

Abstract
Inhibitors of histone deacetylase (HDAC) proteins such as suberoylanilide hydroxamic acid (SAHA) have emerged as effective therapeutic anti-cancer agents. To better understand the structural requirements of HDAC inhibitors, a small molecule library with a variety of substituents attached adjacent to the metal binding hydroxamic acid of SAHA was synthesized. The presence of a substituent adjacent to the hydroxamic acid led to an 800 to 5000-fold decrease in inhibition compared to SAHA. The observed results have implications for drug design, suggesting that HDAC inhibitors with substituents near the metal binding moiety will have inhibitory activities in the μM rather than nM range.
Suberoylanilide hydroxamic acid (SAHA, Vorinostat, Zolinza™) recently gained FDA approval for the treatment of advanced cutaneous T-cell lymphoma (CTCL).1 SAHA is an inhibitor of histone deacetylase (HDAC) proteins, which are linked to a variety of cancers.2 While SAHA is the first HDAC inhibitor (HDACi) to meet FDA approval, several other small molecules that inhibit HDAC proteins are currently in clinical trials for cancer treatment.3 Distinguishing characteristics of HDAC inhibitors include a metal binding moiety, a carbon linker, and a capping group (Figure 1). Based on crystallographic analyses, the capping group is solvent-exposed and interacts with amino acids near the entrance of the active site, while the metal binding moiety resides in the protein interior and complexes the metal ion involved in catalysis.4–6 The linker serves to position the capping and metal binding groups appropriately for high affinity interactions with proteins. With a modular framework and application towards cancer treatment, HDAC inhibitors are viable targets for future drug design.
Figure 1.
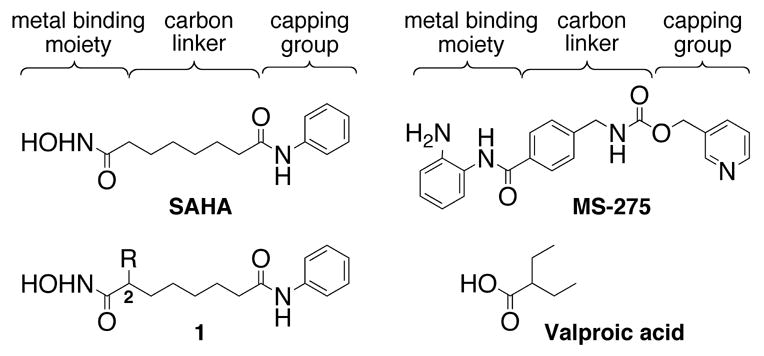
Structures of select HDAC inhibitors.
Previous HDACi design has emphasized modification of the capping group and the metal binding moiety. In the case of the metal binding moiety, SAHA contains a hydroxamic acid while other inhibitors contain thiols, epoxides, carboxylates, or benzamides.7,8 For example, two HDAC inhibitors in clinical trials, MS-275 and valproic acid (Figure 1), contain benzamide and carboxylate metal binding moieties, respectively,9,10 and display IC50 values of 2 μM and 400 μM.11,12 The reduced inhibitory activities compared to SAHA (110–370 nM IC50)13,14 are partially explained by the presence of the benzamide or carboxylic acid group.7,15,16 Interestingly, MS-275 displays modest preference toward select proteins within the eleven member HDAC family.17 Selective HDAC inhibitors would aid in elucidating the role of each individual HDAC protein in cancer and have the potential to be better drugs.18 However, strictly selective HDAC inhibitors have yet to be discovered.
In addition to altering the metal binding moiety towards HDACi design, the hydrocarbon linker has been diversified, focusing on altering chain length, creating points of unsaturation along the chain, and including an aryl or cyclohexyl ring within the chain.19–22 However, few studies have examined the impact of substituents on the linker chain. Recently, small molecules bearing substituents on the linker adjacent to the capping group were shown to not only display nM inhibition, but also modest isoform selectivity.15 In contrast, the incorporation of substituents on the linker adjacent to the metal binding moiety has a variable influence on inhibitory activity. Two reports noted that a methyl substituent near the hydroxamic acid of hydroxamate based libraries led to 120 to 170-fold decreased HDAC inhibition compared to the unsubstituted analog, although the potency was in the μM range.23,24 In addition, the short chain fatty acid valproic acid contains an ethyl group adjacent to the carboxylate and inhibits in the μM range. In contrast, small molecules bearing an intra-chain aryl group near the hydroxamic acids display nM HDAC inhibition.20,21 For example, MS-275 bears an aryl group adjacent to the benzamide group and displays potent HDAC inhibition.11 Therefore, the influence of substituents on the linker adjacent to the hydroxamic acid remains unclear. The structural requirements of HDAC inhibitors, particularly on the linker chain, are an interesting and relatively unexplored area of study. Modification of known HDAC inhibitors is necessary to identify the structural factors influencing inhibitor potency and provide insight for designing new inhibitors.
