (Fig. 4 A). However, the small currents elicited by application of 0.01 μM glycine with 1 mM NMDA are evidently potentiated by 10–300 μM FBM (Fig. 4, B and C). These findings are very similar to those in Fig. 2, and are again consistent with the idea of similar interactions between FBM and NMDA and between FBM and glycine in the NMDA channel.
FIGURE 4.

FBM enhancement of the NMDA currents elicited by very low concentrations of glycine. (A and B) NMDA currents were elicited by the same experimental protocol as that in Fig. 3, but the glycine concentration is decreased to 0 or 0.01 μM. FBM does not enhance the very small current in the absence of glycine (1 mM NMDA without glycine, A). However, the peak NMDA current elicited by 0.01 μM glycine is enhanced by FBM in a dose-dependent fashion (B). (C) Cumulative results are obtained from five cells with the experimental protocol described in panel B. The relative current is defined by the ratio between the amplitude of the peak currents in 10–300 μM FBM and that in the absence of FBM (control) in the same cell. The relative currents are 1.07 ± 0.02, 1.14 ± 0.02, and 1.30 ± 0.04 in the presence of 10, 100, and 300 μM FBM, respectively. *p < 0.05, compared with the control data in 0 FBM.
FBM binds to the activated NMDA channel with a simple bimolecular reaction
We have argued that FBM binding allosterically interacts with both NMDA and glycine binding in the NMDA channel, and thus FBM effectively modifies the gating process of the NMDA channel. The overall gating conformational changes in the FBM binding site, however, do not seem to be dramatic. The affinity change of NMDA or glycine binding to the NMDA channel is only approximately twofold by FBM (Figs. 1 A and 3 C), and there is only an approximately two-to-fourfold difference between FBM affinity to the closed and to the open/desensitized NMDA channels (4). To characterize the molecular basis of the seemingly small gating conformational changes in the FBM binding site based on the steady-state data, we explored the kinetics of FBM binding to and unbinding from the NMDA channel. Fig. 5, A and B, demonstrates the kinetics of inhibition and relaxation of 1 mM NMDA-evoked steady-state currents by 100, 300, or 1000 μM FBM. It is evident that the kinetics of inhibition are FBM concentration-dependent, whereas the kinetics of relaxation are not. Fig. 5 C shows a linear correlation between the binding rates (inverses of the binding time constants) and FBM concentrations, indicating that FBM interacts with the NMDA channel by a one-to-one binding process (simple bimolecular reaction) and a macroscopic binding rate constant of 4.6 × 104 M−1 s−1. Moreover, the y-intercept of the linear regression fit to the macroscopic binding rate data in Fig. 5 C is ∼3.5 s−1, which is well consistent with the inverses of current relaxation (unbinding rate) time constants (∼3.1 s−1), a value independent of FBM concentrations from Fig. 5 B. From the binding and unbinding rate constants, an apparent dissociation constant of ∼70 μM could be calculated for FBM binding to the activated NMDA channel (including both open and desensitized states of the channel, as it is difficult to completely separate the two states with our experiment approaches here). However, if the ratio between the steady-state occupancy of the desensitized state and the open state is ∼4 (i.e., m = 4 in Fig. 1), one may roughly estimate the dissociation constant of FBM binding to either the open (Kf,o) or desensitized (Kf,d) NMDA channel with simple rules of weighted average of the affinity (inverse of the dissociation constant). We may then have Kf,o and Kf,d values of ∼120 and ∼60 μM, respectively. In any case, a dissociation constant value of ∼70 μM to the mixed open and desensitized states of NMDA channels or a Kf,o of ∼120 μM and a Kf,d of ∼60 μM are very much consistent with the previously reported dissociation constant of FBM to the open and desensitized NMDA channels with completely different approaches (∼110 and ∼55 μM, respectively) (4).
