

Abstract
Orthologous mammalian introns contain many highly conserved sequences. Of these sequences, many are likely to represent protein binding sites that are under strong positive selection. In order to identify conserved protein binding sites that are important for splicing, we analyzed the composition of intronic sequences that are conserved between human and six eutherian mammals. We focused on all completely conserved sequences of seven or more nucleotides located in the regions adjacent to splice-junctions. We found that these conserved intronic sequences are enriched in specific motifs, and that many of these motifs are statistically associated with either alternative or constitutive splicing. In validation of our methods, we identified several motifs that are known to play important roles in alternative splicing. In addition, we identified several novel motifs containing GCT that are abundant and are associated with alternative splicing. Furthermore, we demonstrate that, for some of these motifs, conservation is a strong indicator of potential functionality since conserved instances are associated with alternative splicing while nonconserved instances are not. A surprising outcome of this analysis was the identification of a large number of AT-rich motifs that are strongly associated with constitutive splicing. Many of these appear to be novel and may represent conserved intronic splicing enhancers (ISEs). Together these data show that conservation provides important insights into the identification and possible roles of cis-acting intronic sequences important for alternative and constitutive splicing.
The majority of mammalian mRNAs are interrupted by multiple noncoding intronic sequences that must be removed before translation. A large ribonucleoprotein complex known as the spliceosome carries out the recognition and removal of introns (for reviews, see Burge et al. 1999; Brow 2002; Stark and Luhrmann 2006). Vertebrate introns contain characteristic, but degenerate, splice-junction sequences at either end (Burge et al. 1999). Although the information contained in the splice-junction is typically essential for splicing, it is not generally sufficient for accurate splice-junction definition (Cartegni et al. 2002; Faustino and Cooper 2003). Splice-junction usage, in multicellular organisms at least, can be influenced by the presence of a variety of sequence motifs that are typically located near the splice-junction. Identification of such accessory splicing signals (which we will refer to generally as cis-splicing elements) is an active area of research, and many sequences known to influence splice-junction usage have been identified through the use of experimental and computational methods. Based upon their observed effect on splicing and their location relative to the splice-junction, cis-splicing elements are typically divided into several categories that include exonic and intronic splicing enhancers (ESEs and ISEs) and exonic and intronic splicing suppressors/silencers (ESSs and ISSs) (for reviews, see Blencowe 2000; Cartegni et al. 2002; Ladd and Cooper 2002; Faustino and Cooper 2003; Matlin et al. 2005; Pozzoli and Sironi 2005; Zheng 2004). In a manner analogous to promoter elements that bind transcription factors that then influence formation of a productive transcription initiation complex, it appears that cis-splicing signals serve as binding sites for specific proteins that, when bound, influence splice-junction recognition by the spliceosome.
In addition to experimental approaches, several computational approaches for identifying exonic and intronic cis-elements have been described (for review, see Zhang et al. 2005). In order to establish background frequencies for the statistics, many of these studies relied upon properties internal to a single genome, such as overrepresentation of short sequences (n-mers) within tissue-specific splice isoforms (Brudno et al. 2001; Sugnet et al. 2006), in strong versus weak splice-junctions (Fairbrother et al. 2002; Yeo et al. 2004), in intron-containing versus intron-lacking exons (Fedorov et al. 2001), and in real versus pseudo splice-junctions (Zhang et al. 2003; Zhang and Chasin 2004).
The increasing wealth of genomic sequence information has made possible the development of computational methods that rely upon comparative genomic methods. The rationale for comparative approaches is founded in the well-established observation that informationally important sequences tend to be evolutionarily conserved. Several studies using conservation of sequence as a criterion for identifying potential intronic cis-splicing elements have been published (Yeo et al. 2005; Goren et al. 2006; Kabat et al. 2006). Comparative genomic studies have demonstrated that the regions surrounding human splice-junctions often contain sequences conserved among other mammals, and high levels of conservation are especially apparent in the regions surrounding alternatively spliced junctions (Sorek and Ast 2003; Sugnet et al. 2004, 2006; Sironi et al. 2005; Yeo et al. 2005). It seems likely that these regions are conserved because of pressures to maintain sequences involved in splice-junction usage.
Although alternative splice-junctions often display higher levels of conservation than constitutively spliced junctions (Yeo et al. 2005), alignments between orthologous introns reveal that most introns, even constitutively spliced introns, contain one or more regions that are highly conserved (for an example, see Fig. 1). Many of the conserved regions found within the flanks of constitutively spliced introns are much shorter than the highly conserved regions found in alternatively spliced introns and may simply represent regions that have not yet diverged due to random mutational drift. However, it is also possible that some of this conservation results from selective pressures to maintain functionally relevant signals that play important roles in cellular processes such as transcription, poly-adenylation, chromatin remodeling, mRNA trafficking, and splicing.
Figure 1.

Example of mammalian genomic alignment showing conserved exonic and intronic sequences. Shown is a small portion of the human gene LMO7 aligned against the orthologous region of the six mammalian genomes used in this study. (A) Four exons (represented as boxes) and their corresponding introns. The central graphic indicates the observed splicing events. Below that, the conservation is represented as a histogram where the height is proportional to the degree of conservation. (B) An expanded view of the sequence flanking the 5′ splice-junction of the second, alternatively spliced, exon. The actual sequence is displayed below the conservation histogram. Boxes are placed around conserved sequences (CSs). The original graphics were drawn using the UCSC Genome Browser (http://genome.ucsc.edu; Kent et al. 2002).
We present the results of a comprehensive characterization of the composition of sequences that are conserved among orthologous mammalian introns. Our ultimate goal was to characterize conserved cis-splicing elements. Since these are likely to represent protein binding sites and since many RNA binding proteins recognize short (6–10 nt) patterns, we included all conserved sequences (CSs) that are at least 7 nt in length. It should be noted that this analysis therefore differs from studies of much longer conserved nongenic sequences (known as CNGs) (for reviews, see Dermitzakis et al. 2005; Bird et al. 2006). While CNGs tend to be located in gene-sparse regions, we focused exclusively on the intronic regions immediately flanking splice-junctions. Our analysis also differs from other computational studies designed to characterize cis-splicing elements that have largely focused on identifying motifs enriched in alternatively spliced introns (Brudno et al. 2001; Yeo et al. 2005; Sugnet et al. 2006). We designed this study to include all intronic splice-junctions, regardless of whether they are constitutively or alternatively spliced. The approach we developed utilizes a multiple sequence alignment between human and six eutherian mammalian genomes. From this alignment, all contiguous stretches of at least 7 nt of identical sequence were extracted and characterized. In order to identify relevant motifs, we developed a graph clustering method to cluster overrepresented conserved n-mers containing similar substrings.
This analysis produced several interesting observations regarding conserved intronic sequences and splicing. (1) We show that CSs are enriched in specific motifs. This demonstrates that many CSs are the result of positive selective pressures on a limited set of putative cis-acting sequences and are not simply due to chance conservation. (2) Many conserved intronic motifs are associated with either alternative or constitutive splicing. This suggests that these motifs play important roles in splicing. (3) As validation for our methods, we found that several of the motifs associated with alternative splicing resemble motifs previously demonstrated to play important roles in regulated splicing. (4) We identified several novel motifs containing GCT that are associated with alternative splicing. (5) We identified a large number of conserved motifs that are associated with constitutive splicing, most of which have not been previously computationally identified. (6) Lastly, we demonstrate that conservation is an important indicator of functionality by showing that conserved instances of some n-mers are highly associated with alternative splicing, but nonconserved instances are not.
Results
Extraction of conserved intronic sequences
In order to identify conserved motifs representing putative trans-factor binding sites, we wanted to extract all intronic sequences conserved between human and several closely related mammals. We created a database of U2-dependent intronic sequences based upon the RefSeq annotation (Pruitt et al. 2005). We chose this annotation since it is a conservative assessment of human genes and limits pseudo-genes and pseudo-exons. Mammalian introns vary greatly in length (from ∼100 to >400,000 bp), and the signals that govern splicing of shorter (<200 bases) introns may differ from those governing splicing of longer introns (Fox-Walsh et al. 2005). For this analysis, we chose to focus exclusively on introns that are >199 nt. Such introns compose the bulk of human introns. We reasoned that signals involved in splicing are likely to be located near splice-junctions; therefore, we focused on the 100-nt intronic regions adjacent to the splice-junctions. Since we were interested in characterizing intronic sequences that lie outside of the splice-junctions themselves, we excluded the first 7 nt of the donor side and the last 3 nt of the acceptor side of introns (Fig. 2).
Figure 2.

