

Abstract
The pore-forming subunit of the large-conductance Ca2+-dependent K+ (Slo1) channel is encoded by one gene. However, the functional properties of Slo1 channels are diverse in part because of their numerous regulatory mechanisms including posttranslational modification and alternative splicing. In particular, multiple splice variants of the pore-forming subunit have been reported but their significance is only beginning to be elucidated. Here we examined the cell biological properties of the three common C-terminal isoforms that differ in the last 8 (Slo1_ERL and Slo1_VYR) or 61 residues (Slo1_DEC). We found that Slo1_DEC, the longest isoform, shows dramatically reduced surface expression compared to that of Slo1_ERL or Slo1_VYR. Immunocytochemistry revealed that a large fraction of Slo1_DEC remains localized in endoplasmic reticulum (ER). Using a GST fusion protein containing the Slo1_DEC-specific sequence, affinity purification was carried out to isolate interacting proteins. The identified proteins include protein phosphatases 2A (PP2A-A), , actin, and tubulin. The PP2A-A interaction is specific to Slo1_DEC and causes a significant reduction of phosphorylation in Slo1_DEC but not Slo1_ERL or Slo1_VYR. The results together support the notion that Slo1_DEC nucleates isoform-specific protein complexes and possesses a cis element(s) for regulating trafficking of the Slo1 channels.
INTRODUCTION
Large-conductance Ca2+-dependent K+ channels (also known as Slo1, Maxi-K, BK and KCa1.1 channels), a member of the voltage-gated K+ channel superfamily [1,2], are activated by membrane depolarization and elevated intracellular [Ca2+] in an allosteric manner [3,4]. This dualistic activation of BK channels is exemplified in vascular smooth muscle cells [5] and also in neurons [6] where the channels open to hyperpolarize the cell membrane in response to a local increase in intracellular [Ca2+], thus providing a negative feedback influence on cellular excitability.
Functional properties of native Slo1/BK channels are diverse, such that their properties in different tissues or even neighboring cells may be markedly different [7–14]. This remarkable functional diversity arises by multiple molecular mechanisms, including association with auxiliary subunits and posttranslation modification, such as phosphorylation, and extensive alternative splicing [15,16]. The enzymatic modifications of Slo1/BK channel, including those by phosphorylation, are particularly prominent [17,18], but the specific motifs to anchor enzymatic assembly are largely unknown.
A functional Slo1 channel is composed of four pore-forming subunits each of which is encoded by a single gene (KCNMA1) [19,20]. The gene produces Slo1 proteins with seven putative transmembrane segments, S0 through S6, in addition to a large C-terminal cytoplasmic domain with divalent cation binding sites [21]. The C-terminus is in turn considered to contain two modules important for the channel gating, RCK1 (regulator of conductance for K+) and RCK2 [22]. Within this topological organization of Slo1, several alternative splicing sites have been reported [13,15,16]. A splice insertion in the cytoplasmic S0–S1 linker segment, referred to as SV1 (or n1 [13]), influences trafficking of the protein to the surface membrane [23,24] and also some aspects of the channel gating [25]. The potential interaction between the identified motif and cellular proteins remains a subject of further investigation. The cytoplasmic C-terminal domain also contains multiple splice sites that profoundly influence the gating properties of the channel [15,26–28]. For example, insertion of a cysteine-rich sequence (“STREX”) between RCK1 and RCK2 at the c2 site regulated in part by stress hormones [28], alters the channel gating and its modulation by phosphorylation [29]. In addition to the splice sites in the core of the cytoplasmic region, the extreme C-terminus of Slo1 also harbors a splice site (Figure 1). The amino-acid sequence difference at the C-terminus caused by splicing at this site was apparent when the full length cDNAs for mouse and human Slo1 were first reported [15,16,30,31]. However, the functional significance of different C-termini as a result of this alternative splicing remains unknown.
Figure 1.
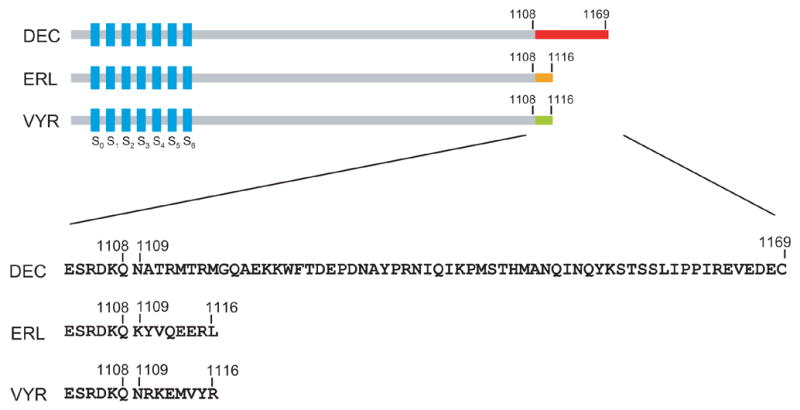
Schematic diagram of three common isoforms differing at the C-terminal sequences as indicated.
As a first step toward elucidating the biological significance of splicing at the extreme C-terminus, we expressed three C-terminal variants, Slo1_DEC, Slo1_ERL and Slo1_VYR (Figure 1), in heterologous expression systems and found that the isoforms differ markedly in surface expression. Further proteomic analyses have yielded a list of candidate interacting proteins, setting the stage for future investigations to the hypothesis that these cis- and trans- elements confer the differences in both trafficking and biophysical properties of these splicing variants.
