

Abstract
The HIV-1 transactivation response RNA element (TAR), which is essential to the lifecycle of the virus, has been suggested, based on NMR and hydrodynamic measurements, to undergo substantial, collective, structural dynamics that are important for its function. To deal with the significant coupling between overall diffusional rotation and internal motion expected to exist in TAR, here we utilize an isotropic reorientational eigenmode dynamics analysis of simulated molecular trajectories to obtain a detailed description of TAR dynamics and an accurately quantified pattern of dynamical correlations. The analysis demonstrates the inseparability of internal and overall motional modes, confirms the existence and reveals the nature of collective domain dynamics, and additionally reveals that the hinge for these motions is centered on residues U23, C24, and C41. Results also indicate the existence of long-range communication between the loop and the core of the RNA, and between the loop and the bulge. Additionally, the isotropic reorientational eigenmode dynamics analysis explains, from a dynamical perspective, several existing biochemical mutational studies and suggests new mutations for future structural dynamics studies.
INTRODUCTION
The human immunodeficiency virus type 1 (HIV-1) transactivation response element (TAR) is a structure found at the 5′ end of the untranslated leader region of the RNA in every HIV-1 transcript (1). TAR is a 59-nucleotide hairpin RNA of which residues 18–44 have been identified as important for protein binding and transactivation (2). This sequence is shown in Fig. 1 a (with an additional terminal GC basepair) and will be referred to as TAR throughout the remainder of this text. TAR RNA contains a uridine-rich bulge and a CUGGGA hexaloop, with the remaining RNA adopting two standard A-form helical stems (3,4). The viral protein Tat and the cellular complex P-TEFb, containing proteins Cyclin T1 (CycT1) and CDK9, bind to TAR after its transcription, and enable the phosphorylation of the polymerase, RNAPII, which enhances transcriptional elongation (5–9). It has been found that the binding of the Tat and CycT1 proteins to TAR is a highly cooperative process (5). Residues in the bulge and basepairs directly adjacent to the bulge have been identified as essential for Tat-CycT1 binding and transactivation, as have the sequence and structure of the loop, and the distance between the bulge and loop (5,10–17). Inhibition of the Tat-TAR interaction leads to premature termination of transcription, interrupting the viral life cycle (18). Thus, TAR is a viable therapeutic target and there have been numerous studies to elucidate its structure, dynamics, and binding properties using crystallographic, NMR, biochemical, and computational techniques (2–4,19–24). Additionally, a number of studies have focused on the design of small molecules to inhibit the proper binding and function of TAR (25–28). Structural dynamics studies employing transient electric birefringence (29) and NMR spectroscopy (4) have found that TAR has a significant bend in the direction of its two helical domain axes (∼45° away from co-linear stacking), induced by the three-nucleotide bulge. Additionally, NMR residual dipolar coupling (RDC) and relaxation studies strongly suggest that the two domains are collectively dynamic with respect to each other around this mean (4,30) with a motional amplitude of up to ±45°, though the collective nature of the motions could not be conclusively determined from the studies. This is relevant for RNA recognition because large-scale interhelical motions in the free state may sample conformational substates that are similar to those in the protein-bound complex. Characterization of TAR through molecular dynamics (MD) simulations could lead to a greater understanding of the mechanism of these motions and thus give important insight as to the function of the RNA.
FIGURE 1.

The secondary structure of HIV-1 TAR RNA (a). The loop and stem II are commonly referred to as domain II in the article, and stem I constitutes domain I. Also shown is the rmsd of the heavy atoms for and when overlaying the entire RNA (b), the loop (c), stem I (d), stem II (e), and the bulge (f).
Advances in simulation methods, including force-field development, the addition of explicit solvent and ions, and the proper treatment of long-range electrostatics have led to more stable and reliable trajectories of nucleic acids (31–33). These simulations have led to significant insight concerning the structure, dynamics, and interactions of the systems studied, the details of which are often inaccessible by experiment (31–37,75). In addition to base flexibility on the timescale of nanoseconds (39–42), several simulations, such as those on the ribosomal RNA kink-turn elements and the UnaL2 RNA hairpin (43,44) have revealed significant global flexibility and hinge-like motions on the nanosecond timescale. Characterization of these motions is extremely important as they often play a role in the function of the molecule, as has been suggested for TAR. Here we further examine the dynamics of TAR and assess the dynamical correlations through MD simulations, utilizing a recent linear-algebraic approach developed for the covariance analysis of angular lattice functions that describe NMR relaxation (45), and which is particularly suited for very flexible molecules.
An understanding of the dynamical correlations within a biomolecular construct provides insight into collective domain dynamics, hinge sites for large-scale collective, “breathing” motions, as well as possible mechanisms for allosteric communication. Chief among the experimental methods that describe molecular motion quantitatively at atomic resolution is solution-state NMR, which can probe the relaxation-active dynamics responsible for the reorientational motion of the lattice part of nuclear spin interactions (46). However, because of the short-range character of the nuclear spin interactions, the ability of NMR to gauge directly large-scale collective motions that span the entire molecule is limited. A useful complement to addressing such long-range correlations is offered by molecular dynamics simulations (47). A typical tool for analyzing correlated motions in MD trajectories involves the textbook strategy of calculating the off-diagonal terms of the covariance matrix of atomic fluctuations (48). However, this necessitates the assumption that the rigid body motions (i.e., overall rotation and translation) can be easily separated from internal motions. In fact, this separability assumption generally underlies the analysis of MD simulations for the calculation of dynamical parameters that are to be compared with relevant experiments that can probe motion. These include heteronuclear NMR relaxation parameters (i.e., spin-lattice and spin-spin time constants, T1 and T2, and the nuclear Overhauser enhancement factor (NOE) (49,50)), and model free parameters for the amplitude (S2) and timescale (τ) of bond-vector motion (51,52). Additionally, quasiharmonic analysis calculations of conformational entropy (53,54), or principle component analysis (PCA) of atomic motion (55), also rely on this separability. In all such analyses, the overall rotational diffusion (overall tumbling) is removed in the MD trajectories by overlaying each trajectory snapshot onto a single reference frame by minimization of the root-mean-square displacement (rmsd) of atoms (56). This strategy is useful, for example, for globular proteins with compact folds, which are unaffected by internal motions, and hence for which the moments of inertia can be considered to be constant in time. However, for highly flexible molecules such as TAR, the assumption that overall rotation can be decoupled from internal motions often does not hold (30,39,45,57,58). This thereby restricts the ability of rmsd-minimization-based analyses to assess motion either qualitatively or quantitatively, and calls for novel approaches to describe the underlying dynamics.