Schematic representation of mammalian introns detailing the regions used in this study. The positions of the 5′ and 3′ splice-junctions are indicated as 5′ SJ and 3′ SJ. Sequence logos, composed from 5000 randomly sampled human introns, are used to show the frequency composition of the splice-junctions. The intronic regions that are the basis of this study are indicated as DI (donor intronic) and AI (acceptor intronic).
We defined a CS to be a contiguous run of at least 7 nt of identity in a multiple sequence alignment between human and six eutherian mammals: chimp, rhesus monkey, mouse, rat, dog, and cow (see Methods). We chose 7 nt as the lower length cutoff since many RNA binding proteins have binding site sizes of 6–10 nt. In order to emphasize the most significant portion of the cis signals and to reduce noise, we chose not to allow any sequence mismatches. For sequence and motif analysis, we extracted and categorized CSs from the donor intronic (DI) and acceptor intronic (AI) regions (see Fig. 2). Details concerning the identification and extraction of CSs are presented in the Methods.
Many thousands of introns containing CSs were identified. Specifically 16,548 introns (11%) contained CSs in the donor side, and 20,342 introns (14%) contained CSs in the acceptor side. Since CSs were extracted from a multiple-sequence alignment, which requires that the relative positions of a sequence be somewhat conserved, it is possible that these numbers underestimate the number of functionally relevant conserved intronic sequences. Nevertheless, we believe this approach produces a conservative sampling that will allow us to identify functionally relevant sequences. The lengths of CSs varied from the minimum of 7 bases to the full-length of 100 bases. The actual distributions are shown in Figure 3, A and B.
Figure 3.

Distribution of total lengths of CSs found in donor and acceptor intronic regions and associations between CS length and alternative splicing. (A,B) Distributions of the lengths of the CSs found in the donor or acceptor intronic regions. In cases where more than one CS was found in a particular intron, the lengths were combined (total length). For each data set, the bin width is equivalent to two bases. The number of CSs in each bin is indicated along the Y-axis. The inset plots are the same data displayed using a log scaled Y-axis to better visualize the longer CSs. (C) The relationship between the percentage of introns that are alternatively spliced and the total length of CS found within the intron. The horizontal bar indicates the average percentage of splice-junctions that are alternatively spliced (3%). (D) The number (and corresponding percentages) of all alternatively spliced (Alt+) or constitutively spliced (Alt−) splice-junctions that contain a CS in the donor (DI) or acceptor (AI) intronic flanking sequence.
Conserved intronic sequences are found adjacent to both constitutive and alternatively spliced junctions
In order to explore the relationships between alternative splicing and CSs, splice-junctions containing a CS were cross-referenced against the alternative splice events annotated in the UCSC ExonWalk database (http://hgdownload.cse.ucsc.edu/goldenPath/hg17/database/). A splice-junction was annotated as alternative if it was involved in either a skipped-exon or alternative adjacent splice event. Using these data, we found that ∼3% of introns lacking any CS were annotated as being alternative. In contrast, we found that 8% of introns containing a CS were involved in an alternative event. Since the lengths of CSs varied greatly, we wanted to establish the relationship between CS length and degree of alternative splicing. This analysis (Fig. 3C) revealed that the degree of alternative splicing increases with the total length of CS found within the intron. In particular, we observed that intron flanks containing between 7 and 25 nt of CS are twice more likely to be involved in alternative events than were introns without CSs, and flanking regions containing >50 nt of CS are eight times more likely to be involved in alternative events. These observations are consistent with earlier studies showing that intronic regions flanking alternatively spliced junctions tend to be highly conserved (Sorek and Ast 2003; Sugnet et al. 2004, 2006; Yeo et al. 2005), and suggests that important cis-splicing elements are found in these intronic flanks. It is also important to note that, although short CSs have a weaker association with alternative splicing, they are far more abundant than longer CSs and are still twice more likely to be associated with alternative events. These shorter CSs might represent the most important portions of protein binding sites.
To explore the distribution of CSs between alternatively or constitutively spliced junctions, we determined the percentage of either category that contains one or more CSs (Fig. 3D). Consistent with previous studies and with our analysis above, we found that a higher proportion of alternatively spliced junctions contain a CS than constitutively spliced junctions. We did not explore the more complex associations between CSs across exons so we do not know the percentage of exons that have CSs in just one or in both intronic flanks. However, these data demonstrate that, although CSs are enriched in alternatively spliced junctions, the majority of human alternatively spliced junctions do not contain a CS in the immediate vicinity of the alternative junction. This observation is consistent with studies suggesting that the majority of human alternatively spliced events are not conserved within mammals (Sorek et al. 2006).
Though intronic CSs are enriched in introns flanking alternatively spliced junctions, the great majority are located within junctions that are constitutively spliced (Fig. 3D). Cis-splicing elements that are involved in constitutive splicing have generally received less attention than those involved in alternative splicing. In order to identify putative cis-splicing elements that may play important roles in alternative and/or constitutive splicing, we wanted to identify n-mers that are enriched in CS sequences.
Conserved intronic sequences are enriched in specific n-mers
Many observations have implicated the importance of auxiliary motifs located within the intronic regions flanking splice-junctions (for reviews, see Ladd and Cooper 2002; Matlin et al. 2005). The hypothesis that highly conserved intronic sequences (CSs) represent conserved protein binding sites predicts that CSs would be enriched in motifs representing such binding sites. In order to determine whether or not CSs are enriched in specific sequences, we chose to perform an enumerative analysis of their sequence composition. Enumerative methods typically begin with counting the occurrences of short sequences (n-mers) within the subject sequence sample. n-Mers that are enriched within the subject sample can be found by comparing these counts against background expectations. Since the effect on the final score that a putative binding site will have on an n-mer is related to both the size of the binding site and the length of the n-mer, we felt it was important to examine a range of n-mer lengths. For this analysis we chose to examine all n-mers from 4–7 nt.
We counted n-mers in the conserved donor (DI-CS) and conserved acceptor (AI-CS) samples using a sliding-window with an overlapping word count. In order to determine the n-mers that are enriched within the CS sequences, we had to establish the background probability for random occurrence for each n-mer within the region. This is complicated by the fact that the sequence composition of introns is nonhomogeneous as one moves away from the splice-junction; therefore, the probability of occurrence for an n-mer may vary at each position within the region. To account for this, we implemented a random sampling strategy that incorporates position as a factor. For each CS identified, we also extracted 100 additional analogous (e.g., having the same splice-junction relative starting position and the same length) sequences as the CS from introns randomly chosen from the original data set of all human introns. These sequences made up the random sequence (RS) pool. Background frequencies were calculated using the entire RS sample. Enrichment was determined using a confidence interval for the binomial distribution (Agresti and Coull 1998) using the probabilities derived from the RS sample and a sample size proportional to the CS samples (see Supplemental Materials and Methods). In this manner, we determined the likelihood that the counts observed in the CS sample could have occurred by chance in a sample the size of the DI-CS or AI-CS data sets.
Figure 4, A and C, demonstrates scatter-plots for the counts of all n-mers found in the CS samples relative to the counts obtained from the RS samples. n-Mers that are significantly enriched in the CS samples (α1-tailed = 0.01) are indicated. The DI-CS sample contained 819 significantly enriched n-mers, while the AI-CS sample contained 1007 (the full set of CS enriched n-mers is available in Supplemental Tables 1, 2). To assess these results, we obtained an additional randomly sampled data set for both the DI-CS and the AI-CS samples (referred to as DI-pseudo and AI-pseudo, respectively). The pseudo data sets were of the same size and were analogous (as described above for the RS sample) to the DI-CS and AI-CS samples. In contrast to the CS samples, only 55 DI-pseudo and 39 AI-pseudo n-mers scored as significant (Table 1). These results demonstrate that CSs are compositionally distinct from intronic flanks in general and that they are enriched in specific n-mers at a level that is much greater than could be expected by chance.
Figure 4.

Scatter-plots for the counts of all n-mers (4–7 nt) in the CS samples (NCS) vs. the counts in the corresponding random samples (NRS). Overlaid on plots A and C are the n-mers that were significantly enriched (according to the confidence intervals described in the Supplemental Materials and Methods) in the donor intronic (DI) and acceptor intronic (AI) regions. Overlaid on plots B and D are all n-mers containing the substrings indicated. These substrings are examples of substrings that are enriched in the corresponding regions.
Table 1.