RESULTS
The three C-terminal splice variants differ in the extreme terminal residues, hence we termed Slo1_DEC, Slo1_ERL and Slo1_VYR. The Slo1_DEC isoform is the longest with 61 residues distinct from the other two isoforms. The Slo1_ERL and Slo1_VYR isoforms have identical length but differ at the last eight residues (Figure 1). As expected, these isoforms form electrophysiologically functional channels activated by depolarization and Ca2+ (Figure 2A) when heterologously expressed. To determine the cell biological significance of the C-terminal residues, we measured their whole-cell macroscopic currents. Peak currents were normalized to the cell capacitance to estimate the relative electrophysiological expression efficiencies of the isoforms (Experimental Procedures). Furthermore, in each transfection batch, the current densities of the Slo1_DEC and Slo1_VYR are normalized to the average current density of the commonly studied Slo1_ERL isoform. Figure 2B compares the relative electrophysiological expression levels measured at 24–26 h after transfection at 200 mV where open probability is essentially saturated. The recording solutions contained 0 Ca2+ to avoid confounding with any differential Ca2+ sensitivity among the isoforms. It is clear that transfection with the three isoform DNAs produced markedly different current densities.
Figure 2.
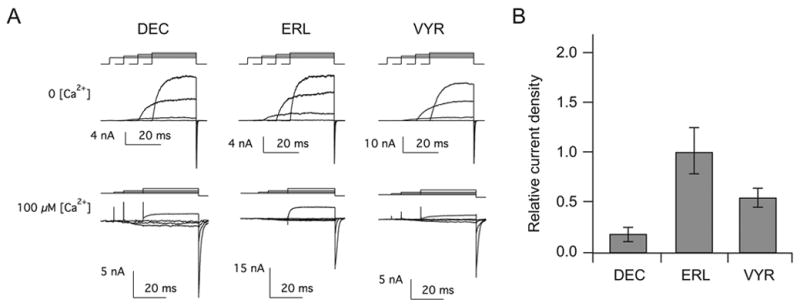
Differential functional expression of Slo1 isoforms. A. Representative macroscopic currents recorded in the inside-out configuration in the virtual absence of Ca2+ at 80, 120, 160, and 100 mV (top) and at 100 μM [Ca2+] at −80, −50, −20, and 40 mV (bottom). B. Histogram shows the relative current densities at 200 mV for the three isoforms expressed in HEK293 cells and measured by voltage-clamp recording using 20 mM K+ in the virtual absence of Ca2+ as outlined in Experimental Procedures. n=8 – 12. ANOVA P<0.009.
To test whether the differential current density is caused by actual different levels of protein expression on the cell surface, we fused mycepitope to the N-termini of the three isoforms. Such protein tags do not induce any detectable effects on surface trafficking [23,24]. The surface expression of the fusion proteins were measured by live cell staining using anti-myc antibody followed by flow cytometry (Figure 3A). Both Slo1_ERL and Slo1_VYR showed considerably higher surface fluorescence signals than Slo1_DEC. Furthermore, immunoprecipitation experiments of the surfaced Slo1 channel proteins were carried out to compare the relative surface expression of the isoforms (Experimental Procedure). The surface protein levels for Slo1_ERL and Slo1_VYR are more abundant than that of Slo1_DEC, even though the total proteins of the three isoforms are comparable (Figure 3B), indicative that the differential surface expression was not caused by difference in total protein production.
Figure 3.
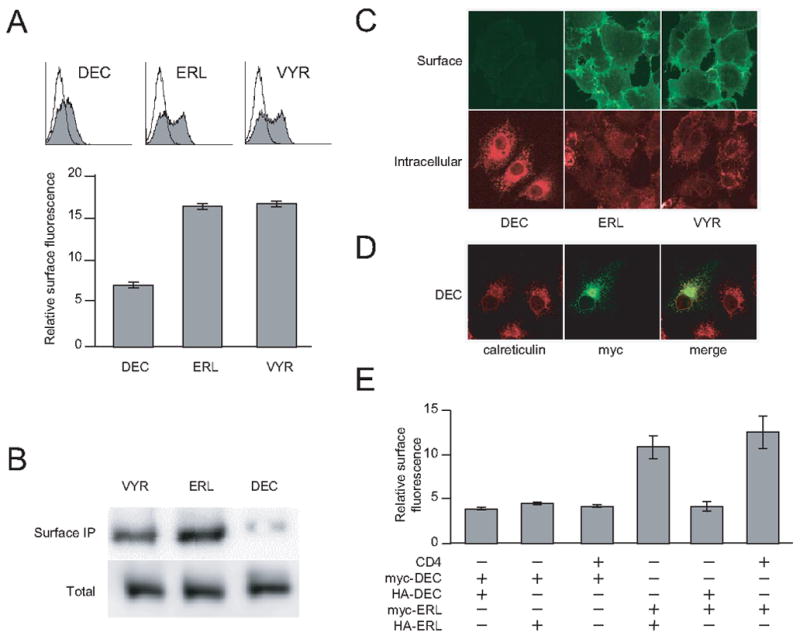
Differential surface expression of Slo1 isoforms. A. Histogram of flow cytometry analyses of myc-tagged Slo1 isoforms that were expressed in COS7 cells (upper panels). Surface expression was quantified using geometric mean fluorescence and plotted as relative surface expression levels (lower panel). B. Immunoprecipitation of Slo1 proteins on cell surface (upper panel) and in total cell lysates (lower panel). Indicated isoforms of Slo1 channel proteins were immunopreciated by anti-myc antibody and blotted by anti-Slo1 antibody. C. Immunofluorescent imaging of cells labeled with anti-myc antibody. COS7 cells were transfected with three myc-Slo1 isoforms. The transfected cells were labeled with anti-myc antibody without detergent treatment (upper panels) or after detergent treatment (lower panels). D. Confocal imaging of Slo1_DEC proteins. COS7 cells were transfected with Slo1_DEC and stained with both mouse anti-myc antibody and rabbit anti-calreticulin antibody. The corresponding secondary antibodies labeled with Alexa Fluor 488 (Slo1) or Alexa Fluor 568 (calreticulin) were then applied to visualize the primary antibodies. Their images were then merged (right). E. Surface expression of Slo1 heteromultimers formed by different isoforms. Indicated combinations of CD4, myc-Slo1_DEC, HA-Slo1_DEC, myc-Slo1_ERL and HA-Slo1_ERL constructs were transfected in COS7 cells. The surface expression for each transfection was monitored by anti-myc antibody and quantified according to the geometric mean fluorescence of flow cytometry histograms.