Isotropic reorientational eigenmode dynamics (iRED) analysis, introduced by Prompers and Bruschweiler (45), provides a novel method for determining dynamical correlations from a molecular dynamics simulation when the elimination of rigid-body motion is not feasible, as the analysis is carried out without relying on the assumption that overall and internal motions are separable. In the iRED approach, one diagonalizes a covariance matrix comprised of the lattice functions that describe spin relaxation. In calculating this matrix, each snapshot of the molecular dynamics trajectory is given an isotropic overall distribution. Rather than removing rotation for each trajectory snapshot as in typical MD analyses, each snapshot is thereby allowed to sample all possible rotational orientations. The eigenvalues and eigenvectors obtained from the iRED matrix give information on the dynamical correlations throughout the molecule. To the authors' knowledge, this method has previously been applied to only two RNA molecules, the IRE RNA (39) and the U1 snRNA SL2 in complex with the U1A RBD1 protein (59), both small, single domain stem-loop structures. Here we implement iRED in the analysis of a 20-ns MD simulation of HIV-1 TAR RNA. Our interest in performing an iRED analysis of TAR is threefold. First is the obvious interest in the nature of the underlying structural dynamics of TAR, hypothesized to be central to its function. Second, unlike the two RNAs studied previously with iRED, TAR is a multidomain (stem-bulge-stem-loop) structure believed to undergo significant interdomain dynamics, and we are interested in the utility of iRED for the detection of these more complex motions. Third, a very useful result of iRED analysis is a measure of the separability of internal and overall motions. Because there is experimental evidence that motions in TAR are inseparable (30), this construct provides a good test of the iRED detection of inseparability.
METHODS
Molecular dynamics simulation
Details of this simulation, reported previously (60), are summarized in this section. A 20-ns MD simulation of wild-type TAR was performed using the CHARMM simulation package (61), with force-field parameter set 27 (62). Structure 3 of the family of free TAR NMR structures (PDB 1ANR) (19) was used as the starting coordinate, chosen because it yields the best agreement with previous residual dipolar coupling measurements (4). The RNA was neutralized using Na+ counterions and solvated in a 35-Å sphere of TIP3P water (63). A stochastic boundary potential was used (64), allowing for >9 Å distance between the surface of the sphere and all RNA atoms. The spherical (nonperiodic) boundary conditions were chosen both for: i), computational expediency over periodic boundary conditions (PBC); and ii), for the generally less appreciated fact that PBC do not conserve total angular momentum (65). Particularly for our case of isotropic averaging, analyzing overall TAR rotation together with local motion, it was not clear how abrupt changes in the angular momentum due to PBC might affect the simulated results. The number of water molecules (15,909) in the spherical boundary potential was chosen such that an average pressure of 1 atm was achieved. The solvated system was minimized and heated to 300 K while harmonically constraining the heavy atoms of the RNA with a force constant of 62 kcal/mol/Å2 for 100 ps, after which the constraints were removed. The system was then preliminarily equilibrated for 1 ns, and a production-run trajectory was followed up to 20 ns. A Nosé-Hoover thermostat using a coupling constant of 50 ps−1 (66,67) was employed to maintain a constant temperature of 300 K throughout the simulation, with a 1-fs time step. The production-run trajectory was saved every 1.0 ps, yielding 20,000 structures that constituted the snapshots used in the iRED formalism (see below).
Isotropic reorientational eigenmode dynamics analysis
The iRED formalism is derived from nuclear spin relaxation theory, a method for detecting molecular motions through the angular fluctuations of spin interactions within the molecule. For biomolecules such as proteins and nucleic acids, 15N and 13C are the predominantly studied nuclei. Their relaxation, measured by parameters such as the spin-lattice relaxation time (T1), spin-spin relaxation time (T2), and the NOE factor, is dominated by the fluctuation of the nuclei's chemical shift anisotropy (CSA) and of the dipolar interaction with a directly bonded 1H nucleus. These fluctuations arise from the overall reorientation of the molecule, as well as from internal fluctuations of chemical groups. The effect of these fluctuations on the relaxation of a nucleus are governed by the lattice functions, expressed in terms of spherical harmonics |YL,M(θ, φ)〉 of rank L = 2, following the theory by Wangsness and Bloch (49) and Redfield (50). Reorientational eigenmode dynamics (RED) (68), a predecessor of the method used here, utilizes an analysis of a covariance matrix of the |Y2,M(θ, φ)〉 lattice functions to study the dynamics of a molecule. This covariance matrix M has the components
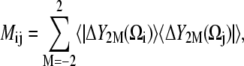 |
(1) |
where Ωi(t) = (θi(t), φi(t)) is the solid-angle direction of the principal axis of interaction i, with i = 1, 2, .., n for n interactions, i.e., in our case, for n vectors connecting the chosen 13C or 15N nuclei with their directly bonded protons. In the above formula, Eq. 1, |ΔY2M〉 = |Y2M(t)〉 − 〈|Y2M〉〉, and the outer brackets denote an average over all trajectory snapshots, whereas the bra (〈⋯|) is the complex conjugate of the ket (|⋯〉) notated function. In the RED formalism, all overall motions must be removed through overlay of each trajectory snapshot onto a single reference frame. In the iRED formalism (45), the reliance on the separability of internal and overall motions is eliminated. Instead, an isotropic distribution of overall orientations is imposed on each snapshot (i.e., each snapshot is rotated uniformly as a rigid body). This distribution is accounted for analytically (see the appendix in Prompers and Bruschweiler (45) for the details of the derivation), leading to the iRED covariance matrix with components
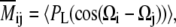 |
(2) |
where PL is the Legendre polynomial of rank L, (Ωi − Ωj) is the angular difference between two directions Ωi and Ωj (in the same trajectory snapshot), and the overbar denotes the isotropic averaging, while the brackets denote, as above, averaging over all snapshots. (For L = 2, 〈P2(cos(Ωi − Ωj))〉 should not be mistaken for the average of the orientational time correlation function 〈P2(cos(Ωi(k) − Ωi(l)))〉k,l typically used to calculate order parameters for one vector i in two distinct snapshots, k, l, averaged over all k, l pairs.)
Diagonalization of the 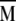 matrix yields its eigenvectors, |m〉, and their corresponding eigenvalues, λm, solutions of the equation
matrix yields its eigenvectors, |m〉, and their corresponding eigenvalues, λm, solutions of the equation 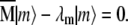 The eigenvectors contain information on which interactions reorient in a correlated manner for a given eigenmode, and the eigenvalues correspond to the variances of the fluctuations along the respective eigenmode and provide a measure of the amplitude of reorientation. Because all diagonal elements of the
The eigenvectors contain information on which interactions reorient in a correlated manner for a given eigenmode, and the eigenvalues correspond to the variances of the fluctuations along the respective eigenmode and provide a measure of the amplitude of reorientation. Because all diagonal elements of the 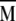 matrix are unity, the sum of all eigenvalues is always equal to n.
matrix are unity, the sum of all eigenvalues is always equal to n.