Summary of the numbers of significantly enriched n-mers found in the DI and AI and of the GCCS clustering performance
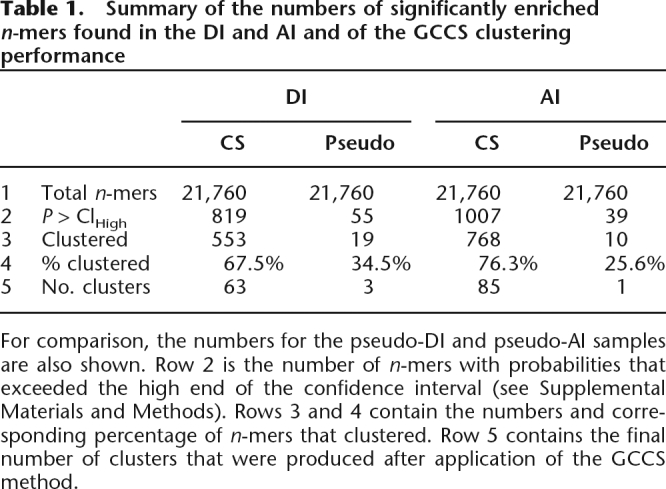
For comparison, the numbers for the pseudo-DI and pseudo-AI samples are also shown. Row 2 is the number of n-mers with probabilities that exceeded the high end of the confidence interval (see Supplemental Materials and Methods). Rows 3 and 4 contain the numbers and corresponding percentage of n-mers that clustered. Row 5 contains the final number of clusters that were produced after application of the GCCS method.
Visual inspection of the CS-enriched n-mers revealed that many contain common substrings. This would be expected if CSs were enriched in specific motifs since when using a sliding-window enumeration, a single conserved motif would spawn many related n-mers containing portions of the motif in different frames. Examples of the distributions of two n-mers containing substrings found by visual inspection to be common to CS-enriched n-mers are shown in Figure 4, B and D. Shown are the distributions for n-mers containing the substring GCATG (as found in the DI sample) and the substring TTCTG (as found in the AI sample). In both cases, it is clear that these substrings confer a distributional bias to n-mers containing these substrings. It is interesting to note that the substring GCATG is identical to the binding site for the Fox family of splicing factors that are known to play important roles in alternative splicing (discussed in greater detail below). The TTCTG substring does not exactly match any described binding sites and is discussed in greater detail below.
Graph based clustering of similar n-mers and construction of CS motifs
Visual inspection of the enriched n-mers can be used to identify particularly common substrings that may represent enriched sequence motifs. However, visual inspection alone cannot be used to thoroughly mine the large number of n-mers in the samples of interest. In order to identify significantly enriched substrings and to construct CS motifs, we developed a graph-clustering and motif reconstruction method that we refer to as Graph Clustering by Common Substrings (GCCS). Reconstruction of motifs from decomposed counts of n-mers is a problem for which many solutions have been proposed (Tompa et al. 2005). The GCCS method utilizes graph clustering to group compositionally similar n-mers in a manner such that clusters tend to form around n-mers with high Z-scores (for details, see Supplemental Materials and Methods). In our approach, we required that all n-mers in a cluster share a common substring with each other. Although positional degeneracy can be incorporated into the GCCS method, we chose not to allow mismatches between substrings. It is difficult to build a general degeneracy model that fits all RNA binding proteins. Some proteins display strong affinities for very specific sites, while others appear to be more promiscuous and have similar affinities for a range of compositionally related sites. Since we wanted to detect all conserved RNA elements and have no a priori knowledge about binding affinities for putative trans-factors, we therefore only allow degeneracy at the ends of the motifs. The effect of this choice is that the number of unique motifs we identify may overestimate the number of distinct protein binding sites since a given protein may equally recognize more than one motif. However, we hoped that by requiring exact matches in the common substring, we would identify the most highly conserved portions of cis-signals. Finally, we required that all final clusters be composed of at least six n-mers. As discussed above, real motifs are expected to generate multiple n-mers with common substrings in different phases. Requiring that an n-mer share a substring with other n-mers allows the GCCS method to take advantage of this phenomenon, and effectively reduces the n-mers that occur in the population by chance.
We refer to the CS motifs as CSMs. An example of two clusters and corresponding CSMs that were obtained from the DI region are shown in Figure 5. The CSMs in this example closely resemble binding sites for two known splicing factors, Fox-1/Fox-2 and QKI (both discussed in greater detail below). Since each of these proteins have well-characterized binding sites and are known splicing factors, the fact that we identified CSMs matching these sites helps validate our methods. In addition, it is especially interesting to note that the GCS for the putative QKI motif centers over the high affinity portion of the QKI site identified biochemically (Galarneau and Richard 2005).
Figure 5.

Samples of GCCS clusters derived from the donor intronic (DI) region. Shown are the graph clusters representing the clustered n-mers used to construct the CSMs for the putative Fox and QKI protein binding sites (Fig. 7, DI-1 and DI-2, respectively). Vertices are colored according to their conservation Z-score (see color key). The graphs were drawn using GraphViz (http://www.graphviz.org/).
Table 1 details the clustering results for the CS and pseudo-CS n-mers. The DI-CS sample yielded 63 clusters, while the AI-CS sample yielded 85 clusters (available in Supplemental Tables 3, 4). Meanwhile only three clusters were obtained from the DI-pseudo sample, and only one was obtained from the AI-pseudo sample. In both cases the percentage of n-mers that clustered was significantly higher in the CS-derived samples compared with the pseudo samples. This demonstrates that GCCS clustering successfully filters out n-mers that, despite showing enrichment, are likely to be due to chance.
Many CSMs are statistically associated with constitutive or alternative splicing events
We have shown that conserved intronic sequences flanking splice-junctions (CSs) are enriched in specific motifs that may represent protein binding sites. Since it is possible that some of these putative binding sites could be important for processes other than splicing, we wanted to identify CSMs that are statistically associated with either alternative or constitutive splicing events.
Using the same database of alternatively spliced junctions that we used above, we counted the occurrences, for all n-mers (4–7 nt), within CSs and categorized them according to their being located adjacent to an alternatively spliced or constitutively spliced junction. The G-test was used to determine significant associations (see Supplemental Materials and Methods). The probabilities for the association were transformed to a value that we refer to as a TA-score (see Supplemental Materials and Methods). A positive TA-score indicates an association with alternative splicing, and a negative TA-score indicates an association with constitutive splicing. Since each CSM is composed of several n-mers, the association for the CSM was determined by comparing the means (using Student’s t-test) of the TA-scores for the CSM versus the mean for all n-mers. An example of the distribution of TA-scores for several CSMs is shown in Figure 6 (for the complete analysis, see Supplemental Figs. 1, 2). This analysis revealed that some motifs were significantly associated with alternative splicing (Pt-test < 0.01) and some with constitutive splicing, and some showed no significant association either way. It is important to point out that the clustering procedure we used does not incorporate any knowledge regarding alternative splicing; yet many of the motifs are clearly enriched in n-mers that show similar biases, which demonstrates that the common substrings are responsible for the observed bias. After removing redundant examples of compositionally similar motifs, we found that five DI-CSMs and five AI-CSMs are significantly associated with alternative splicing, while 18 DI-CSMs and 18 AI-CSMs are significantly associated with constitutive splicing. The CSMs showing a statistically significant association with alternative or constitutive splicing are shown in Figure 7. We also determined the number of splice-junction flanks that contain at least one instance of a conserved n-mer matching each of these CSMs. We found that most CSMs are found in hundreds to more than 1000 individual introns (Supplemental Fig. 3). The combined observation that DI-CSs and AI-CSs are enriched in specific n-mers and that many of these n-mers are statistically associated with alternative or constitutive splicing strongly suggests that they represent motifs that are under positive selective pressures because they play important roles in splicing.
Figure 6.

Box-plots showing the distributions of TA-scores observed for several representative CSMs. The greatest common substrings (GCS) for each CSM are shown to the left. CSMs that were significantly enriched in n-mers associated with alternative splicing are shown in red, those significantly associated with constitutive splicing are in blue, and no association is shown in yellow.
Figure 7.

Intronic conserved sequence motifs (CSMs) showing significant associations with alternative (above line) or constitutive splicing (below line) are shown against a schematic representation of an intron to indicate the region within which they are located. Motifs marked with an asterisk are compositionally similar to the equivalently numbered motif in the other region.
There are many more CSMs that are not as significantly associated with either alternative or constitutive splicing but display a bias toward either category, and there are others that show no bias at all (see Supplemental Figs. 1, 2). Some of these may represent motifs that are important for splicing but are utilized in a context independent of regulated or constitutive splicing. It is also possible that some of these represent motifs that are under selective pressures but play roles in other processes such as mRNA trafficking, maturation, degradation, or poly-adenylation. For further characterizations, we chose to focus only on those motifs with the strongest biases.