To directly visualize the subcellular localization of the three isoforms, transfected COS7 cells were stained with anti-myc antibody. Figure 3C (upper panels) reveals significant surface signals for both the ERL and VYR isoforms but not the DEC isoform. In contrast, when the cells were stained after membrane permeabilization by detergent treatment, considerable antibody binding signals were found intracellularly for Slo1_DEC (Figure 3C, lower panel). Further analyses by double stain with both anti-myc and anti-calreticulin revealed that intracellularly localized Slo1_DEC overlapped noticeably with signals of calreticulin, a well known ER protein (Figure 3D), providing the evidence that the Slo1_DEC is mainly localized in ER.
Slo1_DEC forms functional homomeric channels (Figure 2) and there is evidence for overlapping expression of C-terminal isoforms [32]. To determine the surface expression level upon interaction with other isoforms, we coexpressed Slo1_DEC with Slo1_ERL or control membrane protein CD4. Figure 3E summarizes the anti-myc staining intensities with different co-expression combinations. Indeed, co-expression of Slo1_DEC with Slo1_ERL caused a significant reduction in surface expression measured by either the level of myc-Slo1_DEC or myc-Slo1_ERL. This observation provides the evidence that the isoforms are capable of interacting with each other and that the C-terminal region specific for Slo1_DEC isoform has dominant suppressive activity on channel surface expression.
To determine whether there is a specific motif responsible for the trafficking behavior, we constructed a series of Slo1_DEC deletion mutants (Figure 4, left panel). The surface expression of these mutants was then measured by flow cytometry. The geometric mean fluorescence was used to quantify the relative surface expression (Figure 4, right panel). Intriguingly, the progressive deletion gradually increased the surface expression. The complete removal of DEC-specific region, spanning from aa 1109 to 1169, allowed for a complete recovery of surface expression to a level similar to that of Slo1_ERL or Slo1_VYR (Figure 4, right panel).
Figure 4.
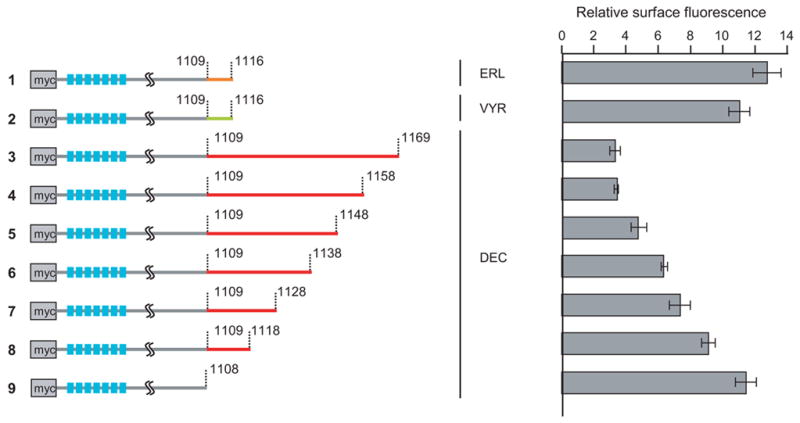
Deletion analyses of Slo1_DEC isoform. Individual deletions with indicated amino acid positions were constructed (left panel) and expressed in COS7 cells. The indicated constructs were then tested for the surface expression using anti-myc antibody. Quadruplicate experiments were performed and quantified according to the geometric mean fluorescence of flow cytometry histograms (right panel).
Slo1_DEC isoform may differ in other aspects in addition to slower surface expression, we constructed a GST-DEC fusion protein spanning from aa1056 to aa1169 (GST-DEC1056/1169). This covers the entire Slo1_DEC-specific region and a fragment of upstream sequence shared among the three isoforms. Using affinity purification, brain proteins specific for GST-DEC1056/1169 but not GST alone have been purified (Figure 5A). Several candidate interacting proteins have been identified by matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) (see Experimental Procedures; Table 1).
Figure 5.