To probe the structural requirements of HDAC inhibitors, analogs of SAHA with substituents adjacent to the hydroxamic acid were tested. Specifically, we synthesized a small library of SAHA analogues (1) bearing a variety of hydrophobic substituents at the C2 position.25 Hydrophobic substituents were selected because crystallographic analysis of HDAC proteins indicates that the active site residues surrounding the linker chain are hydrophobic.4–6 The synthetic route for the small molecule library is outlined in Scheme 1. ε-Caprolactone (2) was opened with aniline to give anilide alcohol 3. The alcohol was activated prior to incubation with the anion of dimethyl malonate to give diester 4. The diester was deprotonated and treated with a variety of alkyl halides to afford compounds 5a–g. Krapcho type decarboxylation26 and subsequent saponification gave compounds 6a–g. Carboxylic acids 6a–d were converted directly to the hydroxamic acids 1a–d with ethyl chloroformate and a hydroxylamine solution. However, the low yields encouraged utilization of a second strategy where hydroxamic acids 1e–g were synthesized via the benzyl protected hydroxamic acids 7e–g followed by deprotection by either H2/Pd-C for compound 1g, or BCl327 for unsaturated compounds 1e and 1f. Yields after the two-step hydroxamic acid installation/benzyl deprotection were superior to direct conversion (38% and 64% compared to 10–24%). Although reported for benzyl ethers, we note that use of BCl3 to remove a benzyl group on a hydroxamic acid is unestablished to the best of our knowledge.28 A more thorough exploration of the scope and limitations of the BCl3 deprotection reaction is currently under investigation.
Scheme 1.
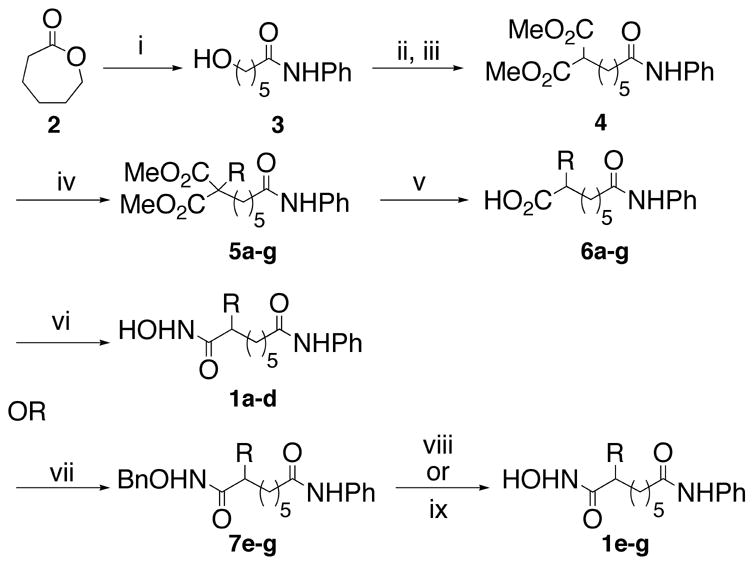
Reagents and conditions for the racemic synthesis of compounds 1a–g: (i) PhNH2, AlMe3, THF, 98%; (ii) MsCl, TEA, CH2Cl2, 99%; (iii) (a) NaH, Dimethylmalonate, THF; (b) mesylate from ii, THF, reflux, 90%; (iv) NaH, RX, THF, reflux, 33–98%; (v) (a) LiCl, H2O, DMSO, reflux; (b) NaOH, MeOH, reflux, 67–83%; (vi) Ethyl chloroformate, N-methylmorpholine, NH2OH, MeOH, 10–24%; (vii) CDI, TEA, NH2OBn, THF, reflux, 75–91%; (viii) H2, Pd/C, MeOH, 48%; (ix) BCl3, CH2Cl2, 84–85%.