Identification of motifs known to be associated with alternative splicing
Several of the CSMs that are strongly associated with alternative splicing are similar to known splicing factor binding sites. These include DI-1 and AI-1 (Fig. 7), which are exceptionally good matches to the binding site TGCATG for members of the Fox family of RNA binding proteins that have been demonstrated to be important for alternative splicing of some human introns (Huh and Hynes 1994; Lim and Sharp 1998; Jin et al. 2003; Nakahata and Kawamoto 2005; Baraniak et al. 2006; Ponthier et al. 2006; Zhou et al. 2007). Fox binding sites have been shown to be enriched in introns adjacent to brain specific alternatively spliced introns (Brudno et al. 2001; Sugnet et al. 2006), and Fox binding sites have been shown to be conserved between vertebrates (Minovitsky et al. 2005). Furthermore, the Fox binding site hexamer was found to be overrepresented in intronic sequences conserved between the nematodes Caenorhabditis elegans and Caenorhabditis briggsae (Kabat et al. 2006).
Meanwhile, the motifs DI-2 and AI-2 are both matches to the Quaking protein (QKI) binding site CTAAC (Wu et al. 2002; Ryder and Williamson 2004; Galarneau and Richard 2005) and to the SF1/BBP protein binding site (Berglund et al. 1997; Garrey et al. 2006). Both of these proteins are members of the STAR-KH RNA binding family, and both are known to be involved in splicing (Berglund et al. 1998a, b; Wu et al. 2002). Recently it was demonstrated that an equivalent motif is enriched in donor intronic regions flanking alternatively included exons in mouse heart and skeletal muscle (Sugnet et al. 2006). Given the similar binding affinities for these proteins, these motifs may represent binding sites for either of these splicing factors.
The acceptor side motifs, AI-3 (TTCTG) and AI-5 (TGTT), are abundant (Supplemental Fig. 3) and may represent conserved targets for members of the CELF/BRUNO-like family (for review, see Barreau et al. 2006). These proteins are known to play roles in alternative splicing, and one member, CUG-BP1, has been shown to bind TGT containing motifs (Marquis et al. 2006). A similar motif was also found in the donor region DI-22 (TTGT) (Supplemental Fig. 1). This donor side motif shows an association with alternative splicing but is not as strongly associated as the acceptor motif.
A previous computational analysis of alternative events conserved between mouse and human identified 4–5 base n-mers enriched in intronic sequences flanking skipped-exons (Yeo et al. 2005). We compared the n-mers reported in Yeo et al. (2005) that were conserved (according to alignment between mouse and human intronic sequences) with the CS-enriched n-mers from this study (Supplemental Table 5). Both studies identified n-mers matching Fox and QKI binding sites, and in both analyses, these were found to be associated with alternative splice events. In addition, both studies identified several n-mers with no obvious trans-factor. These include the n-mers TTGC (enriched in both the DI and AI regions), GTTTG (enriched in DI), and CAAAT (enriched in DI). Our analysis also revealed several possibly novel motifs (e.g., DI-3, DI-5, and AI-3, AI-4, and AI-5) (Fig. 7) not identified in Yeo. et al. (2005). It should be noted that whereas Yeo. et al. (2005) focused exclusively on conserved skipped-exon events, our study involved all CSs regardless of their possible association with alternative splicing. Given this and other differences in the design of these two studies (including statistical methods, length of n-mers evaluated, number of genomes compared, and method for developing background probabilities), differences in the results are not surprising and demonstrate that different methods have different strengths.
Conserved cryptic splice-junctions are associated with alternative splicing
The donor side motif DI-3 (Fig. 7) is strongly associated with alternative splicing and is a perfect match for the canonical 5′ splice-junction AG|GTGAGT. Since splice-junctions were excluded from this analysis; these are unlikely to represent actual splice-junctions. In order to verify whether or not these represent splice-junctions, we examined several individual instances using the UCSC genome browser and found that although some appear to represent alternatively used splice-junctions, the majority are found near alternative and skipped splice-junctions but show no evidence (based upon ESTs and mRNAs) of serving as splice-junctions (data not shown). Interestingly, previous analyses found that similar cryptic 5′ splice-junction sequences can act as ESSs (Wang et al. 2004, 2006) and have been implicated in regulated alternative splicing (Lou et al. 1995). Our results extend these observations by showing that many, apparently cryptic, 5′ splice-junctions located near active 5′ splice-sites are highly conserved, which implies that they are functionally important for regulated splicing at the adjacent splice-junction. Whether these motifs are recognized by the U1 snRNP or are recognized by other factors is currently unknown.
Identification of putative novel GCT motifs associated with alternative splicing
We can only speculate about the identities of the trans-factors that interact with the remaining motifs associated with alternative splicing (DI-4, DI-5, and AI-4) (Fig. 7). In terms of overall representation, these motifs are nearly as abundant as the Fox-1/Fox-2 motifs (Supplemental Fig. 3). None of these exactly match well-characterized binding sites. However, it should be stressed that the binding sites for many splicing factors have not been well characterized or are too degenerate to be identified with certainty.
These three motifs all contain a core substring of GCT. The most likely candidate trans-acting factors for these motifs are members of the MBNL family of RNA binding proteins (Pascual et al. 2006). MBNL proteins are known to play an important role in alternative splicing (for review, see Osborne and Thornton 2006) and were identified because of their binding affinity for long repeats of CTG (Osborne and Thornton 2006; Pascual et al. 2006). The natural sites by which MBNL mediates regulated splicing have not been well defined but are not thought to be long CTG-repeats. Several intronic MBNL targets have been defined and these contain a common motif consisting of YGCT[T/G]Y (Ho et al. 2004). This putative MBNL binding site closely matches motifs DI-4, DI-5, and AI-4. Whether or not these motifs represent conserved MBNL binding sites or are binding sites for other factors remains to be determined. However, this analysis demonstrates that many sites similar to the putative MBNL binding site are highly conserved and are associated with alternative splicing.
The donor side motif DI-4 (GCTTG) (Fig. 7) is similar to a motif, TGYTTTC, enriched in introns flanking included alternative exons in brain (Sugnet et al. 2006). However, several observations from our study suggest that these two motifs are physiologically different. In agreement with Sugnet et al. (2006), many n-mers containing GYTT (including both TGCTTTC and TGTTTC) are enriched in conserved donor intronic sequences (Supplemental Table 1). However, of the 5-mers containing GYTTN or of the 6-mers containing TGYTTN, only those containing GCTTG are both enriched in CSs and are positively associated with alternative splicing (Supplemental Table 3; Supplemental Fig. 1, cf. clusters 4, 21, and 53). These observations are consistent with those of Sugnet et al. (2006), since both analyses indicate that TGYTT-like motifs are enriched in conserved regions. But our additional observations suggest that TGCTTG motifs may play a larger role in alternative splicing. Clarification of this point will require identification and characterization of the trans-acting factors.
Conserved motifs associated with constitutive splicing are abundant and AT-rich
Given the association between the presence of conserved intronic sequences and alternative splicing, we expected to identify specific motifs that were associated with alternative splicing. A rather surprising outcome of this analysis was the large number of motifs that are strongly associated with constitutive splicing (Fig. 7). To our knowledge, this is the first computational identification of conserved intronic motifs that were shown to be associated with constitutive splicing. These motifs are generally more abundant than those that are associated with alternative splicing (Supplemental Fig. 3). An interesting feature of these motifs is that most have nearly exactly matching counterparts on both sides of introns, and they are especially AT rich.
Several families of RNA binding proteins are known to bind sequences similar to motifs in this group. Two well-characterized proteins, TIA1 and TIAL1 (also known as TIAR), are known to bind T-rich sequences and play important roles in splicing (Dember et al. 1996; Del Gatto-Konczak et al. 2000; Forch et al. 2000; Le Guiner et al. 2001; Zhu et al. 2003). Several of the constitutively associated motifs closely resemble sequences known to bind TIA1 and TIAL1, including DI/AI-8, DI/AI-14, and DI-18 (Fig. 7). These motifs may also represent binding sites for members of the Hu RNA binding proteins. Like TIA1 and TIAL1, these proteins are known to bind AT-rich sequences. In particular, interactions between Hu proteins and AT-rich sequences (known as AREs) located in 3′-UTRs are known to be important for regulating mRNA stability (Barreau et al. 2005). One of the conserved motifs, DI/AI-8 (ATTTA), is a close match to a known ARE (Barreau et al. 2005). When we examined the location of ATTTA motifs to see if they are overrepresented in 3′ UTRs, we found that only 3.6% of the conserved instances are located in annotated 3′ UTRs, and the bulk was located within CDSs. Recently, it was shown that Hu proteins may also have an important role in splicing, since they were shown to compete with TIA1/TIAL1 binding, and that this competition is important for establishing neuronal versus non-neuronal ratios of exon skipping for exon 4 of the calcitonin gene (Zhu et al. 2006).