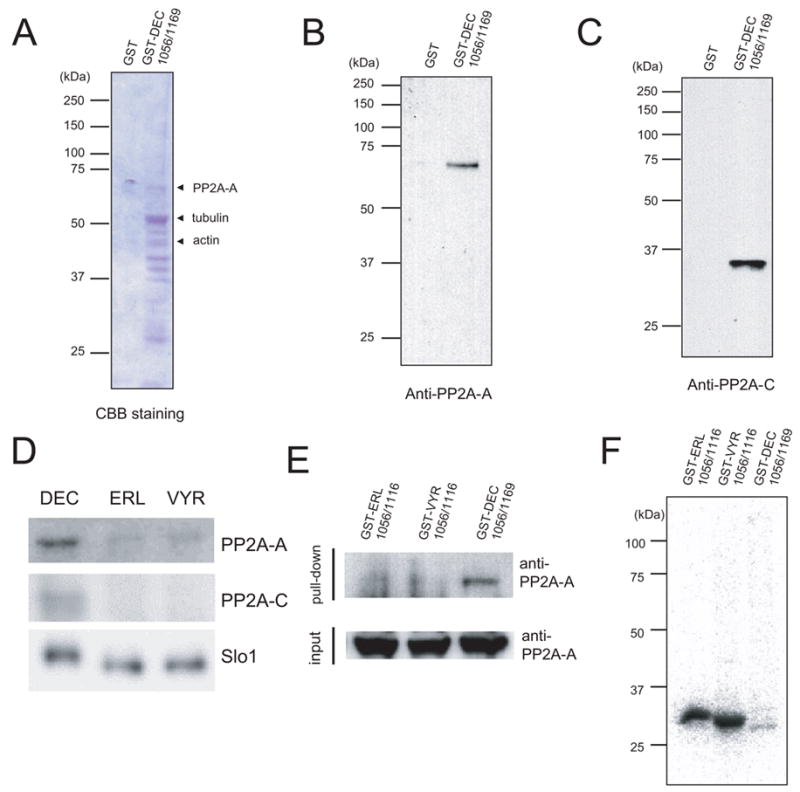
Proteins interacting with the C-terminal domain of Slo1_DEC. A. Commassie stain image of affinity-purified proteins interacting with GST-DEC 1056/1169 fusion. Indicated proteins are positively identified by MALDI-TOF analyses (Table 1). B & C. Affinity pull-down proteins by either GST or GST-DEC 1056/1169 were immunobloted with anti-PP2A-A (B) or anti-PP2A-C antibodies (C). D. Immunoprecipitation of full-length myc-Slo1 isoforms. The immunoprecipitated materials were probed with anti-PP2A-A (upper), anti-PP2A-C (middle) and anti-myc (lower) antibodies. E & F. Metabolic labeling of GST fusion proteins expressed in COS7 cells. The indicated GST fusion proteins were expressed in COS7 cells and metabolically labeled with 32P orthophosphate (Experimental Procedures). The GST fusion proteins were precipitated by glutathione sepharose and fractionated by SDS-PAGE. Bound materials were probed with anti-PP2A-A (E, upper panel) and the input protein lysates of transfected cells were probed with anti-PP2A-A antibody (E, lower panel). The metabolically labeled GST proteins in each transfection were monitored by autoradiography after eluted from the glutathione sepharose (F). Different fusion constructs are shown on the top of each panel. Molecular weight standards in kDa are as indicated. All experiments have been replicated at least three times.
Table I .
Proteomic identification of BK_DEC associated proteins
The positively identified proteins included protein phosphatase 2A A subunit (PP2A-A), and cytoskeletal proteins (both actin and tubulin). Table 2 lists all the digested peptides that match the theoretical tryptic peptides calculated for the PP2A-A. The estimated molecular weight of PP2A-A is around 65.3 kDa. The core enzyme of the protein phosphatase 2A is a dimer, which consists of a 65 kDa structural subunit and a 36 kDa catalytic subunit (PP2A-C). To biochemically validate the mass spectral analysis and detect the existence of the catalytic subunit, the eluates from both GST and GST-DEC (1056–1169) affinity columns were immunoblotted by the anti-PP2A-A and anti-PP2A-C antibodies. From the immunoblot analysis, it is clear that both PP2A-A and PP2A-C subunits were only present in the eluate of GST-DEC (1056–1169) affinity column, but absent in the eluate of GST column (Figure 5, B & C). The preferential PP2A-A binding was further determined by coimmunoprecipitation experiments using full length Slo1 α-subunit. Figure 5D shows immunoblots of anti-myc precipitates probed with antibodies for PP2A-A or PP2A-C subunits. While the precipitated Slo1 isoforms were comparable, PP2A-A signal was considerably stronger in the Slo1_DEC lane. To test for any effect of PP2A association on the Slo1 channel proteins, we expressed GST-fusion proteins for all three isoforms in COS7 cells and performed metabolic labeling using [32P]-orthophosphate. The 32P level of the GST-DEC fusion protein is remarkably lower than the other two GST fusion proteins, GST-ERL and GST-VYR (Figure 5F). This finding is well correlated with PP2A-A binding (Figure 5E). Together, these results support the notion that the C-terminal domain of Slo1_DEC is capable of recruiting a large protein complex with posttranslational enzymatic activities.
Table II .
PP2A-A peptides identified from the MALDI-TOF mass spectrometry
| Measured mass | Matching mass | Δ Mass (p.p.m.) | Position | Missed cleavage | Peptide |
|---|---|---|---|---|---|
| 873.4056 | 873.4430 | −43 | 22–28 | 0 | NEDVQLR |
| 929.4519 | 929.5308 | −85 | 195–202 | 0 | VLELDNVK |
| 1109.4708 | 1109.5380 | −61 | 135–144 | 0 | LAGGDWFTSR |
| 1160.5555 | 1160.5799 | −21 | 567–576 | 0 | LTQDQDVDVK |
| 1165.5657 | 1165.6370 | −61 | 476–485 | 0 | EWAHATIIPK |
| 1242.6527 | 1242.7422 | −72 | 35–46 | 0 | LSTIALALGVER |
| 1376.6373 | 1376.7248 | −64 | 281–292 | 0 | TDLVPAFQNLMK |
| 1472.6996 | 1472.7320 | −22 | 486–498 | 0 | VLAMSGDPNYLHR |
| 1472.6996 | 1472.7459 | −31 | 261–272 | 0 | YMVADKFTELQK |
| 2137.9544 | 2138.0076 | −25 | 203–221 | 0 | SEIIPMFSNLASDEQDSVR |
| 2194.1911 | 2194.2335 | −19 | 547–566 | 0 | IGPILDNSTLQSEVKPILEK |
| 2213.0960 | 2213.1355 | −18 | 114–133 | 0 | AISHEHSPSDLEAHFVPLVK |
DISCUSSION
The difference in primary sequence at the extreme C-terminus for cloned Slo1 channels was immediately noticed after the initial cloning. In this report, we have provided evidence that Slo1_DEC, the longest form, displays considerably lower but functional surface expression. Further investigation showed that a large fraction, but not all, of the Slo1_DEC isoform is localized in ER, consistent with the idea that this isoform has an ER localization activity or confer an overall reduction in forward transport activity. Furthermore, the strong ER localization behavior of the Slo1_DEC isoform is “dominant” when it is co-assembled with the normally high-expressing Slo1_ERL isoform.