The rate of FBM binding to the resting NMDA channel is much slower than that to the activated channel
In Fig. 2 B, we have seen that 300 μM FBM significantly enhances the current evoked by a very low concentration (4 μM) of NMDA. Here we chose 4 μM NMDA-evoked currents and monitored the enhancement effect of FBM to study the rates of FBM binding to and unbinding from the resting state of the NMDA channel. An NMDA pulse length (NMDA exposure time) of 0.3 s was carefully chosen for two reasons: 1), 0.3 s is long enough to record clearly discernible currents; and 2), most NMDA channels should still stay in the resting state and thus the contaminating interaction between FBM and activated NMDA channels is minimized. It is evident that the NMDA current gets larger and then saturates as the preincubation with FBM gets longer (Fig. 6 A). The normalized data points in Fig. 6 B can be reasonably fitted with single-exponential functions, and give binding rate time constants of 14.5, 8.6, and 4.2 s for 100, 300, and 1000 μM FBM, respectively. In addition, the NMDA current is enhanced to 1.5-, 2.3-, and 2.1-fold the size of the control current by 100, 300, and 1000 μM FBM, respectively. The enhancement effect in 1000 μM FBM is slightly smaller than that in 300 μM FBM probably because 1000 μM FBM may start to have mild additional pore blocking effect on the NMDA channel (4). Fig. 6 C shows that the macroscopic binding rates increase linearly with the FBM concentration, again indicating a simple bimolecular reaction between FBM and the NMDA channel. However, the linear regression fit to the data gives a binding rate constant of only 187.5 M−1 s−1, a value much smaller than the binding rate constant to the activated NMDA channel (4.6 × 104 M−1 s−1, Fig. 5 C).
FIGURE 6.

Binding rate of FBM to the resting NMDA channel. (A) The neuron was repeatedly exposed to a 4 μM NMDA pulse 0.3 s in length at a 60 s interval until stable responses were achieved. FBM (300 μM) was then applied to the cell for different periods of time (the preincubation time) before the next NMDA exposure. The numbers above the raw sweeps represent the FBM preincubation time. It is evident that the NMDA current gets larger (and then saturates) as the preincubation with FBM gets longer. (B) The mean peak NMDA currents with different FBM preincubation time are normalized to the peak control currents (obtained without FBM preincubation), and plotted against the FBM preincubation time (n = 5–8). The lines are single-exponential fits of the form: relative current = 1 + 0.5 × (1 − exp(−t/14.5)) (t denotes length of the preincubation time in seconds, the horizontal axis), 1 + 1.3 × (1 − exp(−t/8.6)), and 1 + 1.1 × (1 − exp(−t/4.2)) for 100, 300, and 1000 μM FBM, respectively. (C) The inverses of the time constants in panel B are plotted against the FBM concentration. The line is a linear regression fit to the data points with a slope and an intercept of 187.5 M−1 s−1 and 5.4 × 10−2 s−1, respectively.
The rate of FBM unbinding from the resting NMDA channel is also much slower than that from the activated channel
It is surprising that the binding rate of FBM to the resting NMDA channel is ∼250-fold slower than that to the activated NMDA channel. This is in sharp contrast to the approximately two-to-fourfold difference in the overall binding affinity of FBM to the resting and to the activated NMDA channels. These data altogether strongly imply that there should also be a large difference in the unbinding kinetics of FBM from the resting and the activated NMDA channels. The possibility of a very slow unbinding rate from the resting channel is actually also suggested by the y-intercept of Fig. 6 C (5.4 × 10−2 s−1). We therefore examine the rate of FBM unbinding from the resting NMDA channel in more detail with a two-pulse protocol (Fig. 7). After a fixed 40-s preincubation with 300 μM FBM, the first NMDA pulse (4 μM, 0.3 s) was given to elicit NMDA currents as a reference. The second NMDA pulse identical to the first one was given after different intervals of wash-off with FBM- and NMDA-free Tyrode's solution for the removal of FBM from the resting NMDA channel. As the wash-off interval lengthened, the NMDA current in the second pulse gradually decreased. The peak NMDA current in the second NMDA pulse relative to the reference current is plotted against duration of the wash-off interval to constitute the time course of FBM unbinding form the resting channel. The course can be reasonably fitted with a single-exponential function, giving a time constant of 16.2 s. The unbinding rate constant of FBM from the resting channel is thus 1/16.2 or 6.2 × 10−2 s−1. This value is very close to the y-intercept of Fig. 6 C (5.4 × 10−2 s−1), and is indeed ∼60-fold slower than the unbinding rate constant of FBM from the activated NMDA channel (3.1–3.5 s−1, Fig. 5). An apparent dissociation constant of ∼300 μM could be calculated for FBM binding to the resting NMDA channel (i.e., Kf,c = ∼300 μM) from the binding and unbinding rate constants. This Kf,c value is also consistent with what was previously reported based on a completely different approach (∼200 μM) (4).