The RNA binding protein Sam68 has been shown to be involved in splicing and has been shown to preferentially bind to the sequence TAAA (Lin et al. 1997; Itoh et al. 2002; Matter et al. 2002; Paronetto et al. 2007). Several of the constitutively associated motifs contain this sequence and may represent binding sites for Sam-68 (e.g., DI/AI-9 through DI/AI-11 and DI/AI-12).
Conservation of certain CSMs is associated with functionality
The primary goal of this analysis was to use computational methods to identify motifs that might be important for splicing in mammals. The next step in the characterization of novel motifs would necessarily involve experimental methods. The first step in the design of such experiments involves choosing appropriate candidate introns from the many thousands of human introns, and analysis of a particular motif requires choosing introns for which the subject motif is functionally relevant. Since the motifs identified in this study are relatively short (4–6 bases), there is a high probability that sequences similar to these motifs would occur simply by chance. It is, therefore, important to be able to distinguish functional motifs from compositionally indistinguishable, but functionally irrelevant, sequences that are the result of chance.
It is likely that conservation would increase the likelihood that a particular motif is functionally relevant. If this were true, we would expect to see a stronger correlation between alternative splicing, for instance, and conserved instances of an n-mer versus instances of the n-mer that are not conserved. In order to test this hypothesis, we examined the relationship between alternative splicing for several pentamers matching CSMs associated with either alternative splicing or with constitutive splicing. We also included one pentamer that was not enriched in CSs and was not associated strongly with either alternative or constitutive splicing. We found that the pentamers that are highly enriched in CSs and are associated with alternative splicing are much more likely to be associated with an alternative event (Fig. 8, pentamers 1–4). Meanwhile, the same pentamers are no more likely to be associated with alternative splicing when they occurred in non-CSs than background levels. Importantly, it should be noted that this observation is not simply due to there being an enrichment of these pentamers in CSs. In fact, the great majority of these pentamers occurred in non-CSs (e.g., the pentamer GCATG occurred 330 times in CSs but occurred 12,449 times in non-CSs). In contrast and in agreement with their TA-scores, two pentamers that are enriched in CSs but are significantly associated with constitutive splicing (Fig. 8, pentamers 5 and 6) are much less likely to be associated with alternative splicing when they occurred in either CS or non-CS sequences (in either context the association is less than average). Interestingly, these pentamers still have a higher association with alternative splicing when they occurred in CSs than in non-CSs. This suggests that although they are generally associated with constitutive splicing, they may play important roles in alternative splicing in some introns. Last, a pentamer that showed no enrichment in CSs nor association with alternative splicing is no more likely to be associated with alternative splicing than predicted by chance in either context (Fig. 8, pentamer 7).
Figure 8.

Association between motif conservation and alternative splicing for several 5-mers. Vertical bars represent the percentage of occurrences of each 5-mer that was observed in the DI region of an alternatively spliced intron. The lower horizontal bar indicates the average for all non-CS 5-mers. The upper horizontal bar indicates the average for all CS 5-mers.
These results demonstrate that conservation of an n-mer near an alternatively spliced junction is likely to be an important predictor of functionality, and also suggests that the mere presence of a sequence matching a particular binding site doesn’t indicate that the sequence represents a functional site. A likely explanation for this phenomenon is that the local context (i.e., surrounding sequences) of conserved instances is different from the nonconserved instances, and implies that additional cis- and trans-elements are required for functionality. A more comprehensive analysis of these relationships merits future attention.
Discussion
Evolutionary conservation is a well-established metric for distinguishing signals from noise in genomic sequences (Cooper and Sidow 2003). Previous studies have shown that sequences flanking alternatively spliced junctions tend to be highly conserved (Sorek and Ast 2003; Sugnet et al. 2004, 2006; Yeo et al. 2005), and that these regions are compositionally distinct from constitutively spliced introns (Yeo et al. 2005; Wang et al. 2006). It has also been shown that many experimentally identified intronic cis-elements are highly conserved (Sironi et al. 2005). However, there has been no previous analysis of the makeup of conserved intronic sequences in general, and little attention has been given to sequences conserved in constitutively spliced introns in particular. Here we presented the results of a study designed to provide a comprehensive picture of the makeup of sequences conserved between orthologous introns from seven mammals. The present study provides additional evidence that comparative genomic methods can reveal intronic motifs that are under selective pressures, and that many of these motifs appear to be involved in splicing.
In order to identify such motifs, we carried out an analysis of conserved intronic sequences flanking the splice-junctions. Comparative analysis of mammalian genomes has revealed that many mammalian introns contain stretches of CS. These islands of conservation can be readily visualized by comparing aligned orthologous sequences (see Fig. 1). Prior to this analysis, it was unclear whether or not the many, typically short, CSs simply represent noninformative regions that have not been subject to mutational divergence since the last common ancestor. If this were true, we would expect the sequence composition of CSs to be equivalent to the composition of introns in general. However, as we have shown, we found this not to be the case. Instead, the population of conserved intronic sequences is clearly enriched in specific n-mers. Using a novel graph-clustering algorithm, we show that these n-mers can be clustered into distinct sequence motifs (CSMs). Furthermore, we showed that many of the CSMs show a marked association with either alternative or constitutive splicing. This linkage between splice-type and conservation supports the notion that the selective pressures responsible for conservation of many of the CSMs is likely to be related to splicing.
A variety of auxiliary splicing factors have been identified; however, for the majority of these proteins, the optimal binding sites have either not been well characterized or the observed binding sites are not discrete enough to be distinguishable by sequence composition alone. Thus, we can only speculate about which splicing factors are likely to be the binding partners for many of the mammalian CSMs. Future experimental studies will be required to identify trans-factors for many of the CSMs identified in this analysis. The splicing factors Fox-1/Fox-2 and QKI have well-characterized and distinctive binding sites, and their connections with alternative splicing have been well documented. Motifs matching the binding sites for these proteins were found to be both highly enriched in CSMs and were highly associated with alternative splicing. Interestingly, a comparative analysis to define n-mers that are enriched in conserved alternatively spliced introns in the nematodes C. elegans and C. briggsae also revealed these same motifs (Kabat et al. 2006), indicating their ancient origins. However, most of the motifs identified in this study were not identified in nematodes, suggesting that some of them may be mammalian specific.
Our analysis revealed several GCT-containing motifs that are associated with alternative splicing. To our knowledge these motifs have not been previously predicted using computational methods. These motifs are as abundant as the Fox and QKI motifs, suggesting that they play important roles in alternative splicing of many exons. We are not aware of any known splicing factors that are obvious candidates for binding these motifs. However, these motifs are a close match to the proposed model of the MBNL binding site (Ho et al. 2004). Future analysis of these motifs will be necessary for clarifying their role in alternative splicing.
An interesting outcome of this analysis was the large number of previously unrecognized conserved motifs that are strongly associated with constitutive splicing. These motifs are largely A and T rich. Among these motifs are sequences that resemble Sam68, TIA1/TIAL1, and Hu protein binding sites. Whether or not these motifs represent conserved binding sites for any of these proteins remains to be determined. These proteins have been typically studied in the context of alternative splicing. Considering that TIA1/TIAL1 have been shown to promote splicing via interaction with U1 snRNP (Forch et al. 2000; Zhu et al. 2003), it is possible that these proteins play a more general role in promoting splicing, whether it is constitutive or alternative. Our data suggest that these motifs represent a large class of conserved AT-rich ISEs.
Although we identified several motifs similar to known splicing factor binding sites, several well-known splicing factor sites were not found. Notably absent, for instance, are CSMs matching Nova protein binding sites. Nova has been shown to be involved in alternative splicing and appears to bind clusters of YCAY motifs (Jensen et al. 2000a, b; Dredge and Darnell 2003; Ule et al. 2003, 2006; Dredge et al. 2005). Since these Nova sites are typically conserved between mouse and human (Ule et al. 2006), we might expect them to be found in CSs. However, analysis of Nova targets also revealed that the pyrimidine positions flanking the core CA show a large amount of degeneracy (Ule et al. 2003). Therefore, the fact that CSMs matching these sites were not found is likely due to the high level of stringency that we employed. Future analysis, allowing for degeneracy, may therefore prove useful.