HDAC inhibitory activities of the SAHA analogues were measured using the Fluor de Lyse™ in vitro fluorescence activity assay kit.29 IC50 values were obtained by fitting the data to a sigmoidal dose response curve (Figure 2). The structure-activity relationship of compounds 1a–g is summarized in Table 1. All of the SAHA analogs inhibited HDAC activity in the μM range. The butyl variant 1d was the most potent analog displaying an IC50 of 72 μM, while the ethyl variant 1b displayed the weakest inhibitory activity of 449 μM. Interestingly, the analogs containing the smallest (1a-methyl) or the largest (1g- benzyl) substituents displayed HDAC inhibitory activities between those of 1d and 1b. In addition, the propargyl analog 1f inhibited to almost the same level as the butyl analog 1d (87 μM and 72 μM, respectively). The results indicate steric considerations alone cannot predict the inhibitory activities of the C2-substituted analogs.
Figure 2.
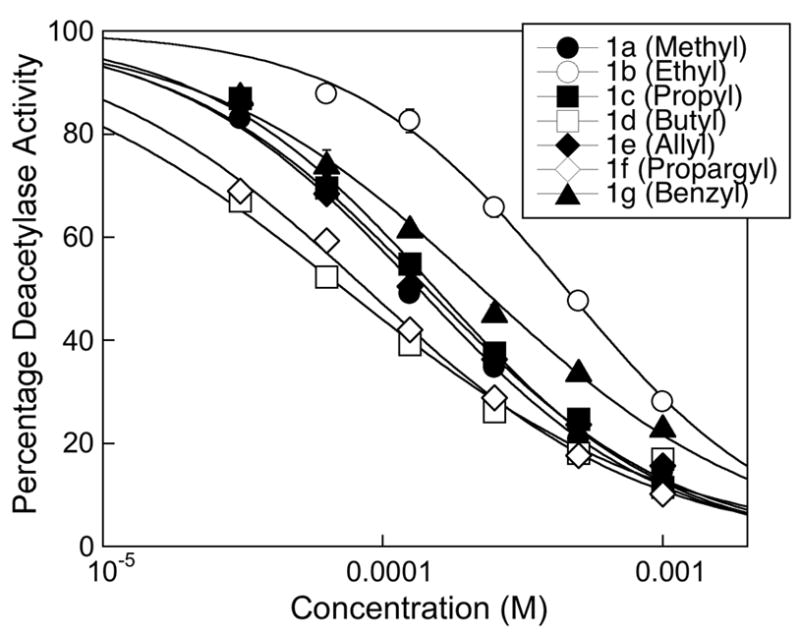
Dose response curves of SAHA analogues 1a–1g from three independent trials with error bars indicating standard error.
Table 1.
HDAC inhibition by compounds 1a–g, SAHA, and MS-275.
| Compounds | R | IC50, μMa |
|---|---|---|
| SAHA | 0.090 (± 0.004) | |
| MS-275 | 3.2 (± 0.1) | |
| 1a | methyl | 134 (± 6) |
| 1b | ethyl | 449 (± 17) |
| 1c | n-propyl | 154 (± 7) |
| 1d | n-butyl | 72 (± 6) |
| 1e | allyl | 144 (± 9) |
| 1f | propargyl | 87 (± 5) |
| 1g | benzyl | 226 (± 11) |
Values are the means of three experiments with standard error given in parentheses.
While the SAHA analogs displayed inhibitory activities in the μM range, all had significantly decreased inhibitory activities when compared to those of SAHA (90 nM) or MS-275 (3.2 μM). The most potent butyl variant 1d demonstrated 800-fold and 20-fold decreased activity compared to SAHA and MS-275, respectively. The weakest ethyl variant 1b displayed 5000-fold and 128-fold decreased inhibition compared to SAHA and MS-275. The results suggest that any group, regardless of size, incorporated adjacent to the hydroxamic acid will result in decreased inhibitory activity compared to the unsubstituted analog.
Several HDAC inhibitors maintain similar nM potency compared to SAHA yet contain a ring within the carbon linker adjacent to the metal binding moiety.20,21 The fact that SAHA analogs with substituents at the C2 position display μM IC50 values indicates that only modest steric bulk near the hydroxamic acid is tolerated for nM inhibitory activity. Therefore, the results suggest that the steric environment near the hydroxamic acid in the HDAC active site is significantly confined. The results have implications for anti-cancer drug design, predicting that HDAC inhibitors with substituents near the hydroxamic acid will have inhibitory activities in the μM range.