FIGURE 7.

Unbinding rate of FBM from the resting NMDA channel. The unbinding rate of FBM from the resting NMDA channel was assessed by a two-pulse protocol with a fixed 40-s preincubation of 300 μM FBM. The current was elicited by the first NMDA pulse (4 μM, 0.3 s), and then a similar NMDA pulse was given after different intervals of FBM wash-off. The peak NMDA current in the second pulse is normalized to that in the first NMDA pulse and is plotted against the duration of the wash-off interval (n = 7). The line is a single-exponential fit of the form: relative current = 1–0.65 × (1 − exp(−t/16.2)) (t denotes length of wash-off time in seconds, the horizontal axis).
The unbinding rate of FBM from the resting NMDA channel is confirmed with a different experimental approach
Because FBM binds more strongly to the open than to the closed NMDA channels, channel deactivation could also be slowed by FBM. We examined the deactivation of the NMDA channel in control and in 300 μM FBM with rapid solution change by the use of theta-glass (Fig. 8). The decay phase of the current upon switching the NMDA-containing external solution back to the NMDA-free solution is fitted with single-exponential functions, giving deactivation rates (the inverses of the current decay time constants) of 26.3 ± 1.3 and 20.7 ± 1.2 s−1 (p < 0.05, n = 4) in the absence and presence of FBM, respectively (Fig. 8 B). Although the slowing of NMDA channel deactivation in the presence of 300 μM FBM is not dramatic, it is definite and consistent with the slightly stronger binding of FBM to the open than to the closed NMDA channels (e.g., Kf,o/Kf,c = 120 μM/300 μM). It seems that the dissociation of NMDA (NMDA channel deactivation) is significantly faster than the dissociation of FBM from the activated NMDA channel (rates = ∼20 vs. ∼3 s−1, Figs. 5 C and 8 B). Based on this attribute, we tried to verify the very slow unbinding rate of FBM from the resting NMDA channel with one more different experimental approach (Fig. 8, C and D). FBM was applied to the steady-state activated NMDA channel. NMDA was then washed off for 4 s, when most FBM-bound activated channels would be converted to either FBM-free channels or FBM-bound resting channels. The course of current change subsequent to 4 s therefore would mostly reflect the dissociation of FBM from the FBM-bound resting channel. In other words, as the wash time is lengthened from 4 to 32 s, more and more FBM would unbind from the (resting) channel. The second NMDA current thus gradually increased and finally reached a similar amplitude to the control current in the first pulse (Fig. 8 C). The course of current increase after 4 s can be reasonably fitted with a single-exponential function, which gives a time constant of 17.4 s (Fig. 8 D), or an unbinding rate of 5.8 × 10−2 s−1 for FBM from the resting (closed) NMDA channel. This value is again consistent with the unbinding rates of FBM from the resting NMDA channel in Figs. 6 and 7 (5.4–6.2 × 10−2 s−1).
DISCUSSION
FBM modifies NMDA channel gating via a one-to-one binding stoichiometry
We have seen that the rates of FBM binding to either the resting (closed) or activated (open/desensitized) NMDA channels are linearly correlated with FBM concentrations (Figs. 5 C and 6 C), strongly suggesting that FBM binds to the NMDA channel via a one-to-one binding process (i.e., a simple bimolecular reaction). It has been shown that the formation of a functional NMDA channel in mammalian cells requires four subunits, or a heterotetrameric combination of two NR1 and two NR2 subunits (24–26), but see (27)), with the NMDA (or glutamate) and glycine binding sites located in the NR2 and NR1 subunits, respectively (24,28–30). The probability of NMDA channel activation would be greatly increased only when all of the four sites, each in one of the four subunits, are occupied by NMDA and glycine (two for NMDA and two for glycine). In this regard, it is intriguing that FBM effectively modulates the affinity of both NMDA and glycine (and thus the opening probability) of the NMDA channel with a simple bimolecular reaction (i.e., a stoichiometry of one-to-one binding to the channel). The subtle but significant difference in the basic features of FBM and NMDA (or glycine) binding to the NMDA channel may suggest that structurally the FBM binding site in the NMDA channel is formed by more than one subunit. This would also be very much consistent with the idea that the major effect of FBM on the gating process is probably at a point where the allosteric effects of NMDA and glycine binding on NMDA channel gating have coalesced (see below).