Lastly, we demonstrated that there is a strong association between conservation of specific n-mers and apparent functionality since conserved occurrences of these n-mers are statistically associated with alternative splicing while nonconserved occurrences are not. This strongly suggests that there is a fundamental difference between random occurrences of n-mers and functional occurrences. The most likely explanation for this phenomenon is that higher order associations exist between functionally relevant instances of a potential binding site and the local context (e.g., other cis-elements or RNA secondary structure). Future analysis to elucidate such associations may be important for uncovering the higher order language of splice-site definition.
Methods
Extraction of conserved and RS populations
A database of human introns was constructed using sequences obtained from the May 2004 GenBank release build 35 and gene predictions from the NCBI RefSeq project (http://hgdownload.cse.ucsc.edu/goldenPath/hg17/database/). Predicted introns that did not begin with GY and end with AG were discarded. We should note that this would not exclude the relatively small population of GT-AG U12-dependent introns (Sharp and Burge 1997; Dietrich et al. 2005); however, these are few when compared with the large population of U2-dependent introns. As discussed in the Results section, we chose to focus exclusively on long introns (i.e., >199 bases in length). After excluding introns that did not meet the established criteria, we were left with 145,325 unique donor splice-junction sequences and 144,884 acceptor splice-junction sequences.
Custom software (available upon request) was used to extract CSs from the intronic sequences flanking splice-junctions (Fig. 2). A CS was defined to be a contiguous run of at least 7 nt of identity from an alignment between human and six eutherian mammals: Pan troglodytes (chimp), Macaca mulatta (rhesus monkey), Mus musculus (house mouse), Rattus norvegicus (house rat), Canis lupus familiaris (domestic dog), and Bos taurus (domestic cow). The alignment used was the UCSC alignment of 17 vertebrate genomes (hg17, March 2004, http://hgdownload.cse.ucsc.edu/goldenPath/hg17/multiz17way/). Intronic sequences were extracted from the first 7–100 nt for the donor (DI) and the last 100 to last 4 nt for the acceptor (AI) region (see Fig. 2). Since we included only introns that were >199 bases in length, this value eliminated overlap between the donor and acceptor sides of the intron. Extracted CSs were categorized according to the region from which they were recovered. The splice-junction database and CS sequences are available upon request.
For each CS identified, we also extracted 100 additional analogous (e.g., having the same splice-junction relative starting position and the same length) sequences as the CS from introns randomly chosen from the original data set of all human introns. These sequences (equivalent to 100 times the size of the CS samples) made up the RS pool.
Acknowledgments
We thank those who gave valuable feedback during preparation of this manuscript: Stephen M. Garrey, Marcus J. Lanskey, Amy Mahady, Scott Mahady, Kristy Henscheid, Jill I. Murray, Emily Goers, Pascale M. Voelker, and Alice Barkan. This work was partially supported by the American Heart Association grant 0420073Z to R.B.V. and grants from NSF (MCB-0616264) and NIH (AR053903) to J.A.B.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6017807
References
- Agresti A., Coull B.A., Coull B.A. Approximate is better than exact for interval estimation of binomial proportions. Am. Stat. 1998;52:119–126. [Google Scholar]
- Baraniak A.P., Chen J.R., Garcia-Blanco M.A., Chen J.R., Garcia-Blanco M.A., Garcia-Blanco M.A. Fox-2 mediates epithelial cell-specific fibroblast growth factor receptor 2 exon choice. Mol. Cell. Biol. 2006;26:1209–1222. doi: 10.1128/MCB.26.4.1209-1222.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau C., Paillard L., Osborne H.B., Paillard L., Osborne H.B., Osborne H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2005;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau C., Paillard L., Mereau A., Osborne H.B., Paillard L., Mereau A., Osborne H.B., Mereau A., Osborne H.B., Osborne H.B. Mammalian CELF/Bruno-like RNA-binding proteins: Molecular characteristics and biological functions. Biochimie. 2006;88:515–525. doi: 10.1016/j.biochi.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Berglund J.A., Chua K., Abovich N., Reed R., Rosbash M., Chua K., Abovich N., Reed R., Rosbash M., Abovich N., Reed R., Rosbash M., Reed R., Rosbash M., Rosbash M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell. 1997;89:781–787. doi: 10.1016/s0092-8674(00)80261-5. [DOI] [PubMed] [Google Scholar]
- Berglund J.A., Abovich N., Rosbash M., Abovich N., Rosbash M., Rosbash M. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes & Dev. 1998a;12:858–867. doi: 10.1101/gad.12.6.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund J.A., Fleming M.L., Rosbash M., Fleming M.L., Rosbash M., Rosbash M. The KH domain of the branchpoint sequence binding protein determines specificity for the pre-mRNA branchpoint sequence. RNA. 1998b;4:998–1006. doi: 10.1017/s1355838298980499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird C.P., Stranger B.E., Dermitzakis E.T., Stranger B.E., Dermitzakis E.T., Dermitzakis E.T. Functional variation and evolution of non-coding DNA. Curr. Opin. Genet. Dev. 2006;16:559–564. doi: 10.1016/j.gde.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Blencowe B.J. Exonic splicing enhancers: Mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci. 2000;25:106–110. doi: 10.1016/s0968-0004(00)01549-8. [DOI] [PubMed] [Google Scholar]
- Brow D.A. Allosteric cascade of spliceosome activation. Annu. Rev. Genet. 2002;36:333–360. doi: 10.1146/annurev.genet.36.043002.091635. [DOI] [PubMed] [Google Scholar]
- Brudno M., Gelfand M.S., Spengler S., Zorn M., Dubchak I., Conboy J.G., Gelfand M.S., Spengler S., Zorn M., Dubchak I., Conboy J.G., Spengler S., Zorn M., Dubchak I., Conboy J.G., Zorn M., Dubchak I., Conboy J.G., Dubchak I., Conboy J.G., Conboy J.G. Computational analysis of candidate intron regulatory elements for tissue-specific alternative pre-mRNA splicing. Nucleic Acids Res. 2001;29:2338–2348. doi: 10.1093/nar/29.11.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C.B., Tuschl T., Sharp P.A., Tuschl T., Sharp P.A., Sharp P.A. Splicing of precursors to mRNAs by the spliceosomes. In: Gesteland R.F., et al., editors. The RNA world. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1999. pp. 525–560. [Google Scholar]
- Cartegni L., Chew S.L., Krainer A.R., Chew S.L., Krainer A.R., Krainer A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- Cooper G.M., Sidow A., Sidow A. Genomic regulatory regions: Insights from comparative sequence analysis. Curr. Opin. Genet. Dev. 2003;13:604–610. doi: 10.1016/j.gde.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Del Gatto-Konczak F., Bourgeois C.F., Le Guiner C., Kister L., Gesnel M.C., Stevenin J., Breathnach R., Bourgeois C.F., Le Guiner C., Kister L., Gesnel M.C., Stevenin J., Breathnach R., Le Guiner C., Kister L., Gesnel M.C., Stevenin J., Breathnach R., Kister L., Gesnel M.C., Stevenin J., Breathnach R., Gesnel M.C., Stevenin J., Breathnach R., Stevenin J., Breathnach R., Breathnach R. The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5′ splice site. Mol. Cell. Biol. 2000;20:6287–6299. doi: 10.1128/mcb.20.17.6287-6299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dember L.M., Kim N.D., Liu K.Q., Anderson P., Kim N.D., Liu K.Q., Anderson P., Liu K.Q., Anderson P., Anderson P. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J. Biol. Chem. 1996;271:2783–2788. doi: 10.1074/jbc.271.5.2783. [DOI] [PubMed] [Google Scholar]