In the current analysis, all C8H8, C6H6, N1H1, N3H3, and C1′H1′ bond vectors in TAR (n = 74 in total) were used to define the interactions and to generate a second order (L = 2) iRED matrix, leading to a 74 × 74 matrix. This set of interactions provides a representative probe of both the base and sugar dynamics without complicating the analysis with too many parameters. This matrix was diagonalized to obtain the eigenvectors and eigenvalues. The eigenmodes, denoted from here on by e, were sorted in order of eigenvalue from 1 to 74, with e1 representing the mode with the largest eigenvalue. The (Ωi − Ωj) arguments of the Legendre polynomial were obtained from 1 to 20 ns of the 20-ns MD trajectory. Calculation of the matrix and subsequent diagonalization by the Jacobi transformation scheme (69) were performed using in-house C programs.
Three parameters were calculated for the analysis of motional correlations. First, from the eigenvalues (ordered from high to low) and eigenvectors, the separability index was calculated as the ratio of two cumulative contributions to relaxation: that of the total and, respectively, internal modes,
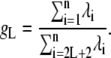 |
(3) |
The index gL provides a measure of the separability of internal and overall modes of motion. In the limit of a rigid molecule, for which there are at most 2L + 1 nonzero eigenvalues (which correspond to overall rotation), gL is infinity. For a flexible molecule, a real number will be obtained providing a measure of how separable the overall motions are from the internal fluctuations. Additionally, collectivity factors for each eigenmode, derived from the information-theory entropy, were calculated as
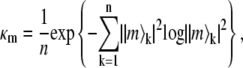 |
(4) |
where |m〉k is the k-th component of the normalized eigenvector |m〉, and which represents the fraction of interactions collectively affected by a given mode m. Lastly, the principal order parameter components for each interaction and for each mode were calculated as
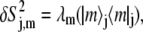 |
(5) |
which gives the contribution of a given mode to the decay of the orientational time autocorrelation function Cj(t) describing the relaxation of the jth heavy nucleus to equilibrium through angular reorientation,
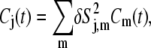 |
(6) |
with Cm being the normalized correlation functions for mode m.
RESULTS AND DISCUSSION
Dynamical trajectory
Shown in Fig. 1 is the rmsd of the heavy atoms of TAR as compared to the starting structure over the course of the 20-ns simulation. When overlaying the entire structure (Fig. 1 b), the rmsd reaches a maximum of 7.2 Å, with an average of ∼4 Å. However, when overlaying each structural component separately (i.e., the two helical stems, the loop, and the bulge) it can be seen that the rmsd for each region is reasonable and comparable to the rmsd in typical RNA and DNA duplex simulations (Fig. 1, c–f). After the period of equilibration (1 ns) the loop and bulge reach the largest mean rmsd values of 4.2 and 3.5 Å, which is to be expected given that these regions would likely be the least resolved in the starting NMR structure. The helical regions reach mean rmsd values (after equilibration) of 2.2 and 1.9 Å for stems I and II, respectively. The loop and bulge also have the highest standard deviations around this mean of 0.44 and 0.54 Å, respectively, as compared to values of 0.28 and 0.38 Å for domains I and II, indicating that the bulge and loop are far more dynamic than the helical domains. Overall, the structure appears stable throughout the 20-ns simulation. Furthermore, the large variance of the rmsd obtained when overlaying the entire RNA (0.82 Å) as compared to the individual regions clearly indicates that there are global dynamics between the locally stable domains, which is consistent with previous NMR findings that suggest that TAR is globally dynamic (4,23,30).
Analysis of basepairs in the helical stems using the program 3DNA (70) reveals that all form standard Watson-Crick basepairs, in agreement with the starting structure. Excluding the G21-C41 basepair and those flanking the bulge and loop all are formed for >90% of the simulation, and have standard deviations in their basepairing parameters that are reasonably close to those reported previously from a survey of helical basepairs for structures in the NDB (data not shown) (60). Of the remaining basepairs C29-G36 (directly below the loop) is formed for 57% of the simulation, G26-C39 (directly above the bulge) is formed for 66% of the simulation, A22-U40 (directly below the bulge) is formed for 40% of the simulation, and G21-C41 is formed for 75% of the simulation. Only the lattermost is surprising and will be discussed in more detail below. The basepairs directly flanking the bulge and the loop would be expected to experience a greater level of dynamics due to the flexibility of the adjacent nonhelical moieties, explaining the lower population of the basepaired state for these pairs. In fact, A22-U40, which is basepaired for only 40% of the simulation is generally not detected in experiment at all including in the starting structural coordinates (3,19).
Residues U23 and C24 in the bulge appear to be stabilized by stacking interactions with A22, whereas U25 is looped out into solution and quite dynamic, consistent with previous experiments (3,19). In the loop, C30 and G34 intermittently form a cross-loop Watson-Crick basepair (formed for 27% of the simulation), which is also consistent with previous studies (71). G34 also forms a Watson-Crick basepair with C29 in stem II for 35% of the simulation (thus, C29 is involved in a basepair with either G34 or G36 for >90% of the simulation). The remaining loop residues A35, U31, G32, and G33 are looped out into solution and flexible, though U31 and G32 are observed to be in a stacked conformation much of the time. The high level of flexibility in the bulge and loop explains the large variance in the rmsd, as well as explaining the low resolution of the starting structure and thus the higher average rmsd of these moieties in the equilibrated structure. The average value of ∼4 Å for the total rmsd (see Fig. 1 b), is consistent with that found in a previous MD simulation of TAR using the AMBER force field on three of the NMR TAR structures (including structure 3 used in our study) (23). That study also reported rmsds >3 Å and as high as 8 Å from the starting structures within the first 250 ps (see Fig. 2 of Nifosi et al. (23)). Specifically for structure 3, an rmsd of 4 Å is seen at 250 ps, which is comparable to the rmsd in our study that is seen to reach 3.8 Å at 250 ps. Moreover, an rmsd in the same range is also seen among the various structures in the NMR ensemble (3).
FIGURE 2.

The interhelical bend and twist (a) as a function of time. Also shown are three snapshots taken from the 20-ns trajectory showing the structural variation in the bend and twist (b). The representative structures have a corresponding range in the bend of 16.6–72.5 and in the twist of −41.4–24.4.
Previous NMR studies have suggested that TAR is dynamic on the global level, with an estimated interhelical bend angle of 45° with a dynamical range of 45° around this mean (4,23,30). However, the collectivity of motions could only be inferred in the NMR relaxation study from the similarity of the amplitudes and timescales of the slow motions detected across different bond vectors in stem II. Similarly, in an RDC study of TAR, the conclusion that there were collective dynamics relied on the assumption that local motions were uniform throughout the helical portions of the RNA (4). These studies could not exclude the formal possibility that there were actually low frequency localized motions of the bases being detected.