FBM has quantitatively very similar enhancement effect on NMDA and glycine binding to the NMDA channel
We have seen that FBM binding modulates both the NMDA and glycine binding to the NMDA channel. Detailed quantitative analysis shows that the increase of affinity is slight but definite (approximately twofold). Most interestingly, the increase of affinity is quantitatively very similar for both NMDA and glycine (based on the apparent dissociation constant or on the opening probability when two molecules of NMDA or glycine have bound to the channel; Figs. 1 A and 3 C). The approximately twofold increase in NMDA or glycine affinity in our study is roughly consistent with the previously reported effect of 1 mM FBM on the NMDA channel despite the different experimental approaches in different studies (e.g., 3.5-fold increase in NMDA affinity, (3); 211% enhancement in [3H]glycine binding, (15)). It is also consistent with the finding that FBM has slightly stronger binding to the open than to the closed NMDA channels (in view of the Kf,o/Kf,c = 120 μM/300 μM, i.e., only 2.5-fold). Considering that NMDA and glycine have separate binding sites in different subunits of the NMDA channel and the one-to-one binding stoichiometry between FBM and the channel, FBM most likely allosterically interacts with both the NMDA and glycine sites on the NMDA channel. The very similar quantitative effect on NMDA and glycine binding may further imply that the major molecular action of FBM on the gating process is downstream of NMDA and glycine binding (i.e., at a point where the effects of NMDA and glycine binding on NMDA channel gating have coalesced). In other words, these findings suggest that NMDA and glycine binding should not each control a completely separate part of the activation gate of the channel (and it is unlikely that the channel is open simply when all of its four separate parts are open). Instead, the conformational changes induced by NMDA and glycine binding in the four subunits seem to be interrelated to lead into a significant conformational change of the activation gate and channel opening. The major action of FBM seems to be at or after this point of intersubunit interaction, and thus it has quantitatively very similar effects on both NMDA and glycine binding to the channel. The FBM binding site thus should be close to or right at the actual activation gate of the NMDA channel. In this regard, it is interesting to note that in contrast to the case at pH 7.4, the pore-blocking effect of FBM on the NMDA channel becomes manifest at pH 8.4 (31). FBM thus is an opportunistic pore blocker of the NMDA channel, very likely with its binding site located in the pore, where the activation gate should also be located to control ion permeation.
FBM is both an agonist and an antagonist of glycine in the NMDA channel
We find that FBM increases the affinity of glycine binding to the NMDA channel (Fig. 3) and dose-dependently enhances the currents when the glycine concentration is extremely low (e.g., 0.01 μM, Fig. 4). These findings, together with the idea that FBM binding to NMDA channels is downstream from the glycine and NMDA binding sites, imply that FBM is a partial allosteric agonist of glycine. On the other hand, FBM effectively modifies NMDA channel gating by enhancement of the activation and especially the desensitization processes, but glycine may decrease NMDA channel desensitization (Fig. 3 A; see also (22,23)). Desensitization of the NMDA channel thus may potentiate FBM binding but attenuate glycine binding, and FBM could have an allosteric antagonistic effect on glycine binding by enhancement of channel desensitization. In other words, FBM is very likely an agonist of glycine for channel activation but an antagonist of glycine for channel desensitization, demonstrating delicate interactions between FBM, glycine, and different gating conformations of the NMDA channel.