- Dermitzakis E.T., Reymond A., Antonarakis S.E., Reymond A., Antonarakis S.E., Antonarakis S.E. Conserved non-genic sequences—an unexpected feature of mammalian genomes. Nat. Rev. Genet. 2005;6:151–157. doi: 10.1038/nrg1527. [DOI] [PubMed] [Google Scholar]
- Dietrich R.C., Fuller J.D., Padgett R.A., Fuller J.D., Padgett R.A., Padgett R.A. A mutational analysis of U12-dependent splice site dinucleotides. RNA. 2005;11:1430–1440. doi: 10.1261/rna.7206305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge B.K., Darnell R.B., Darnell R.B. Nova regulates GABA(A) receptor γ2 alternative splicing via a distal downstream UCAU-rich intronic splicing enhancer. Mol. Cell. Biol. 2003;23:4687–4700. doi: 10.1128/MCB.23.13.4687-4700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge B.K., Stefani G., Engelhard C.C., Darnell R.B., Stefani G., Engelhard C.C., Darnell R.B., Engelhard C.C., Darnell R.B., Darnell R.B. Nova autoregulation reveals dual functions in neuronal splicing. EMBO J. 2005;24:1608–1620. doi: 10.1038/sj.emboj.7600630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother W.G., Yeh R.F., Sharp P.A., Burge C.B., Yeh R.F., Sharp P.A., Burge C.B., Sharp P.A., Burge C.B., Burge C.B. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- Faustino N.A., Cooper T.A., Cooper T.A. Pre-mRNA splicing and human disease. Genes & Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- Fedorov A., Saxonov S., Fedorova L., Daizadeh I., Saxonov S., Fedorova L., Daizadeh I., Fedorova L., Daizadeh I., Daizadeh I. Comparison of intron-containing and intron-lacking human genes elucidates putative exonic splicing enhancers. Nucleic Acids Res. 2001;29:1464–1469. doi: 10.1093/nar/29.7.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forch P., Puig O., Kedersha N., Martinez C., Granneman S., Seraphin B., Anderson P., Valcarcel J., Puig O., Kedersha N., Martinez C., Granneman S., Seraphin B., Anderson P., Valcarcel J., Kedersha N., Martinez C., Granneman S., Seraphin B., Anderson P., Valcarcel J., Martinez C., Granneman S., Seraphin B., Anderson P., Valcarcel J., Granneman S., Seraphin B., Anderson P., Valcarcel J., Seraphin B., Anderson P., Valcarcel J., Anderson P., Valcarcel J., Valcarcel J. The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol. Cell. 2000;6:1089–1098. doi: 10.1016/s1097-2765(00)00107-6. [DOI] [PubMed] [Google Scholar]
- Fox-Walsh K.L., Dou Y., Lam B.J., Hung S.P., Baldi P.F., Hertel K.J., Dou Y., Lam B.J., Hung S.P., Baldi P.F., Hertel K.J., Lam B.J., Hung S.P., Baldi P.F., Hertel K.J., Hung S.P., Baldi P.F., Hertel K.J., Baldi P.F., Hertel K.J., Hertel K.J. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc. Natl. Acad. Sci. 2005;102:16176–16181. doi: 10.1073/pnas.0508489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A., Richard S., Richard S. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat. Struct. Mol. Biol. 2005;12:691–698. doi: 10.1038/nsmb963. [DOI] [PubMed] [Google Scholar]
- Garrey S.M., Voelker R., Berglund J.A., Voelker R., Berglund J.A., Berglund J.A. An extended RNA binding site for the yeast branch point-binding protein and the role of its zinc knuckle domains in RNA binding. J. Biol. Chem. 2006;281:27443–27453. doi: 10.1074/jbc.M603137200. [DOI] [PubMed] [Google Scholar]
- Goren A., Ram O., Amit M., Keren H., Lev-Maor G., Vig I., Pupko T., Ast G., Ram O., Amit M., Keren H., Lev-Maor G., Vig I., Pupko T., Ast G., Amit M., Keren H., Lev-Maor G., Vig I., Pupko T., Ast G., Keren H., Lev-Maor G., Vig I., Pupko T., Ast G., Lev-Maor G., Vig I., Pupko T., Ast G., Vig I., Pupko T., Ast G., Pupko T., Ast G., Ast G. Comparative analysis identifies exonic splicing regulatory sequences—the complex definition of enhancers and silencers. Mol. Cell. 2006;22:769–781. doi: 10.1016/j.molcel.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Ho T.H., Charlet B.N., Poulos M.G., Singh G., Swanson M.S., Cooper T.A., Charlet B.N., Poulos M.G., Singh G., Swanson M.S., Cooper T.A., Poulos M.G., Singh G., Swanson M.S., Cooper T.A., Singh G., Swanson M.S., Cooper T.A., Swanson M.S., Cooper T.A., Cooper T.A. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh G.S., Hynes R.O., Hynes R.O. Regulation of alternative pre-mRNA splicing by a novel repeated hexanucleotide element. Genes & Dev. 1994;8:1561–1574. doi: 10.1101/gad.8.13.1561. [DOI] [PubMed] [Google Scholar]
- Itoh M., Haga I., Li Q.H., Fujisawa J., Haga I., Li Q.H., Fujisawa J., Li Q.H., Fujisawa J., Fujisawa J. Identification of cellular mRNA targets for RNA-binding protein Sam68. Nucleic Acids Res. 2002;30:5452–5464. doi: 10.1093/nar/gkf673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K.B., Dredge B.K., Stefani G., Zhong R., Buckanovich R.J., Okano H.J., Yang Y.Y., Darnell R.B., Dredge B.K., Stefani G., Zhong R., Buckanovich R.J., Okano H.J., Yang Y.Y., Darnell R.B., Stefani G., Zhong R., Buckanovich R.J., Okano H.J., Yang Y.Y., Darnell R.B., Zhong R., Buckanovich R.J., Okano H.J., Yang Y.Y., Darnell R.B., Buckanovich R.J., Okano H.J., Yang Y.Y., Darnell R.B., Okano H.J., Yang Y.Y., Darnell R.B., Yang Y.Y., Darnell R.B., Darnell R.B. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron. 2000a;25:359–371. doi: 10.1016/s0896-6273(00)80900-9. [DOI] [PubMed] [Google Scholar]
- Jensen K.B., Musunuru K., Lewis H.A., Burley S.K., Darnell R.B., Musunuru K., Lewis H.A., Burley S.K., Darnell R.B., Lewis H.A., Burley S.K., Darnell R.B., Burley S.K., Darnell R.B., Darnell R.B. The tetranucleotide UCAY directs the specific recognition of RNA by the Nova K-homology 3 domain. Proc. Natl. Acad. Sci. 2000b;97:5740–5745. doi: 10.1073/pnas.090553997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Suzuki H., Maegawa S., Endo H., Sugano S., Hashimoto K., Yasuda K., Inoue K., Suzuki H., Maegawa S., Endo H., Sugano S., Hashimoto K., Yasuda K., Inoue K., Maegawa S., Endo H., Sugano S., Hashimoto K., Yasuda K., Inoue K., Endo H., Sugano S., Hashimoto K., Yasuda K., Inoue K., Sugano S., Hashimoto K., Yasuda K., Inoue K., Hashimoto K., Yasuda K., Inoue K., Yasuda K., Inoue K., Inoue K. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 2003;22:905–912. doi: 10.1093/emboj/cdg089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat J.L., Barberan-Soler S., McKenna P., Clawson H., Farrer T., Zahler A.M., Barberan-Soler S., McKenna P., Clawson H., Farrer T., Zahler A.M., McKenna P., Clawson H., Farrer T., Zahler A.M., Clawson H., Farrer T., Zahler A.M., Farrer T., Zahler A.M., Zahler A.M. Intronic alternative splicing regulators identified by comparative genomics in nematodes. PLoS Comput. Biol. 2006;2 doi: 10.1371/journal.pcbi.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Pringle T.H., Zahler A.M., Haussler D., Zahler A.M., Haussler D., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd A.N., Cooper T.A., Cooper T.A. Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol. 2002 doi: 10.1186/gb-2002-3-11-reviews0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guiner C., Lejeune F., Galiana D., Kister L., Breathnach R., Stevenin J., Del Gatto-Konczak F., Lejeune F., Galiana D., Kister L., Breathnach R., Stevenin J., Del Gatto-Konczak F., Galiana D., Kister L., Breathnach R., Stevenin J., Del Gatto-Konczak F., Kister L., Breathnach R., Stevenin J., Del Gatto-Konczak F., Breathnach R., Stevenin J., Del Gatto-Konczak F., Stevenin J., Del Gatto-Konczak F., Del Gatto-Konczak F. TIA-1 and TIAR activate splicing of alternative exons with weak 5′ splice sites followed by a U-rich stretch on their own pre-mRNAs. J. Biol. Chem. 2001;276:40638–40646. doi: 10.1074/jbc.M105642200. [DOI] [PubMed] [Google Scholar]
- Lim L.P., Sharp P.A., Sharp P.A. Alternative splicing of the fibronectin EIIIB exon depends on specific TGCATG repeats. Mol. Cell. Biol. 1998;18:3900–3906. doi: 10.1128/mcb.18.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q., Taylor S.J., Shalloway D., Taylor S.J., Shalloway D., Shalloway D. Specificity and determinants of Sam68 RNA binding. Implications for the biological function of K homology domains. J. Biol. Chem. 1997;272:27274–27280. doi: 10.1074/jbc.272.43.27274. [DOI] [PubMed] [Google Scholar]