To further characterize the motions in TAR, the bend and twist angles relating stem I and stem II throughout the trajectory were obtained using the program eulerRNAAform (72). This program finds the rotation (expressed through Euler angles α, β, and γ) that transforms one stem from where it would be if it were in a co-linear conformation with the other stem to its actual orientation using a best fit overlay of atoms. From these angles the twist of stem I and stem II (α and γ, respectively), the interhelical twist (α + γ) and the interhelical bend (β) can be obtained. Seen in Fig. 2 a are the interhelical bend and twist angles as a function of time. The large variance in these angles unequivocally shows that TAR is dynamic on the global level, experiencing hinge-like motions between the two stable helical stems. These results give definitive evidence that the motions detected in the NMR studies were indeed collective motions of the stems. The interhelical bend ranges between 5.3° and 72.5° with an average of 41.7° and the interhelical twist ranges between −55.3° and 64.3°, with an average of 14.3°. Shown in Fig. 2 b are tertiary representations of the bending and twisting motions, and the Supplementary Material features further description of the three-dimensional structural dynamics in the form of an mpeg-format movie of the entire simulated trajectory.
iRED analysis
Separability of motions
Generally for biomolecular systems, providing proof for the inseparability of motion by solution-state NMR alone is very difficult, as the separability of motions can only be inferred from the measured timescales, which, if they are indeed inseparable, will not be accurately and/or fully detected. However, a recent NMR experiment on an elongated TAR construct obtained convincing evidence that the internal and overall motional modes are inseparable (30). In that study, relaxation parameters were obtained on a TAR-like construct in which the lower helical domain had been elongated significantly, thereby slowing down the overall rotational diffusion by a factor of ∼3. Relaxation parameters obtained on the elongated TAR revealed internal motional modes on the timescale of the overall rotation of the nonelongated construct, which were not detected in the nonelongated TAR due to their coupling with the overall rotation (30). The overall rotational tumbling time of TAR has been determined, through hydrodynamic calculations and NMR spectroscopy, to be ∼6 ns (30), and thus the relevant motions should be accessible on the timescale of our simulation.
Shown in Fig. 3 are the eigenmode collectivity factors (κm; see Eq. 4) as a function of the eigenvalue (λm), where κm gives the percentage of interactions significantly affected by a given eigenmode, |m〉. For systems in which the internal and overall motions are separable, a gap would be observed between the five largest modes (for L = 2) and the remaining modes for both the eigenvalues and corresponding collectivities. In such a case, the five largest modes represent only the overall motion, whereas the remaining modes represent internal motions. As can clearly be seen in Fig. 3, such a gap is not observed for TAR. The separability factor, g2 (see Eq. 3), is 2.6. This is comparable to the separability factor observed for the partially unstructured A-state of ubiquitin (g2 = 2.5), for which the overall and internal motions were previously observed to be inseparable by iRED (45). The pattern of separability is also similar to that of the IRE RNA, although no separability factor in that study was reported. Thus, the coupling of motions detected in TAR through the elongation experiment (see above) is accurately reproduced in the simulation and detected by the iRED analysis. Additionally, this confirms that indeed a single reference frame cannot be defined for analysis of the MD trajectory, precluding, for such a flexible system, many traditional methods of dynamical analysis.
FIGURE 3.

The collectivity factor, κ (see Eq. 4), as a function of eigenvalue, λ. The lack of a gap in λ and κ between the largest five modes and the remaining modes demonstrates that the internal and overall motions are inseparable. Eigenmodes with the five largest eigenvalues, which would correspond to the overall rotation for a molecule in which the internal and overall motions are separable, are marked by the dotted ellipse.
Large amplitude motional modes
The principal order parameter components (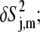 see Eq. 5) represent the contribution of a given mode with corresponding eigenvector |m〉 to the decay of the correlation function for a given interaction j. (Although this should not be confused with the Lipari-Szabo order parameter of that interaction (
see Eq. 5) represent the contribution of a given mode with corresponding eigenvector |m〉 to the decay of the correlation function for a given interaction j. (Although this should not be confused with the Lipari-Szabo order parameter of that interaction (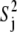 ), if the overall and internal motions are separable, then the sum over all internal modes of the order parameter components for a given vector will yield 1 −
), if the overall and internal motions are separable, then the sum over all internal modes of the order parameter components for a given vector will yield 1 − 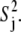 ) In general, an analysis of the
) In general, an analysis of the 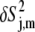 values for each mode leads to significant insight concerning the dynamics of the RNA, and especially the dynamical correlations between interactions throughout the construct. Shown in Fig. 4 are the principle order parameter components for the largest amplitude motions, eigenmodes e1 − e10, as a function of residue. With the exception of mode e9 (see below), these modes are highly collective in nature, with κ = 28–58%, all affecting different portions of the RNA.
values for each mode leads to significant insight concerning the dynamics of the RNA, and especially the dynamical correlations between interactions throughout the construct. Shown in Fig. 4 are the principle order parameter components for the largest amplitude motions, eigenmodes e1 − e10, as a function of residue. With the exception of mode e9 (see below), these modes are highly collective in nature, with κ = 28–58%, all affecting different portions of the RNA.
FIGURE 4.

δS2-values (see Eq. 5) as a function of residue for modes e1–e10. Circles represent base interactions and squares represent C1′H1′ interactions. Also shown are the κ-values (see Eq. 4) for each mode.
A detailed description of the large-scale motional modes is presented as follows.
Modes e1–e3: interdomain dynamics
Mode e1 is the largest amplitude mode and the only mode that significantly affects interactions in the entire RNA, although effects in the bulge and loop are noticeably lower. This mode likely has the most overall motional character.
Mode e2, the second largest amplitude mode, is highly collective with κ = 58%, and most significantly affects interactions in residues 17–21 and 42–45, comprising most of domain I. Similarly mode e3 has κ = 58%, and predominantly affects interactions in residues 26–39, with the exception of residues 31, 32, and 35 in the loop. This comprises most of domain II, with the exception of the most flexible residues. The large amplitude associated with these modes indicates that they cannot be attributed to correlations of small base fluctuations. Rather, these modes clearly represent the collective motions of each domain. This confirms the ability of the iRED analysis to detect collective domain motions such as those seen here in TAR. It is interesting to note that even though each mode affects one domain most significantly, interactions in the other domain, although far less significant, are still affected. This implies that the domain motions are correlated. This correlation can be examined further through the Euler angles obtained using the program eulerRNAAform (72) as described in the “Dynamical trajectory” section. Shown in Fig. 5 is a plot of the twist of stem I (α) versus the twist of stem II (γ), which reveals that these angles are significantly correlated. This explains the correlation between the stem I and stem II bond vectors observed in iRED modes e2 and e3. No correlation is observed between the twisting and bending motions (data not shown).