There are much larger gating conformational changes at the entrance zone than in the binding region(s) of the FBM binding site
Our previous study has demonstrated that FBM has an approximately two-to-fourfold higher affinity to the activated (open/desensitized) than to the resting (closed) states of NMDA channels (4). In this study we further examined the kinetics of FBM binding to and unbinding from the NMDA channel in detail. The binding rates of FBM to the resting and to the activated NMDA channels are 187.5 and 4.6 × 104 M−1 s−1, respectively (Figs. 5 and 6). The unbinding rates of FBM from the resting and from the activated NMDA channels are 5.4–6.2 × 10−2 and 3.1–3.5 s−1, respectively (Figs. 5 and 6). Harty and Rogawski (32) also measured the binding and unbinding rate constants of FBM in the cloned NMDA channels composed of NR1-2B subunits, but they had focused on the NMDA-bound channels (i.e., not resting channels) and reported binding and unbinding rate constants of 1.6 × 104 M−1 s−1 and 5.0 s−1, respectively. These data are roughly consistent with our results obtained from neonatal (7–14-day-old) rat hippocampal neurons, in which the main population of NMDA channels probably also contains NR1-2B subunit (33). On the other hand, to the best of our knowledge, there has been no precedent on the kinetics of FBM binding to and unbinding from the resting NMDA channel. Based on the binding and unbinding rate constants, apparent dissociation constants of ∼300 and ∼70 μM could be calculated for FBM binding to the resting and the activated NMDA channels, respectively. The slight (approximately fourfold) difference in FBM binding affinity to the resting and to the activated NMDA channels thus is actually composed of much larger differences in the binding and unbinding kinetics (∼250- and ∼60-fold difference, respectively). These findings strongly suggest that the entrance zone of the FBM binding site undergoes a much larger conformational change along the gating process than the binding region(s) of the binding site. In other words, the FBM binding site becomes much more accessible to FBM with NMDA channel activation (and FBM could therefore more easily bind to and unbind from its binding site in the activated than in the resting NMDA channel), but the spatial configurations of the binding ligand(s) for FBM themselves are not altered so much along the gating process. It would be desirable to further differentiate the interactions of FBM with open and desensitized NMDA channels, which may provide more molecular insight into the different gating states of the channel. Recently, the molecular mechanisms of different types of NMDA channel desensitization have been characterized in more detail (36–38). This could be achieved when the structure-function relationship of NMDA channel desensitization becomes sufficiently clear, and then one may study the interactions between FBM and different mutant NMDA channels with different open and/or desensitization properties, possibly with the help of photolabile transmitters (39) or other technical advances to minimize the diffusion time of the applied transmitters and thus the resolution of different gating states. In any case, as we have argued that the FBM binding site is located close to or right at the activation gate in the NMDA channel (see above), it would be especially interesting to see in the future if the gated access of FBM to its binding site is actually controlled by the activation gate of the NMDA channel.
Acknowledgments
This work was supported by grant No. NSC 94-2320-B-002-062 from the National Science Council and grant No. NHRI-EX96-9606NI from the National Health Research Institutes, Taiwan. Huai-Ren Chang is a recipient of the MD-PhD Predoctoral Fellowship No. DD9303C91 from the National Health Research Institutes, Taiwan.
Editor: David S. Weiss.
References
- 1.Rogawski, M. A. 1992. The NMDA receptor, NMDA antagonists and epilepsy therapy. A status report. Drugs. 44:279–292. [DOI] [PubMed] [Google Scholar]
- 2.Rogawski, M. A. 1998. Mechanism-specific pathways for new antiepileptic drug discovery. Adv. Neurol. 76:11–27. [PubMed] [Google Scholar]