SAHA analogues with substituents adjacent to the capping group were potent nM inhibitors.15 In contrast, SAHA analogs with substituents adjacent to the hydroxamic acid demonstrated μM IC50 values. The combined data suggest that substituents are tolerated along the linker chain but potency diminishes when positioned near the metal binding moiety. Because modest isoform selectivity has been reported with HDAC inhibitors bearing substituents along the linker,15,17 a systematic assessment of substituent tolerance along the linker chain will guide future HDACi design. The effect of incorporating substituents at additional positions along the linker chain is currently under investigation.
Acknowledgments
We thank the National Institute of Health (GM067657) and Wayne State University for funding, Derek A. Pflum for advice, and Emily Aubie, Fabiola Bittencourt, Paulina Karwowska-Desaulniers, and Kevin Kells for comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference and Notes
- 1.Grant S, Easley C, Kirkpatrick P. Nat Rev Drug Discov. 2007;6:21. doi: 10.1038/nrd2227. [DOI] [PubMed] [Google Scholar]
- 2.Marks PA, Breslow R. Nat Biotech. 2007;25:84. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 3.Bolden JE, Peart MJ, Johnstone RW. Nat Rev Drug Discov. 2006;5:769. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 4.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Pavletich NP. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 5.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structure. 2004;12:1325. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Proc Natl Acad Sci U S A. 2004;101:15064. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T, Kouketsu A, Matsuura A, Kohara A, Ninomiya S, Kohda K, Miyata N. Bioorg Med Chem Lett. 2004;14:3313. doi: 10.1016/j.bmcl.2004.03.063. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki T, Nagano Y, Kouketsu A, Matsuura A, Maruyama S, Kurotaki M, Nakagawa H, Miyata N. J Med Chem. 2005;48:1019. doi: 10.1021/jm049207j. [DOI] [PubMed] [Google Scholar]
- 9.Atmaca A, Maurer A, Heinzel T, Gottlicher M, Neumann A, Al-Batran SE, Martin E, Bartsch I, Knuth A, Jaeger E. J Clin Oncol (Meeting Abstracts) 2004;22:3169. [Google Scholar]
- 10.Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, Figg WD, Hwang K, Chung EJ, Murgo A, Melillo G, Elsayed Y, Monga M, Kalnitskiy M, Zwiebel J, Sausville EA. J Clin Oncol. 2005;23:3912. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- 11.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. Proc Natl Acad Sci USA. 1999;96:4592. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. J Biol Chem. 2001;276:36734. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 13.Kapustin GV, Fejer G, Gronlund JL, McCafferty DG, Seto E, Etzkorn FA. Org Lett. 2003;5:3053. doi: 10.1021/ol035056n. [DOI] [PubMed] [Google Scholar]
- 14.Gu W, Nusinzon I, Smith JRD, Horvath CM, Silverman RB. Bioorg Med Chem. 2006;14:3320. doi: 10.1016/j.bmc.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 15.Jones P, Altamura S, Chakravarty PK, Cecchetti O, Francesco RD, Gallinari P, Ingenito R, Meinke PT, Petrocchi A, Rowley M. Bioorg Med Chem Lett. 2006;16:5948. doi: 10.1016/j.bmcl.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Park JH, Jung Y, Kim TY, Kim SG, Jong HS, Lee JW, Kim DK, Lee JS, Kim NK, Kim TY, Bang YJ. Clin Cancer Res. 2004;10:5271. doi: 10.1158/1078-0432.CCR-03-0709. [DOI] [PubMed] [Google Scholar]