FIGURE 5.

The correlation between the twist of stem I (α) and stem II (γ) as obtained from the program eulerRNAAform.
Modes e4–e5: internucleotide correlations
Modes e4 and e5 have collectivity factors, κ, of 54% and 56%, and most significantly affect the 3′ and 5′ strands, respectively, showing correlation of motion from nucleotide to nucleotide along the backbone. This correlation is interrupted at residue U25 in the 5′ strand, and for each strand dissipates through the loop, showing that the highly dynamic regions interrupt this correlation. Each strand also has some correlation with the opposing strand, most often through the bases. A similar mode of motion was seen in the iRED analysis of the IRE RNA element (39), suggesting that this may be a universal mode of motion that all RNA molecules experience. It has been shown that in A-form RNA as compared to B-form DNA the local motions tend to propagate to the global level rather then manifesting as flexibility on the local level (38,73,74). In other words, RNA appears locally more stiff as compared to DNA but globally more flexible. The correlation seen here along each strand from nucleotide to nucleotide and across basepairs could represent this propagation of local motion to a more global flexibility. It is tempting to speculate that the break in correlation introduced by the highly dynamic regions, which are also the protein binding sites, are important for protein/ligand accessibility, which might otherwise be blocked by the local “stiffness” in RNA.
Mode e6: the hinge and long-range bulge/loop interactions
Although it is clear that the hinge for the interdomain motions in TAR must be in, or around, the bulge, the specific residues that constitute the hinge, and the detailed mechanism of hinge motion are yet to be determined. Mode e6 gives significant insight into this question. As can be seen in Fig. 4, mode e6 most significantly affects residues U23 and C24 in the bulge and the opposing strand residue C41. The large amplitude associated with this mode suggests that it is also representative of large-scale motions. Given that the residues most affected are in and around the bulge, it is quite possible that these residues constitute the hinge for the collective motions. Additional evidence for this is found in referencing modes e2 and e3, which represent the collective domain motions. If residues U23, C24, and C41 are acting as a hinge for the collective motions, one would expect the same interactions that are largely affected by mode e6 to be far less affected by modes e2 and e3 as compared to the domain residues. Similarly, the domain residues should be far less affected by mode e6. This pattern of dynamics is indeed observed for all three modes, strongly suggesting that the hinge is comprised of residues 23, 24, and 41.
Returning to the analysis of the global motions as described in the “Dynamical trajectory” section we can obtain further insight into the details of the hinge dynamics. The opening parameter of the G21-C41 basepair and the backbone dihedral angles ε and ζ of residue C41 (obtained using the program CURVES) have corresponding standard deviations over the trajectory that are significantly larger than observed for the same parameters for the other helical residues (data not shown). Plotting these parameters alongside the interhelical twist (see Fig. 6 a) reveals a clear correlation over the trajectory, demonstrating the functional role of C41 in the hinge for the global motions. Similarly, though most of the backbone dihedrals for residues U23 and C24 have corresponding standard deviations similar to the stable helical residues, the angles ε and ζ show significantly larger variations across the simulation. The angle ζ for C24 demonstrates periodic motions on the nanosecond timescale consistent with the interhelical bending motions as seen in Fig. 6 b. Though such a correlation for ε and ζ of U23 and ε of C24 is not so straightforward, the large variation of these angles over the trajectory suggests that these torsions play a large role in the interhelical bending and twisting motions as well.
FIGURE 6.

The correlation between the base and backbone parameters for residue C41 with the interhelical twist angle (a). Specifically, the opening basepair parameter (circles) and the backbone parameters ε and ζ (inverted triangles and triangles) are shown for residue C41 with the interhelical twist angle (squares) as a function of time. Also shown (b) is the interhelical bend (squares) and the backbone dihedral ζ for C24 (circles) as a function of time.
Residue U31 in the loop, the C1′H1′ of residue 32 and, less so, the adjacent residues in the loop are also affected by mode e6. The correlation of the hinge residues with the loop is very interesting. A similar coupling was previously proposed from phylogenetic studies claiming that U31 forms a base triple with the A22-U40 basepair in free TAR (76), though there has been no experimental evidence for this. Although residue U31 is seen to be solvent exposed in the simulation and is very dynamic, it does not come into close enough contact with either residue U23, C24, or C41 to have a direct interaction with any of these residues at any time during the simulation, and the same can be said of G32. Thus, barring finite-time sampling limitations in the simulation, the correlation of dynamics seen in this mode would have to be a long-range interaction. Mutational studies show that the identity of residue 31 does not significantly affect Tat-CycT1 binding to TAR (12), however mutation from a U to a C decreases the viral activity by approximately one-half (76). Additionally, cross-linking studies show a significant interaction between CycT1 and residue 31, and indicate that this interaction mediates an interaction between Tat and residue 34, a residue that is essential for binding (11). It is also interesting to note that, in our results, residue U31 shows a strong correlation with the base of residue G32 (mode e7) which is also seen to be essential for Tat-CycT1 binding. Correlation between interactions in the loop and bulge are also seen in modes e12, e13, and e28. In these modes, residues 23/25/35/41, 25/32/35/40, and 22/34/41 are significantly affected, respectively (data not shown). The cooperative binding of Tat to the bulge and CycT1 to the loop along with the CycT1 mediated interaction of Tat with the loop, implies a strong functional coupling between the two motifs. It is possible that this long-range correlation detected between the bulge and loop is important for this functional coupling.
Mode e7, e8, and e10: loop dynamics
The three modes e7, e8, and e10 most significantly affect residues in the loop. Mode e7 affects base interactions of residues U31 and G32, mode e8 affects base interactions of residues G32 and G33, and mode e10 affects base interactions of residue G33 and the C1′H1′ interactions of residues 34, 35, and 36. Interestingly, these modes are highly collective with mode e7 having κ = 28% and both modes e8 and e10 having κ > 50%. This demonstrates that the motion of these residues, especially G32 and G33, is correlated with a large number of interactions throughout the RNA, which are fairly uniformly distributed. This is significant as clearly mutations to the loop that affect the dynamics of these residues could have an effect on the dynamics of the entire RNA, though from this data it cannot be determined how significant or what the mechanistic details of this effect would be. However, this correlation points to the necessity for care in drawing conclusions from mutational studies alone.
Mode e9: dynamics of residue 25
In contrast to modes e7, e8, and e10, mode e9 affects the base interactions of residue U25 alone, with no correlation to any other interaction in the RNA. Residue U25 is in the UCU bulge, and has been observed to be looped out into solution in the free form of the RNA. The lack of correlation with any other interaction in the RNA indicates that mutation of this residue should have little to no effect on the dynamics of the overall RNA. In fact, mutational experiments on U25 have shown that its identity is not important for the function of TAR (20), and it can even be deleted (which simply yields the HIV-2 TAR analog).