- 3.Kleckner, N. W., J. C. Glazewski, C. C. Chen, and T. D. Moscrip. 1999. Subtype-selective antagonism of N-methyl-d-aspartate receptors by felbamate: insights into the mechanism of action. J. Pharmacol. Exp. Ther. 289:886–894. [PubMed] [Google Scholar]
- 4.Kuo, C.-C., B.-J. Lin, H.-R. Chang, and C.-P. Hsieh. 2004. Use-dependent inhibition of the N-methyl-d-aspartate currents by felbamate: a gating modifier with selective binding to the desensitized channels. Mol. Pharmacol. 65:370–380. [DOI] [PubMed] [Google Scholar]
- 5.Rho, J. M., S. D. Donevan, and M. A. Rogawski. 1994. Mechanism of action of the anticonvulsant felbamate: opposing effects on N-methyl-d-aspartate and γ-aminobutyric acid A receptors. Ann. Neurol. 35:229–234. [DOI] [PubMed] [Google Scholar]
- 6.Subramaniam, S., J. M. Rho, L. Penix, S. D. Donevan, R. P. Fielding, and M. A. Rogawski. 1995. Felbamate block of the N-methyl-d-aspartate receptor. J. Pharmacol. Exp. Ther. 273:878–886. [PubMed] [Google Scholar]
- 7.Borowicz, K. K., B. Piskorska, Z. Kimber-Trojnar, R. Malek, G. Sobieszek, and S. J. Czuczwar. 2004. Is there any future for felbamate treatment? Pol. J. Pharmacol. 56:289–294. [PubMed] [Google Scholar]
- 8.Pellock, J. M., E. Faught, I. E. Leppik, S. Shinnar, and M. L. Zupanc. 2006. Felbamate: consensus of current clinical experience. Epilepsy Res. 71:89–101. [DOI] [PubMed] [Google Scholar]
- 9.Rogawski, M. A., and W. Loscher. 2004. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 5:553–564. [DOI] [PubMed] [Google Scholar]
- 10.McCabe, R. T., C. G. Wasterlain, N. Kucharczyk, R. D. Sofia, and J. R. Vogel. 1993. Evidence for anticonvulsant and neuroprotectant action of felbamate mediated by strychnine-insensitive glycine receptors. J. Pharmacol. Exp. Ther. 264:1248–1252. [PubMed] [Google Scholar]
- 11.Wamsley, J. K., R. D. Sofia, R. L. Faull, N. Narang, T. Ary, and R. T. McCabe. 1994. Interaction of felbamate with [3H]DCKA-labeled strychnine-insensitive glycine receptors in human postmortem brain. Exp. Neurol. 129:244–250. [DOI] [PubMed] [Google Scholar]
- 12.Coffin, V., M. Cohen-Williams, and A. Barnett. 1994. Selective antagonism of the anticonvulsant effects of felbamate by glycine. Eur. J. Pharmacol. 256:R9–10. [DOI] [PubMed] [Google Scholar]
- 13.White, H. S., W. L. Harmsworth, R. D. Sofia, and H. H. Wolf. 1995. Felbamate modulates the strychnine-insensitive glycine receptor. Epilepsy Res. 20:41–48. [DOI] [PubMed] [Google Scholar]
- 14.Kanthasamy, A. G., R. R. Matsumoto, P. G. Gunasekar, and D. D. Trunong. 1995. Excitoprotective effect of felbamate in cultured cortical neurons. Brain Res. 705:97–104. [DOI] [PubMed] [Google Scholar]
- 15.McCabe, R. T., R. D. Sofia, R. T. Layer, K. A. Leiner, R. L. Faull, N. Narang, and J. K. Wamsley. 1998. Felbamate increases [3H]glycine binding in rat brain and sections of human postmortem brain. J. Pharmacol. Exp. Ther. 286:991–999. [PubMed] [Google Scholar]
- 16.The Felbamate Study Group in Lennox-Gastaut Syndrome. 1993. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). N. Engl. J. Med. 328:29–33. [DOI] [PubMed] [Google Scholar]
- 17.Adusumalli, V. E., J. K. Wichmann, N. Kucharczyk, M. Kamin, R. D. Sofia, J. French, M. Sperling, B. Bourgeois, O. Devinsky, and F. E. Dreifuss. 1994. Drug concentrations in human brain tissue samples from epileptic patients treated with felbamate. Drug Metab. Dispos. 22:168–170. [PubMed] [Google Scholar]
- 18.Johnson, J. W., and P. Ascher. 1987. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature (Lond.). 325:529–531. [DOI] [PubMed] [Google Scholar]
- 19.Kleckner, N. W., and R. D. Dingledine. 1988. Requirement for glycine in activation of NMDA receptors expressed in Xenopus oocytes. Science. 241:835–837. [DOI] [PubMed] [Google Scholar]