The mechanism by the DEC-specific amino-acid residues at the extreme C-terminus promotes retention of the channel in ER is not entirely clear. However, our deletion analysis suggests that the ER retention propensity is approximately proportional to the number of the DEC-specific residues present at the C-terminus in an apparently graded manner. As few as 10 DEC-specific residues at the C-terminus can appreciably interfere with surface expression (Figure 4), consistent with a high degree of sequence specificity. ER localization signals are typically small peptide motifs interacting with COPI complex [33,34]. Intriguingly, our deletion analyses instead suggest the DEC-specific residues serve the ER localization in an apparently graded manner (Figure 4). One possibility causing the DEC-specific behavior is the formation of a unique protein complex. The potential causal effect by assembled enzymes requires thorough investigation.
The DEC C-terminus is capable of intimately interacting with actin, tubulin, and PP2A-A, as revealed by our affinity purification and proteomics analyses. The DEC C-terminus thus acts to nucleate this large protein complex involving multiple polypeptides. The isoform-specific association of the PP2A with Slo1_DEC raises the intriguing possibility that the enzymatic activity in one subunit in a tetrameric channel may regulate its own state of phosphorlylation as well as those of the neighboring subunits. This intra- and inter-subunit regulation may then contribute to the isoform specificity including the channel surface expression. Phosphorylation-dependent surface expression of proteins represents a new emerging concept in the cell biology of ion channel regulation. Recent examples include activity-dependent surface expression mediated by 14-3-3 [35]. Whether PP2A-mediated reduction of phosphorylation causes the lower surface expression of the Slo1_DEC is an interesting subject of future investigation.
While the overall expression level is low, some Slo1_DEC channels along with the interacting protein partners are indeed found in the plasma membrane as a macromolecular signaling complex. Indeed, Slo1 channels function as a large macromolecular complex [36]. Functional interactions with nucleotide-dependent protein kinase and phosphatase activities have been reported and that the enzymatic activities regulate functional properties of the channel complex allows local and spatially directed intracellular signaling [17,18]. Thus, functional properties of a Slo1_DEC complex, such as the allosteric voltage- and Ca2+-dependent gating [37], may be regulated by PP2-A present within the same complex.. Whether the Slo1_DEC channel complex is regulated by PP2A in concert with other reported kinase activities waits further investigation. Their identity and validation of interactions are under way. The purified actin and tubulin are consistent with the recent reports [38–40]. Accumulating evidence indicates that Slo1 channel macromolecular complexes may involve not only enzymes and cytoskeletal elements but also other ion channel and receptor proteins [6,41]. Thus, that enzymatic components in the DEC isoform influence the neighboring channels or receptors within one macromolecular complex is an interesting possibility.
In summary, our results demonstrate that the Slo1 channel variants produced by alternative splicing differing in the extreme C-terminal sequence show different trafficking properties. Furthermore, the Slo1 channel variants form distinct macromolecular signaling complexes with different phosphorylation and dephosphorylation capabilities, uncovering additional layers of modulatory potential and diversity of Slo1 channels. Alternative splicing of the Slo1 channel pre-mRNA is plastic and regulated by specific hormone stimulation or during different stages of pregnancy [28,42,43]. Thus, it would be interesting to see if the splicing at the C-terminus is subject to regulation in vivo.
EXPERIMENTAL PROCEDURES
Plasmid construction
Slo1_DEC, which is the mbr5 clone of mouse brain mslo1 [15], was obtained from Dr. Jianmin Cui (Washington University, St Louis, MO). The other two isoforms, Slo1_ERL and Slo1_VYR, were constructed by PCR amplification and mutagenesis using mbr5 clone as a template. For expression of full-length Slo1 channel protein or their partial C-terminal fragments in mammalian cells, the inserts were cloned at SalI and NotI sites into cytomegalovirus (CMV) promoter-based expression vectors with N-terminal tag of myc, HA, and GST. For bacterial GST fusion proteins, pGEX4T2 vector (GE Healthcare, Piscataway, NJ) was used to clone Slo1 inserts at SalI and NotI.
Cell culture and transfection
COS7 and HEK293 cells were cultured in 50%DMEM/50%F12 medium supplemented with 10% fetal bovine serum, L-glutamine and penicillin-streptomycin. The plasmid DNA was transfected using FuGene6 (Roche, Indianapolis, IN) according to the manufacturer’s instruction.
Flow cytometry and quantification of surface expression level of Slo1 channel
COS7 cells cultured in 6-well plates were transfected with 1 μg of plasmids. At 36 h after transfection, the cells were harvested by incubation in phosphate-buffered saline (PBS, pH 7.3) containing 0.5 mM ethylenediaminetetraacetic acid (EDTA) for 10 min at 37°C. After washing with Hanks' balanced salt solution (HBSS) supplemented with 2% FBS (staining medium), the cells were incubated with mouse anti-myc monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 5 μg/ml in staining medium for 30 min on ice. After washing three times, the cells were incubated for 10 min with Alexa Fluor 488-conjugated goat anti-mouse antibody (Invitrogen, Carlsbad, CA). After washing the cells three times, the surface expression of Slo1 channel proteins was measured by FACSCalibur (BD Biosciences, Rockville, MD) with CELLQUEST software (BD Biosciences).