- 17.Hu E, Dul E, Sung C, Chen Z, Kirkpatrick R, Zhang G, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, De Los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. J Pharmacol Exp Ther. 2003;307:720. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- 18.Karagiannis TC, El-Osta A. Leukemia. 2006;21:61. doi: 10.1038/sj.leu.2404464. [DOI] [PubMed] [Google Scholar]
- 19.Bouchain G, Delorme D. Curr Med Chem. 2003;10:2359. doi: 10.2174/0929867033456585. [DOI] [PubMed] [Google Scholar]
- 20.Nagaoka Y, Maeda T, Kawai Y, Nakashima D, Oikawa T, Shimoke K, Ikeuchi T, Kuwajima H, Uesato S. Eur J Med Chem. 2006;41:697. doi: 10.1016/j.ejmech.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Uesato S, Kitagawa M, Nagaoka Y, Maeda T, Kuwajima H, Yamori T. Bioorg Med Chem Lett. 2002;12:1347. doi: 10.1016/s0960-894x(02)00175-0. [DOI] [PubMed] [Google Scholar]
- 22.Jung M, Brosch G, Kolle D, Scherf H, Gerhauser C, Loidl P. J Med Chem. 1999;42:4669. doi: 10.1021/jm991091h. [DOI] [PubMed] [Google Scholar]
- 23.Woo SH, Frechette S, Khalil EA, Bouchain G, Vaisburg A, Bernstein N, Moradei O, Leit S, Allan M, Fournel M, Trachy-Bourget MC, Li Z, Besterman JM, Delorme D. J Med Chem. 2002;45:2877. doi: 10.1021/jm020154k. [DOI] [PubMed] [Google Scholar]
- 24.Lavoie R, Bouchain G, Frechette S, Woo SH, Khalil EA, Leit S, Fournel M, Yan PT, Trachy-Bourget MC, Beaulieu C, Li Z, Besterman J, Delorme D. Bioorg Med Chem Lett. 2001;11:2847. doi: 10.1016/s0960-894x(01)00552-2. [DOI] [PubMed] [Google Scholar]
- 25.Hydroxamic acid characterization. No spurious peaks were observed in the NMR spectra of the synthesized compounds. 1a, 33% yield from 6a. mp = 128–130 °C; 1HNMR (500 MHz, CD3OD) δ (ppm): 1.3 (d, 3H), 1.35–1.4(m, 6H), 1.6–1.7 (m, 4 H), 2.2 (m, 1H), 2.4 (t, 2H), 7.1 (t, 1H), 7.3 (t, 2H), 7.5 (t, 2H); 13CNMR (125 MHz, CD3OD) δ (ppm): 17.0, 25.0, 27.0, 29.0, 34.0, 37.0, 38.0, 120.0, 124.0, 129.0, 139.0, 173.0, 175.0; HRMS (ESI-LC-MS, m/z): found [M], 278.1640, calc. for C15H22N2O3, 278.1630. 1b, 14% yield from 6b. mp = 121–123 °C; 1HNMR (500 MHz, CD3OD) δ (ppm): 0.9 (t, 3H), 1.25–1.5 (m, 6H), 1.6 (m, 2H), 1.7 (m, 2H), 1.95 (m, 1H), 2.4 (t, 2H), 7.1 (t, 1H), 7.3 (t, 2H), 7.5 (t, 2H); 13CNMR (125 MHz, CD3OD) δ (ppm): 11.0, 25.8, 25.9, 27.1, 29.0, 32.3, 36.7, 45.0, 120.0, 124.0, 129.0, 139.0, 173.0, 175.0; HRMS (ESI-LC-MS, m/z): found [M - H2O], 274.1687, calc. for C16H22N2O2, 274.1681. 1c, 10% yield from 6c. mp = 164–166 °C; 1HNMR (500 MHz, CD3OD) δ (ppm): 0.9 (t, 3H), 1.25–1.5 (m, 8H), 1.6 (m, 2H), 1.7 (m, 2H), 2.1 (m, 1H), 2.35 (t, 2H), 7.1 (t, 1H), 7.3 (t, 2H), 7.5 (t, 2H); 13CNMR (125 MHz, CD3OD) δ (ppm): 13.0, 20.7, 25.9, 27.0, 29.0, 33.0, 35.0, 37.0, 43.7, 120.0, 124.0, 129.0, 139.0, 173.5, 175.0; HRMS (ESI-LC-MS, m/z): found [M - H2O], 288.1851, calc. for C17H24N2O2, 288.1838. 1d, 24% yield from 6d. mp = 153–156 °C; 1HNMR (500 MHz, CD3OD) δ (ppm): 0.9 (t, 3H), 1.2–1.5 (m, 10H), 1.6 (m, 2H), 1.7 (m, 2H), 2.05 (m, 1H), 2.35 (t, 2H), 7.1 (t, 1H), 7.3 (t, 2H), 7.5 (t, 2H); 13CNMR (125 MHz, CD3OD) δ (ppm): 13.0, 22.5, 25.5, 27.0, 29.0, 30.0, 32.4, 32.5, 37.0, 44.0, 120.0, 124.0, 129.0, 139.0, 173.0, 175.0; HRMS (ESI-LC-MS, m/z): found [M - H2O], 302.2000, calc. for C18H26N2O2, 302.1994. 