- 20.Benveniste, M., J. Clements, J. L. Vyklicky, and M. L. Mayer. 1990. A kinetic analysis of the modulation of N-methyl-d-aspartic acid receptors by glycine in mouse cultured hippocampal neurones. J. Physiol. (Lond.). 428:333–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lester, R. A. J., G. Tong, and C. E. Jahr. 1993. Interactions between the glycine and glutamate binding sites of the NMDA receptor. J. Neurosci. 13:1088–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lerma, J., R. S. Zukin, and M. V. Bennett. 1990. Glycine decreases desensitization of N-methyl-d-aspartate (NMDA) receptors expressed in Xenopus oocytes and is required for NMDA responses. Proc. Natl. Acad. Sci. USA. 87:2354–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayer, M. L., L. J. Vyklicky, and J. Clements. 1989. Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature (Lond.). 338:425–427. [DOI] [PubMed] [Google Scholar]
- 24.Laube, B., H. Hirai, M. Sturgess, H. Betz, and J. Kuhse. 1997. Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron. 18:493–503. [DOI] [PubMed] [Google Scholar]
- 25.Laube, B., J. Kuhse, and H. Betz. 1998. Evidence for a tetrameric structure of recombinant NMDA receptors. J. Neurosci. 18:2954–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schorge, S., and D. Colquhoun. 2003. Studies of NMDA receptor function and stoichiometry with truncated and tandem subunits. J. Neurosci. 23:1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Premkumar, L. S., and A. Auerbach. 1997. Stoichiometry of recombinant N-methyl-d-aspartate receptor channels inferred from single-channel current patterns. J. Gen. Physiol. 110:485–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anson, L. C., P. E. Chen, D. J. Wyllie, D. Colquhoun, and R. Schoepfer. 1998. Identification of amino acid residues of the NR2A subunit that control glutamate potency in recombinant NR1/NR2A NMDA receptors. J. Neurosci. 18:581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen, P. E., M. T. Geballe, P. J. Stansfeld, A. R. Johnston, H. Yuan, A. L. Jacob, J. P. Snyder, S. F. Traynelis, and D. J. Wyllie. 2005. Structural features of the glutamate binding site in recombinant NR1/NR2A N-methyl-d-aspartate receptors determined by site-directed mutagenesis and molecular modeling. Mol. Pharmacol. 67:1470–1484. [DOI] [PubMed] [Google Scholar]
- 30.Kuryatov, A., B. Laube, H. Betz, and J. Kuhse. 1994. Mutational analysis of the glycine-binding site of the NMDA receptor: structural similarity with bacterial amino acid-binding proteins. Neuron. 12:1291–1300. [DOI] [PubMed] [Google Scholar]
- 31.Chang, H.-R., and C.-C. Kuo. 2007. Extracellular proton-modulated pore-blocking effect of the anticonvulsant felbamate on NMDA channels. Biophys. J. (Epub ahead of print). [DOI] [PMC free article] [PubMed]
- 32.Harty, T. P., and M. A. Rogawski. 2000. Felbamate block of recombinant N-methyl-d-aspartate receptors: selectivity for the NR2B subunit. Epilepsy Res. 39:47–55. [DOI] [PubMed] [Google Scholar]
- 33.Monyer, H., N. Burnashev, D. J. Laurie, B. Sakmann, and P. H. Seeburg. 1994. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 12:529–540. [DOI] [PubMed] [Google Scholar]
- 34.Lin, F., and C. F. Stevens. 1994. Both open and closed NMDA receptor channels desensitize. J. Neurosci. 14:2153–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colquhoun, D., and A. G. Hawkes. 1995. Desensitization of N-methyl-d-aspartate receptors: a problem of interpretation. Proc. Natl. Acad. Sci. USA. 92:10327–10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krupp, J. J., B. Vissel, S. F. Heinemann, and G. L. Westbrook. 1998. N-terminal domains in the NR2 subunit control desensitization of NMDA receptors. Neuron. 20:317–327. [DOI] [PubMed] [Google Scholar]
- 37.Erreger, K., and S. F. Traynelis. 2005. Allosteric interaction between zinc and glutamate binding domains on NR2A causes desensitization of NMDA receptors. J. Physiol. (Lond.). 569:381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas, C. G., J. J. Krupp, E. E. Bagley, R. Bauzon, S. F. Heinemann, B. Vissel, and G. L. Westbrook. 2006. Probing N-methyl-d-aspartate receptor desensitization with the substituted-cysteine accessibility method. Mol. Pharmacol. 69:1296–1303. [DOI] [PubMed] [Google Scholar]
- 39.Hess, G. P. 2005. Photochemical release of neurotransmitters—transient kinetic investigations of membrane-bound receptors on the surface of cells in the microsecond-to-millisecond time region. In Dynamic Studies in Biology. M. Goeldner, and R. Givens, editors. Wiley-VCH, Weinheim.