Immunocytochemistry for Slo1 channel localization
COS-7 cells transfected with Slo1 constructs were cultured on cover slips for 24 h. To first stain the surface-localized Slo1 channel proteins, the cells were incubated with anti-myc antibody for 1 h at 4°C. After wash, the cells were fixed with 4% paraformaldehyde for 15 min at 4°C and then permeabilized with 0.1% Triton X-100 for 5 min. The permeabilized cells were labeled with rabbit polyclonal anti-myc antibody (Santa Cruz Biotechnology) for 1 h at 4°C, followed by labeling with Alexa Fluor 488-goat anti-mouse antibody and Cy3-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 30 min to detect surface and intracellular Slo1 proteins, respectively. The Slo1_DEC protein was further examined for subcellular localization. The transfected COS7 cells were fixed and permeabilized as described above. The cells were stained with both mouse anti-myc antibody and rabbit anti-calreticulin (Affinity Bioreagent, Golden, CO) and then with corresponding secondary antibodies conjugated with Alexa Fluor 488 (Slo1) or Alexa Fluor 568 (calreticulin). The staining signal was visualized with Ultraview confocal microscope (Carl Zeiss, Thornwood, NY).
In vivo phosphorylation assay of Slo1 C-terminal proteins
HEK293 cells transfected with GST fusion constructs were re-plated on 6-well plates 1 day after the transfection. On the following day, after incubation in serum- and phosphate-free medium for 1 h, the cells were labeled with 0.3 mCi/ml of [32P]orthophosphate (ICN Biomedicals, Irvine, CA) for 2 h. The cells were then lysed in cell lysis buffer (1% Triton X-100, 25 mM HEPES, pH 7.5, 10% glycerol, 50 mM KCl, and 1x protease inhibitor cocktail (Sigma, St. Louis, MO)) and the GST fusion proteins were pulled down by the glutathione sepharose beads. The pull-down protein samples were fractionated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane. The phosphorylated proteins were then detected by phospho-imaging analysis. The association of PP2A core enzyme was detected by immunoblotting using goat polyclonal anti-PP2A-A (Santa Cruz Biotechnology) and mouse monoclonal anti-PP2A-C (BD Biosciences) followed by corresponding secondary antibodies supplied with SuperSignal chemiluminescence reagent (Pierce Chemical, Rockford, IL).
The amount of pull-down GST fusion proteins was determined by Ponceau S staining or immunoblotting with horse radish peroxidase (HRP)-conjugated anti-GST polyclonal antibody (GE Healthcare).
Immunoprecipitation of myc-tagged Slo1 proteins
The cells transfected with full-length myc-Slo1 constructs were washed with PBS once and lysed with lysis buffer for 20 min at 4°C. After centrifuge for 20 min at 11,000 x g, the supernatant was mixed with Protein G-conjugated agarose beads which were pre-incubated with 1 μg of myc antibody. After 2 h incubation, the beads were washed 3 times with lysis buffer, boiled with 2X SDS sample buffer for 5 min. The eluates were resolved by SDS-PAGE, transferred to the nitrocellulose membrane, and then immunoblotted by rabbit polyclonal anti-Slo1 antibody (Alomone Labs, Jerusalem, Israel) or anti-PP2A antibodies followed by corresponding secondary antibodies as described for GST fusion proteins.
For immunoprecipitation of cell surface Slo1 proteins, the transfected cells were first labeled with anti-myc antibody for 1 h at 4°C. After extensive wash with staining medium, the cells were lysed with lysis buffer and then the centrifuged supernatant was incubated with Protein G beads for 2 h. After wash, the eluates were resolved by SDS-PAGE and immunoblotted with anti-Slo1 antibody. In the parallel experiments, the cell lysates were made from myc-Slo1-transfected cells and the centrifuge supernatants were immunoprecipitated to quantify total Slo1 channel proteins.
Purification of bacterial GST-fused Slo1 proteins
The BL21 E. coli carrying GST or GST fusion constructs were induced for protein expression by isopropylthiogalactoside (IPTG). The bacterial cells were sonicated in PBS on ice, and then incubated in 1% Triton X-100 for 30 min. The lysate was centrifuged at 15,000 x g for 20 min at 4°C and the supernatant was incubated with glutathione sepharose for 30 min. After washing three times with PBS, the beads were resuspended as 50% slurry in PBS.
Affinity purification of Slo1_DEC channel C-terminus-associated proteins
Four grams of mouse brains were homogenized on ice with a glass homogenizer in 10 ml of Buffer A (20 mM HEPES-KOH (pH 7.5), 25mM KCl, 1x protease inhibitor cocktail, 0.5mM dithiothreitol, 10% sucrose). The homogenate was centrifuged at 500 x g for 10 min at 4°C and the supernatant was mixed with equal volume of solubilization buffer (Buffer A added with 4M NaCl) for 1 h at 4°C to extract any soluble proteins. After centrifuge at 27,000 x g for 15 min at 4°C to remove insoluble matters. The supernatant was dialyzed against buffer A without sucrose for three times overnight and then centrifuged at 27,000 x g for 20 min to remove any insoluble matters. The supernatant was pre-cleared twice with glutathione sepharose resin to remove any non-specific binding proteins. The pre-cleared mouse brain lysate samples (equivalent of 20 mg proteins) were subjected to affinity purification with control GST or GST-fused Slo1 proteins that were bound to glutathione sepharose. After incubation with brain samples for 2 h at 4°C, the resins were washed extensively with buffer A. The associated proteins were eluted with 500 mM NaCl in buffer A. The eluates were resolved by SDS-PAGE and visualized by Commassie Brilliant Blue (CBB) staining.