CONCLUDING DISCUSSION
iRED analysis of TAR RNA has confirmed that the internal and overall motions are inseparable, in accord with the relaxation measurements in the NMR experiment on the elongated construct. Our results demonstrate hinge-like collective motions of the stable helical domains of TAR, providing firm evidence for what was previously suggested by NMR relaxation and RDC studies. Additionally, results from the iRED mode analysis and analysis of the basepairing and backbone dihedral parameters show that the hinge for these motions is centered on residues U23 and C24 in the bulge and residue C41 in the opposite strand, of which U23 and C41 are seen to be essential for binding of Tat to Tar and/or transactivation (13). Residues A22 and U40, the basepair directly below the bulge and positioned between the bulge and residue C41, are not seen to be correlated to the hinge residues. However, the very weak hydrogen bond between this AU basepair (3,19) imparts a large degree of flexibility around the bulge region. Thus, it is quite likely that these residues are indirectly involved in the hinge for the interstem motions, perhaps allowing dynamical access to C41.
Mode analysis also indicates a correlation between the hinge residues and two distant sites in the apical loop of domain II: the base of residue U31, and the C1′H1′ of residue G32. Clearly there is not a direct interaction occurring as neither U31 nor G32 come close enough to the bulge residues to undergo such an interaction, indicating that it is a long-range interaction that exists between the two regions. It is not clear at this point what type of interaction may be occurring between the loop and the bulge. However there is clearly a functional coupling between the two regions through the Tat-CycT1-TAR complex, in which cooperative binding and mediated interactions occur between the binding proteins and the bulge and loop. Though extensive mutational studies have been performed on TAR with an interest toward the effect on Tat or CycT1 binding, further structural dynamics studies on mutants involving changes to the bulge and loop could prove to be very valuable in determining what, if any, communication is occurring between these two functional regions and in understanding the binding and function of TAR.
Lastly, with our results showing the detection of the interdomain dynamics, this indicates that iRED is useful in this capacity and likely to aid in the interpretation of dynamical correlations in RNA molecules with even more complex tertiary structures. For example, it would be very interesting to apply iRED to any of the recently reported riboswitch structures (77–80), which contain multiple helical domains, and for which a description of the collective dynamics could give significant insight into their function. Also, the reported ability of iRED to describe motions in complex structures for which internal fluctuations and overall rotations are inseparable, indicates that this approach is likely to be of significant aid in quantifying more complex motions with a severe lack of separability, such as the drastic conformational changes occurring in unstructured ensembles or upon RNA or protein unfolding.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
C.M. thanks Dr. Scott Showalter for assistance in implementing iRED and for insightful conversations. Mpeg-format movie (Supplementary Material) of the simulated trajectory was created with the Visual Molecular Dynamics package (VMD) (81).
C.M. acknowledges support from the National Institutes of Health Molecular Biophysics Training Grant. I.A. acknowledges funds from the National Science Foundation CAREER Award program (CHE-0548047), the Donors of the American Chemical Society PRF-G grant program, and the University of Michigan. H.M.A. acknowledges support from National Institutes of Health grant R01 AI066975-01.
Editor: Kathleen B. Hall.
References
- 1.Muesing, M., D. Smith, and D. Capon. 1987. Regulation of mRNA accumulation by a human immunodeficiency virus trans-activator protein. Cell. 48:691–701. [DOI] [PubMed] [Google Scholar]
- 2.Weeks, K., C. Ampe, S. Schultz, T. Steitz, and D. Crothers. 1990. Fragments of the HIV-1 Tat protein specifically bind TAR RNA. Science. 249:1281–1285. [DOI] [PubMed] [Google Scholar]
- 3.Aboulela, F., J. Karn, and G. Varani. 1995. The structure of the human-immunodeficiency-virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. J. Mol. Biol. 253:313–332. [DOI] [PubMed] [Google Scholar]
- 4.Al-Hashimi, H., Y. Gosser, A. Gorin, W. Hu, A. Majumdar, and D. Patel. 2002. Concerted motions in HIV-1 TAR RNA may allow access to bound state conformations: RNA dynamics from NMR residual dipolar couplings. J. Mol. Biol. 315:95–102. [DOI] [PubMed] [Google Scholar]
- 5.Wei, P., M. Garber, S. Fang, W. Fischer, and K. Jones. 1998. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 92:451–462. [DOI] [PubMed] [Google Scholar]
- 6.Zhu, Y., T. Peery, T. Peng, Y. Ramanathan, N. Marshall, T. Marshall, B. Amendt, M. Mathews, and D. Price. 1997. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 11:2622–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feinberg, M., D. Baltimore, and A. Frankel. 1991. The role of Tat in the human-immunodeficiency-virus life-cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. USA. 88:4045–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marciniak, R., and P. Sharp. 1991. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 10:4189–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou, Q., D. Chen, E. Pierstorff, and K. Luo. 1998. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 17:3681–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng, S., and E. Holland. 1988. Hiv-1 Tat trans-activation requires the loop sequence within TAR. Nature. 334:165–167. [DOI] [PubMed] [Google Scholar]
- 11.Richter, S., Y. Ping, and T. Rana. 2002. TAR RNA loop: a scaffold for the assembly of a regulatory switch in HIV replication. Proc. Natl. Acad. Sci. USA. 99:7928–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richter, S., H. Cao, and T. Rana. 2002. Specific HIV-1 TAR RNA loop sequence and functional groups are required for human cyclin Tl-Tat-TAR ternary complex formation. Biochemistry. 41:6391–6397. [DOI] [PubMed] [Google Scholar]
- 13.Delling, U., L. Reid, R. Barnett, M. Ma, S. Climie, M. Sumnersmith, and N. Sonenberg. 1992. Conserved nucleotides in the TAR RNA stem of human-immunodeficiency-virus type-1 are critical for Tat binding and transactivation: model for TAR RNA tertiary structure. J. Virol. 66:3018–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weeks, K., and D. Crothers. 1991. RNA recognition by Tat-derived peptides: interaction in the major groove. Cell. 66:577–588. [DOI] [PubMed] [Google Scholar]