1e, 84% yield from 7e. mp = 143–146 °C; 1HNMR (500 MHz, CD3OD) δ (ppm): 1.25–1.5 (m, 4H), 1.6 (m, 2H), 1.7 (m, 2H), 2.1 (m, 2H), 2.3 (m, 1H), 2.35 (t, 2H), 5.0 (q, 2H), 5.7 (m, 1H), 7.1 (t, 1H), 7.3 (t, 2H), 7.5 (t, 2H); 13CNMR (125 MHz, CD3OD) δ (ppm): 25.5, 27.0, 29.0, 32.0, 36.5, 37.0, 44.0, 116.0, 120.05, 124.0, 129.0, 136.0, 139.0, 174.0; HRMS (ESI-LC-MS, m/z): found [M], 304.1797, calc. for C17H24N2O3, 304.1787. 1f, 85% yield from 7f. mp = 138–140 °C; 1HNMR (500 MHz, CD3OD) δ (ppm): 1.25–1.5 (m, 4H), 1.6 (m, 2H), 1.7 (m, 2H), 2.2–2.45 (m, 6H), 7.1 (t, 1H), 7.3 (t, 2H), 7.5 (t, 2H); ); 13CNMR (125 MHz, CD3OD) δ (ppm): 22.0, 25.0, 27.0, 29.0, 32.0, 37.0, 43.5, 70.3, 81.3, 120.0, 124.0, 129.0, 139.0, 173.0, 175.0; HRMS (ESI-LC-MS, m/z): found [M], 302.1640, calc. for C17H22N2O3, 302.1630. 1g, 48% yield from 7g. mp = 138–140 °C; 1HNMR (500 MHz, CD3OD) δ (ppm): 1.3 (m, 4H), 1.5 (m, 2H), 1.7 (m, 2H), 2.35 (m, 2H), 2.65 (m, 1H), 2.7 (m, 1H), 2.9 (m, 1H), 7.1 (t, 1H), 7.17 (m, 3H), 7.2 (t, 2H), 7.3 (t, 2H), 7.5 (t, 2H); 13CNMR (125 MHz, CD3OD) δ (ppm): 25.0, 27.0, 29.0, 37.0, 39.0, 46.0, 120.0, 124.0, 126.0, 128.0, 128.5, 129.0, 139.0, 140.0, 173.0, 174.0; HRMS (ESI-LC-MS, m/z): found [M], 354.1957, calc. for C21H26N2O3, 354.1943.
- 26.Krapcho AP, Glynn GA, Grenon BJ. Tetrahedron Lett. 1967;8:215. doi: 10.1016/s0040-4039(00)90519-7. [DOI] [PubMed] [Google Scholar]
- 27.In a representative procedure, 0.12 g (0.3 mmol) of O-benzyl protected hydroxamic acid 7e was dissolved in 3 ml THF (0.1 M solution) and cooled to 0 °C via an ice bath. 1.5 ml (5 eq.) of a 1 M BCl3 solution in CH2Cl2 was then added dropwise. The ice bath was removed after 5 min and the reaction was stirred an additional 3 hours (complete by TLC analysis) at room temperature. The mixture was then quenched with 1 M HCl and extracted with ethyl acetate (3 X 10 ml). The organic layers were pooled, dried over magnesium sulfate, and evaporated onto silica gel. Flash chromatography (10% MeOH/CH2Cl2) with acid-washed (deferrated) silica gel afforded the hydroxamic acid 1e in 84–85% yield. For a representative procedure for obtaining deferrated silica gel, see Guo H, Naser SA, Ghobrial G, Phanstiel O. J Med Chem. 2002;45:2056–2063. doi: 10.1021/jm0104522.
- 28.Farr RA, Bey P, Sunkara PS, Lippert BJ. J Med Chem. 1989;32:1879. doi: 10.1021/jm00128a032. provides an example of an unsuccessful hydroxamic acid O-benzyl deprotection with BCl3. [DOI] [PubMed] [Google Scholar]
- 29.The HDAC activity was measured using Fluor de Lyse™ activity assay (Biomol). Briefly, HeLa lysates (25 μL) were incubated with or without the small molecule inhibitor for 30 minutes at 30 °C with shaking. Fluor de Lys substrate (25 μL, 100 μM) was added and the reaction was incubated at 37°C for 45 minutes with shaking. Fluor de Lys developer (50 μL of 1X) was added and incubated with shaking for 5 minutes. The fluorescence intensity was determined at 465 nm using a Genios Fluorimeter (Tecan). The deacetylase activity was determined by dividing the fluorescence intensity of the reaction in the presence of SAHA analog with the intensity in the absence of inhibitor. At least three determinations were used to calculate the mean and standard error in Figure 1 and Table 1.