Protein identification by MALDI-TOF mass spectrometry
Proteins of interest were excised from CBB-stained gels and digested in gel using trypsin (sequencing grade, Promega, Madison, WI), as previously described [44]. Digest products were completely dehydrated in a vacuum centrifuge and resuspended in 10 μl formic acid (2% v/v), desalted using Zip Tips C18 (Millipore, Bedford, MA), eluted with 2 μl acetonitrile:trifluoroacetic acid, (80:0.1%). The extracted peptides were mixed with the 2,5-dihydroxybenozic acid (10 mg/ml in acetonitrile:trifluoroacetic acid, 50:0.1%) and loaded on the sample plate of the MALDI-TOF mass spectrometer. Analysis was performed on the Voyager DE-STR MALDI-TOF in the Johns Hopkins protein core facility. Mass spectra were acquired as the sum of the ion signals that were generated by irradiation of the sample with a mean of 300 laser pulses. Spectra were analyzed using the Data Explorer software (Applied Biosystems, Foster City, CA). Peptides were selected in the mass range of 800–3500 Da. Identification of proteins was performed by using Mascot software (Matrix Science Inc, Boston, MA). A mass deviation of 100 ppm was allowed for data base interrogation.
Electrophysiology
The three isoforms were transiently expressed in HEK293 cells as described previously [45] and the whole-cell currents were recorded at 24 to 26 h later with an AxoPatch 200B amplifier (Molecular Devices, Sunnyvale, CA). The extracellular solution contained (in mM): 20 KCl, 120 N-methyl-D-glucamine (NMDG), 2 MgCl2, 10 HEPES, pH 7.2. The intracellular (pipette) solution contained (in mM): 20 KCl, 120 NMDG, 2 MgCl2, 11 EGTA, 10 HEPES, pH 7.2. The K+ concentration was lowered to 20 mM to limit the peak current size. Cell capacitance was estimated using 25 mV depolarization pulses. Inside-out macroscopic currents were recorded with different size electrodes to obtain <20 nA currents at 200 mV. The solutions used in the inside-out experiments contained 140 KCl replacing 20 KCl and 120 NMDG. The 100 μM Ca2+ results were obtained using a solution without any chelator. The results obtained in the virtual absence of Ca2+ were corrected for capacitative and leak currents using a P/6 protocol and the high Ca2+ results were not corrected for leak or capacitative currents.
Acknowledgments
We thank our lab members for valuable comments and discussions on this manuscript. The work is supported by grants from the National Institutes of Health (GM70959 to M.L.) and (GM57654 to T.H.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–53. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- 2.Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–9. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 3.Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. Journal of General Physiology. 2002;120:267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marty A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature. 1981;291:497–500. doi: 10.1038/291497a0. [DOI] [PubMed] [Google Scholar]
- 5.Nelson MT, et al. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–7. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 6.Berkefeld H, et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–20. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- 7.Brenner R, et al. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–6. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 8.Du W, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–8. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- 9.Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem. 2004;279:36746–52. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- 10.Sausbier M, et al. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A. 2004;101:9474–8. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Werner ME, Zvara P, Meredith AL, Aldrich RW, Nelson MT. Erectile dysfunction in mice lacking the large-conductance calcium-activated potassium (BK) channel. J Physiol. 2005;567:545–56. doi: 10.1113/jphysiol.2005.093823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, et al. Functionally diverse complement of large conductance calcium- and voltage-activated potassium channel (BK) alpha-subunits generated from a single site of splicing. J Biol Chem. 2005;280:33599–609. doi: 10.1074/jbc.M505383200. [DOI] [PubMed] [Google Scholar]
- 13.Shipston MJ. Alternative splicing of potassium channels: a dynamic switch of cellular excitability. Trends Cell Biol. 2001;11:353–8. doi: 10.1016/s0962-8924(01)02068-2. [DOI] [PubMed] [Google Scholar]
- 14.Ricci AJ, Gray-Keller M, Fettiplace R. Tonotopic variations of calcium signalling in turtle auditory hair cells. J Physiol. 2000;524(Pt 2):423–36. doi: 10.1111/j.1469-7793.2000.00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler A, Tsunoda S, McCobb DP, Wei A, Salkoff L. mSlo, a complex mouse gene encoding "maxi" calcium-activated potassium channels. Science. 1993;261:221–4. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- 16.Tseng-Crank J, et al. Cloning, expression, and distribution of functionally distinct Ca(2+)-activated K+ channel isoforms from human brain. Neuron. 1994;13:1315–30. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- 17.Chung SK, Reinhart PH, Martin BL, Brautigan D, Levitan IB. Protein kinase activity closely associated with a reconstituted calcium-activated potassium channel. Science. 1991;253:560–2. doi: 10.1126/science.1857986. [DOI] [PubMed] [Google Scholar]