- 15.Berkhout, B., and K. Jeang. 1989. Trans-activation of human immunodeficiency virus type-1 is sequence specific for both the single-stranded bulge and loop of the trans-acting-responsive hairpin. A quantitative analysis. J. Virol. 63:5501–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calnan, B., B. Tidor, S. Biancalana, D. Hudson, and A. Frankel. 1991. Arginine-mediated RNA recognition: the arginine fork. Science. 252:1167–1171. [DOI] [PubMed] [Google Scholar]
- 17.Sumnersmith, M., S. Roy, R. Barnett, L. Reid, R. Kuperman, U. Delling, and N. Sonenberg. 1991. Critical chemical-features in trans-acting-responsive RNA are required for interaction with human-immunodeficiency-virus type-1 Tat protein. J. Virol. 65:5196–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kao, S., A. Calman, P. Luciw, and B. Peterlin. 1987. Anti-termination of transcription within the long terminal repeat of HIV-1 by Tat gene-product. Nature. 330:489–493. [DOI] [PubMed] [Google Scholar]
- 19.Aboulela, G., J. Karn, and G. Varani. 1996. Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res. 24:3974–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Churcher, M., C. Lamont, F. Hamy, C. Dingwall, S. Green, A. Lowe, P. Butler, M. Gait, and J. Karn. 1993. High-affinity binding of TAR RNA by the human-immunodeficiency-virus type-1 Tat protein requires base-pairs in the RNA stem and amino-acid-residues flanking the basic region. J. Mol. Biol. 230:90–110. [DOI] [PubMed] [Google Scholar]
- 21.Ippolito, J., and T. Steitz. 1998. A 1.3-angstrom resolution crystal structure of the HIV-1 trans-activation response region RNA stem reveals a metal ion-dependent bulge conformation. Proc. Natl. Acad. Sci. USA. 95:9819–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long, K., and D. Crothers. 1999. Characterization of the solution conformations of unbound and Tat peptide-bound forms of HIV-1 TAR RNA. Biochemistry. 38:10059–10069. [DOI] [PubMed] [Google Scholar]
- 23.Nifosi, R., C. Reyes, and P. Kollman. 2000. Molecular dynamics studies of the HIV-1 TAR and its complex with argininamide. Nucleic Acids Res. 28:4944–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitt, S., A. Majumdar, A. Serganov, D. Patel, and H. Al-Hashimi. 2004. Argininamide binding arrests global motions in HIV-1 TAR RNA: comparison with Mg2+-induced conformational stabilization. J. Mol. Biol. 338:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faber, C., H. Sticht, K. Schweimer, and P. Rosch. 2000. Structural rearrangements of HIV-1 Tat-responsive RNA upon binding of neomycin B. J. Biol. Chem. 275:20660–20666. [DOI] [PubMed] [Google Scholar]
- 26.Olejniczak, M., Z. Gdaniec, A. Fischer, T. Grabarkiewicz, L. Bielecki, and R. Adamiak. 2002. The bulge region of HIV-1 TAR RNA binds metal ions in solution. Nucleic Acids Res. 30:4241–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Litovchick, A., A. Evdokimov, and A. Lapidot. 2000. Aminoglycoside-arginine conjugates that bind TAR RNA: synthesis, characterization, and antiviral activity. Biochemistry. 39:2838–2852. [DOI] [PubMed] [Google Scholar]
- 28.Mu, Y., and G. Stock. 2006. Conformational dynamics of RNA-peptide binding: a molecular dynamics simulation study. Biophys. J. 90:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zacharias, M., and P. Hagerman. 1995. The bend in RNA created by the transactivation response element bulge of human-immunodeficiency-virus is straightened by arginine and by Tat-derived peptide. Proc. Natl. Acad. Sci. USA. 92:6052–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang, Q., X. Sun, E. Watt, and H. Al-Hashimi. 2006. Resolving the motional modes that code for RNA adaptation. Science. 311:653–656. [DOI] [PubMed] [Google Scholar]
- 31.Auffinger, P., and E. Westhof. 1998. Simulations of the molecular dynamics of nucleic acids. Curr. Opin. Struct. Biol. 8:227–236. [DOI] [PubMed] [Google Scholar]
- 32.Beveridge, D., and K. McConnell. 2000. Nucleic acids: theory and computer simulation, Y2K. Curr. Opin. Struct. Biol. 10:182–196. [DOI] [PubMed] [Google Scholar]
- 33.Cheatham, T., and P. Kollman. 2000. Molecular dynamics simulation of nucleic acids. Annu. Rev. Phys. Chem. 51:435–471. [DOI] [PubMed] [Google Scholar]
- 34.Mcdowell, S., N. Špaèkova, J. Šponer, and N. Walter. 2007. Molecular dynamics simulations of RNA: an in silico single molecule approach. Biopolymers. 85:169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Norberg, J., and L. Nilsson. 2002. Molecular dynamics applied to nucleic acids. Acc. Chem. Res. 35:465–472. [DOI] [PubMed] [Google Scholar]
- 36.Rhodes, M. M., K. Reblova, J. Šponer, and N. G. Walter. 2006. Trapped water molecules are essential to structural dynamics and function of a ribozyme. Proc. Natl. Acad. Sci. USA. 103:13380–13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krasovska, M. V., J. Sefcikova, K. Reblova, B. Schneider, N. G. Walter, and J. Šponer. 2006. Cations and hydration in catalytic RNA: molecular dynamics of the hepatitis delta virus ribozyme. Biophys. J. 91:626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noy, A., A. Perez, F. Lankaš, F. Luque, and M. Orozco. 2005. The relative flexibility of DNA and RNA duplexes: a molecular dynamics study. Biophys. J. 89:2939–2949.16100266 [Google Scholar]
- 39.Showalter, S., N. Baker, C. Tang, and K. Hall. 2005. Iron responsive element RNA flexibility described by NMR and isotropic reorientational eigenmode dynamics. J. Biomol. NMR. 32:179–193. [DOI] [PubMed] [Google Scholar]
- 40.Kieken, F., E. Arnoult, F. Barbault, F. Paquet, T. Huynh-Dinh, J. Paoletti, D. Genest, and G. Lancelot. 2002. HIV-1(Lai) genomic RNA: combined used of NMR and molecular dynamics simulation for studying the structure and internal dynamics of a mutated SL1 hairpin. Eur. Biophys. J. 31:521–531. [DOI] [PubMed] [Google Scholar]
- 41.Barthel, A., and M. Zacharias. 2006. Conformational transitions in RNA single uridine and adenosine bulge structures: a molecular dynamics free energy simulation study. Biophys. J. 90:2450–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koplin, J., Y. Mu, C. Richter, H. Schwalbe, and G. Stock. 2005. Structure and dynamics of an RNA tetraloop: a joint molecular dynamics and NMR study. Structure. 13:1255–1267. [DOI] [PubMed] [Google Scholar]
- 43.Razga, F., J. Koèa, J. Šponer, and N. Leontis. 2005. Hinge-like motions in RNA kink-turns: the role of the second A-minor motif and nominally unpaired bases. Biophys. J. 88:3466–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Razga, F., M. Zacharias, K. Reblova, J. Koèa, and J. Šponer. 2006. RNA kink-turns as molecular elbows: hydration, cation binding, and large-scale dynamics. Structure. 14:825–835. [DOI] [PubMed] [Google Scholar]
- 45.Prompers, J., and R. Bruschweiler. 2002. General framework for studying the dynamics of folded and nonfolded proteins by NMR relaxation spectroscopy and MD simulation. J. Am. Chem. Soc. 124:4522–4534. [DOI] [PubMed] [Google Scholar]
- 46.Abragam, A. 1961. Principles of Nuclear Magnetism. Clarendon Press, Oxford, UK.