- 18.Reinhart PH, Chung S, Martin BL, Brautigan DL, Levitan IB. Modulation of calcium-activated potassium channels from rat brain by protein kinase A and phosphatase 2A. J Neurosci. 1991;11:1627–35. doi: 10.1523/JNEUROSCI.11-06-01627.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen KZ, et al. Tetraethylammonium block of Slowpoke calcium-activated potassium channels expressed in Xenopus oocytes: evidence for tetrameric channel formation. Pflugers Arch. 1994;426:440–5. doi: 10.1007/BF00388308. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka Y, Meera P, Song M, Knaus HG, Toro L. Molecular constituents of maxi KCa channels in human coronary smooth muscle: predominant alpha + beta subunit complexes. J Physiol. 1997;502 ( Pt 3):545–57. doi: 10.1111/j.1469-7793.1997.545bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magleby KL. Gating mechanism of BK (Slo1) channels: so near, yet so far. J Gen Physiol. 2003;121:81–96. doi: 10.1085/jgp.20028721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang Y, Pico A, Cadene M, Chait BT, MacKinnon R. Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron. 2001;29:593–601. doi: 10.1016/s0896-6273(01)00236-7. [DOI] [PubMed] [Google Scholar]
- 23.Zarei MM, et al. An endoplasmic reticulum trafficking signal prevents surface expression of a voltage- and Ca2+-activated K+ channel splice variant. Proc Natl Acad Sci U S A. 2004;101:10072–7. doi: 10.1073/pnas.0302919101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarei MM, et al. A novel MaxiK splice variant exhibits dominant-negative properties for surface expression. J Biol Chem. 2001;276:16232–9. doi: 10.1074/jbc.M008852200. [DOI] [PubMed] [Google Scholar]
- 25.Korovkina VP, Fergus DJ, Holdiman AJ, England SK. Characterization of a novel 132-bp exon of the human maxi-K channel. Am J Physiol Cell Physiol. 2001;281:C361–7. doi: 10.1152/ajpcell.2001.281.1.C361. [DOI] [PubMed] [Google Scholar]
- 26.Chen L, et al. Fnctionally diverse complement of calcium-activated BK channel alpha -subunits generated from a single site of splicing. J Biol Chem. 2005 doi: 10.1074/jbc.M505383200. [DOI] [PubMed] [Google Scholar]
- 27.Saito M, Nelson C, Salkoff L, Lingle CJ. A cysteine-rich domain defined by a novel exon in a slo variant in rat adrenal chromaffin cells and PC12 cells. J Biol Chem. 1997;272:11710–7. doi: 10.1074/jbc.272.18.11710. [DOI] [PubMed] [Google Scholar]
- 28.Xie J, McCobb DP. Control of alternative splicing of potassium channels by stress hormones. Science. 1998;280:443–6. doi: 10.1126/science.280.5362.443. [DOI] [PubMed] [Google Scholar]
- 29.Tian L, et al. Alternative splicing switches potassium channel sensitivity to protein phosphorylation. J Biol Chem. 2001;276:7717–20. doi: 10.1074/jbc.C000741200. [DOI] [PubMed] [Google Scholar]
- 30.Dworetzky SI, Trojnacki JT, Gribkoff VK. Cloning and expression of a human large-conductance calcium-activated potassium channel. Brain Res Mol Brain Res. 1994;27:189–93. doi: 10.1016/0169-328x(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 31.Pallanck L, Ganetzky B. Cloning and characterization of human and mouse homologs of the Drosophila calcium-activated potassium channel gene, slowpoke. Hum Mol Genet. 1994;3:1239–43. doi: 10.1093/hmg/3.8.1239. [DOI] [PubMed] [Google Scholar]
- 32.Langer P, Grunder S, Rusch A. Expression of Ca2+-activated BK channel mRNA and its splice variants in the rat cochlea. J Comp Neurol. 2003;455:198–209. doi: 10.1002/cne.10471. [DOI] [PubMed] [Google Scholar]
- 33.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–66. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 34.Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307. [DOI] [PubMed] [Google Scholar]
- 35.Chung JJ, Shikano S, Hanyu Y, Li M. Functional diversity of protein C-termini: more than zipcoding? Trends Cell Biol. 2002;12:146–50. doi: 10.1016/s0962-8924(01)02241-3. [DOI] [PubMed] [Google Scholar]
- 36.Lu R, et al. MaxiK channel partners: physiological impact. J Physiol. 2006;570:65–72. doi: 10.1113/jphysiol.2005.098913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120:267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brainard AM, Miller AJ, Martens JR, England SK. Maxi-K channels localize to caveolae in human myometrium: a role for an actin-channel-caveolin complex in the regulation of myometrial smooth muscle K+ current. Am J Physiol Cell Physiol. 2005;289:C49–57. doi: 10.1152/ajpcell.00399.2004. [DOI] [PubMed] [Google Scholar]
- 39.Douglas RM, et al. The calcium-sensitive large-conductance potassium channel (BK/MAXI K) is present in the inner mitochondrial membrane of rat brain. Neuroscience. 2006;139:1249–61. doi: 10.1016/j.neuroscience.2006.01.061. [DOI] [PubMed] [Google Scholar]
- 40.Misonou H, et al. Immunolocalization of the Ca2+-activated K+ channel Slo1 in axons and nerve terminals of mammalian brain and cultured neurons. J Comp Neurol. 2006;496:289–302. doi: 10.1002/cne.20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu G, et al. Assembly of a Ca2+-dependent BK channel signaling complex by binding to beta2 adrenergic receptor. Embo J. 2004;23:2196–205. doi: 10.1038/sj.emboj.7600228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benkusky NA, Fergus DJ, Zucchero TM, England SK. Regulation of the Ca2+-sensitive domains of the maxi-K channel in the mouse myometrium during gestation. J Biol Chem. 2000;275:27712–9. doi: 10.1074/jbc.M000974200. [DOI] [PubMed] [Google Scholar]
- 43.Eghbali M, Toro L, Stefani E. Diminished surface clustering and increased perinuclear accumulation of large conductance Ca2+-activated K+ channel in mouse myometrium with pregnancy. J Biol Chem. 2003;278:45311–7. doi: 10.1074/jbc.M306564200. [DOI] [PubMed] [Google Scholar]
- 44.Becamel C, et al. A proteomic approach based on peptide affinity chromatography, 2-dimensional electrophoresis and mass spectrometry to identify multiprotein complexes interacting with membrane-bound receptors. Biol Proced Online. 2002;4:94–104. doi: 10.1251/bpo39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santarelli LC, Wassef R, Heinemann SH, Hoshi T. Three methionine residues located within the regulator of conductance for K+ (RCK) domains confer oxidative sensitivity to large-conductance Ca2+-activated K+ channels. J Physiol. 2006;571:329–48. doi: 10.1113/jphysiol.2005.101089. [DOI] [PMC free article] [PubMed] [Google Scholar]