- 47.Karplus, M., and J. A. McCammon. 2002. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9:646–652. [DOI] [PubMed] [Google Scholar]
- 48.Brooks, C., M. Karplus, and B. Pettitt. 1988. Proteins: A Theoretical Perspective of Dynamics, Structure, and Thermodynamics. John Wiley, New York.
- 49.Wangsness, R., and F. Bloch. 1953. The dynamical theory of nuclear induction. Physical Review. 89:728–739. [Google Scholar]
- 50.Redfield, A. 1957. On the theory of relaxation processes. IBM Journal of Research and Development. 1:19–31. [Google Scholar]
- 51.Lipari, G., and A. Szabo. 1982. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules.1. Theory and range of validity. J. Am. Chem. Soc. 104:4546–4559. [Google Scholar]
- 52.Lipari, G., A. Szabo, and R. Levy. 1982. Protein dynamics and NMR relaxation: comparison of simulations with experiment. Nature. 300:197–198. [Google Scholar]
- 53.Karplus, M., and J. Kushick. 1981. Method for estimating the configurational entropy of macromolecules. Macromolecules. 14:325–332. [Google Scholar]
- 54.Andricioaei, I., and M. Karplus. 2001. On the calculation of entropy from covariance matrices of the atomic fluctuations. J. Chem. Phys. 115:6289–6292. [Google Scholar]
- 55.Garcia, A. E. 1992. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 68:2696–2699. [DOI] [PubMed] [Google Scholar]
- 56.Zhou, Y. Q., M. Cook, and M. Karplus. 2000. Protein motions at zero-total angular momentum: the importance of long-range correlations. Biophys. J. 79:2902–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang, Q., R. Throolin, S. Pitt, A. Serganov, and H. Al-Hashimi. 2003. Probing motions between equivalent RNA domains using magnetic field induced residual dipolar couplings: accounting for correlations between motions and alignment. J. Am. Chem. Soc. 125:10530–10531. [DOI] [PubMed] [Google Scholar]
- 58.Vugmeyster, L., D. Raleigh, A. Palmer, and B. Vugmeister. 2003. Beyond the decoupling approximation in the model free approach for the interpretation of NMR relaxation of macromolecules in solution. J. Am. Chem. Soc. 125:8400–8404. [DOI] [PubMed] [Google Scholar]
- 59.Showalter, S., and K. Hall. 2005. Correlated motions in the U1 snRNA stem/loop 2: U1A RBD1 complex. Biophys. J. 89:2046–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Musselman, C., S. Pitt, K. Gulati, L. Foster, I. Andricioaei, and H. Al-Hashimi. 2006. Impact of static and dynamic A-form heterogeneity on the determination of RNA global structural dynamics using NMR residual dipolar couplings. J. Biomol. NMR. 36:235–249. [DOI] [PubMed] [Google Scholar]
- 61.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics. J. Comput. Chem. 4:187–217. [Google Scholar]
- 62.MacKerell, A., N. Banavali, and N. Foloppe. 2000. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 56:257–265. [DOI] [PubMed] [Google Scholar]
- 63.Jorgensen, W., J. Chandrasekhar, J. Madura, R. Impey, and M. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 64.Brooks, C., and M. Karplus. 1983. Deformable stochastic boundaries in molecular-dynamics. J. Chem. Phys. 79:6312–6325. [Google Scholar]
- 65.Hoover, W. 1986. Molecular Dynamics. Springer-Verlag, New York.
- 66.Nosé, S. 1984. A unified formulation of the constant temperature molecular-dynamics methods. J. Chem. Phys. 81:511–519. [Google Scholar]
- 67.Hoover, W. 1985. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 68.Prompers, J., and R. Bruschweiler. 2001. Reorientational eigenmode dynamics: a combined MD/NMR relaxation analysis method for flexible parts in globular proteins. J. Am. Chem. Soc. 123:7305–7313. [DOI] [PubMed] [Google Scholar]
- 69.Press, W. 1992. Numerical Recipes in C: The Art of Computing. Cambridge University Press, New York.
- 70.Lu, X., and W. Olson. 2003. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 31:5108–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kulinski, T., M. Olejniczak, H. Huthoff, L. Bielecki, K. Pachulska-Wieczorek, A. Das, B. Berkhout, and R. Adamiak. 2003. The apical loop of the HIV-1 TAR RNA hairpin is stabilized by a cross-loop base pair. J. Biol. Chem. 278:38892–38901. [DOI] [PubMed] [Google Scholar]
- 72.Bailor, M., C. Musselman, A. Hansen, K. Gulati, D. Patel, and H. Al-Hashimi. 2007. Characterizing the relative orientation and dynamics of RNA A-form helices using NMR residual dipolar couplings. In press. [DOI] [PMC free article] [PubMed]
- 73.Hagerman, P. 1997. Flexibility of RNA. Annu. Rev. Biophys. Biomol. Struct. 26:139–156. [DOI] [PubMed] [Google Scholar]
- 74.Perez, A., A. Noy, F. Lankaš, F. Luque, and M. Orozco. 2004. The relative flexibility of B-DNA and A-RNA duplexes: database analysis. Nucleic Acids Res. 32:6144–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van Wynsberghe, A., and Q. Cui. 2005. Comparison of mode analyses at different resolutions applied to nucleic acid systems. Biophys. J. 89:2939–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huthoff, H., F. Girard, S. Wijmenga, and B. Berkhout. 2004. Evidence for a base triple in the free HIV-1 TAR RNA. RNA. 10:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edwards, T., and A. Ferré-D'Amaré. 2006. Crystal structures of the Thi-box riboswitch bound to thiamine pyrophosphate analogs reveal adaptive RNA-small molecule recognition. Structure. 14:1459–1468. [DOI] [PubMed] [Google Scholar]
- 78.Montange, R., and R. Batey. 2006. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 441:1172–1175. [DOI] [PubMed] [Google Scholar]
- 79.Serganov, A., Y. Yuan, O. Pikovskaya, A. Polonskaia, L. Malinina, A. Phan, C. Hobartner, R. Micura, R. Breaker, and D. Patel. 2004. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 11:1729–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Batey, R., S. Gilbert, and R. Montange. 2004. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 432:411–415. [DOI] [PubMed] [Google Scholar]
- 81.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.