- Lou H., Yang Y., Cote G.J., Berget S.M., Gagel R.F., Yang Y., Cote G.J., Berget S.M., Gagel R.F., Cote G.J., Berget S.M., Gagel R.F., Berget S.M., Gagel R.F., Gagel R.F. An intron enhancer containing a 5′ splice site sequence in the human calcitonin/calcitonin gene-related peptide gene. Mol. Cell. Biol. 1995;15:7135–7142. doi: 10.1128/mcb.15.12.7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis J., Paillard L., Audic Y., Cosson B., Danos O., Le Bec C., Osborne H.B., Paillard L., Audic Y., Cosson B., Danos O., Le Bec C., Osborne H.B., Audic Y., Cosson B., Danos O., Le Bec C., Osborne H.B., Cosson B., Danos O., Le Bec C., Osborne H.B., Danos O., Le Bec C., Osborne H.B., Le Bec C., Osborne H.B., Osborne H.B. CUG-BP1/CELF1 requires UGU-rich sequences for high-affinity binding. Biochem. J. 2006;400:291–301. doi: 10.1042/BJ20060490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin A.J., Clark F., Smith C.W., Clark F., Smith C.W., Smith C.W. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- Matter N., Herrlich P., Konig H., Herrlich P., Konig H., Konig H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature. 2002;420:691–695. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- Minovitsky S., Gee S.L., Schokrpur S., Dubchak I., Conboy J.G., Gee S.L., Schokrpur S., Dubchak I., Conboy J.G., Schokrpur S., Dubchak I., Conboy J.G., Dubchak I., Conboy J.G., Conboy J.G. The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic Acids Res. 2005;33:714–724. doi: 10.1093/nar/gki210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata S., Kawamoto S., Kawamoto S. Tissue-dependent isoforms of mammalian Fox-1 homologs are associated with tissue-specific splicing activities. Nucleic Acids Res. 2005;33:2078–2089. doi: 10.1093/nar/gki338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne R.J., Thornton C.A., Thornton C.A. RNA-dominant diseases. Hum. Mol. Genet. 2006;15:R162–R169. doi: 10.1093/hmg/ddl181. (spec. no. 2) [DOI] [PubMed] [Google Scholar]
- Paronetto M.P., Achsel T., Massiello A., Chalfant C.E., Sette C., Achsel T., Massiello A., Chalfant C.E., Sette C., Massiello A., Chalfant C.E., Sette C., Chalfant C.E., Sette C., Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol. 2007;176:929–939. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M., Vicente M., Monferrer L., Artero R., Vicente M., Monferrer L., Artero R., Monferrer L., Artero R., Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74:65–80. doi: 10.1111/j.1432-0436.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- Ponthier J.L., Schluepen C., Chen W., Lersch R.A., Gee S.L., Hou V.C., Lo A.J., Short S.A., Chasis J.A., Winkelmann J.C., Schluepen C., Chen W., Lersch R.A., Gee S.L., Hou V.C., Lo A.J., Short S.A., Chasis J.A., Winkelmann J.C., Chen W., Lersch R.A., Gee S.L., Hou V.C., Lo A.J., Short S.A., Chasis J.A., Winkelmann J.C., Lersch R.A., Gee S.L., Hou V.C., Lo A.J., Short S.A., Chasis J.A., Winkelmann J.C., Gee S.L., Hou V.C., Lo A.J., Short S.A., Chasis J.A., Winkelmann J.C., Hou V.C., Lo A.J., Short S.A., Chasis J.A., Winkelmann J.C., Lo A.J., Short S.A., Chasis J.A., Winkelmann J.C., Short S.A., Chasis J.A., Winkelmann J.C., Chasis J.A., Winkelmann J.C., Winkelmann J.C., et al. Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. J. Biol. Chem. 2006;281:12468–12474. doi: 10.1074/jbc.M511556200. [DOI] [PubMed] [Google Scholar]
- Pozzoli U., Sironi M., Sironi M. Silencers regulate both constitutive and alternative splicing events in mammals. Cell. Mol. Life Sci. 2005;62:1579–1604. doi: 10.1007/s00018-005-5030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K.D., Tatusova T., Maglott D.R., Tatusova T., Maglott D.R., Maglott D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder S.P., Williamson J.R., Williamson J.R. Specificity of the STAR/GSG domain protein Qk1: implications for the regulation of myelination. RNA. 2004;10:1449–1458. doi: 10.1261/rna.7780504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P.A., Burge C.B., Burge C.B. Classification of introns: U2-type or U12-type. Cell. 1997;91:875–879. doi: 10.1016/s0092-8674(00)80479-1. [DOI] [PubMed] [Google Scholar]
- Sironi M., Menozzi G., Comi G.P., Cagliani R., Bresolin N., Pozzoli U., Menozzi G., Comi G.P., Cagliani R., Bresolin N., Pozzoli U., Comi G.P., Cagliani R., Bresolin N., Pozzoli U., Cagliani R., Bresolin N., Pozzoli U., Bresolin N., Pozzoli U., Pozzoli U. Analysis of intronic conserved elements indicates that functional complexity might represent a major source of negative selection on non-coding sequences. Hum. Mol. Genet. 2005;14:2533–2546. doi: 10.1093/hmg/ddi257. [DOI] [PubMed] [Google Scholar]
- Sorek R., Ast G., Ast G. Intronic sequences flanking alternatively spliced exons are conserved between human and mouse. Genome Res. 2003;13:1631–1637. doi: 10.1101/gr.1208803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R., Dror G., Shamir R., Dror G., Shamir R., Shamir R. Assessing the number of ancestral alternatively spliced exons in the human genome. BMC Genomics. 2006;7:273. doi: 10.1186/1471-2164-7-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark H., Luhrmann R., Luhrmann R. Cryo-electron microscopy of spliceosomal components. Annu. Rev. Biophys. Biomol. Struct. 2006;35:435–457. doi: 10.1146/annurev.biophys.35.040405.101953. [DOI] [PubMed] [Google Scholar]
- Sugnet C.W., Kent W.J., Ares M., Jr., Haussler D., Kent W.J., Ares M., Jr., Haussler D., Ares M., Jr., Haussler D., Haussler D. Transcriptome and genome conservation of alternative splicing events in humans and mice. Pac. Symp. Biocomput. 2004: 66–77. doi: 10.1142/9789812704856_0007. [DOI] [PubMed] [Google Scholar]
- Sugnet C.W., Srinivasan K., Clark T.A., O’Brien G., Cline M.S., Wang H., Williams A., Kulp D., Blume J.E., Haussler D., Srinivasan K., Clark T.A., O’Brien G., Cline M.S., Wang H., Williams A., Kulp D., Blume J.E., Haussler D., Clark T.A., O’Brien G., Cline M.S., Wang H., Williams A., Kulp D., Blume J.E., Haussler D., O’Brien G., Cline M.S., Wang H., Williams A., Kulp D., Blume J.E., Haussler D., Cline M.S., Wang H., Williams A., Kulp D., Blume J.E., Haussler D., Wang H., Williams A., Kulp D., Blume J.E., Haussler D., Williams A., Kulp D., Blume J.E., Haussler D., Kulp D., Blume J.E., Haussler D., Blume J.E., Haussler D., Haussler D., et al. Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLoS Comput. Biol. 2006;2:1–14. doi: 10.1371/journal.pcbi.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa M., Li N., Bailey T.L., Church G.M., De Moor B., Eskin E., Favorov A.V., Frith M.C., Fu Y., Kent W.J., Li N., Bailey T.L., Church G.M., De Moor B., Eskin E., Favorov A.V., Frith M.C., Fu Y., Kent W.J., Bailey T.L., Church G.M., De Moor B., Eskin E., Favorov A.V., Frith M.C., Fu Y., Kent W.J., Church G.M., De Moor B., Eskin E., Favorov A.V., Frith M.C., Fu Y., Kent W.J., De Moor B., Eskin E., Favorov A.V., Frith M.C., Fu Y., Kent W.J., Eskin E., Favorov A.V., Frith M.C., Fu Y., Kent W.J., Favorov A.V., Frith M.C., Fu Y., Kent W.J., Frith M.C., Fu Y., Kent W.J., Fu Y., Kent W.J., Kent W.J., et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B., Ruggiu M., Mele A., Ule A., Darnell R.B., Mele A., Ule A., Darnell R.B., Ule A., Darnell R.B., Darnell R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B., Gaasterland T., Blencowe B.J., Darnell R.B., Blencowe B.J., Darnell R.B., Darnell R.B. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- Wang Z., Rolish M.E., Yeo G., Tung V., Mawson M., Burge C.B., Rolish M.E., Yeo G., Tung V., Mawson M., Burge C.B., Yeo G., Tung V., Mawson M., Burge C.B., Tung V., Mawson M., Burge C.B., Mawson M., Burge C.B., Burge C.B. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Wang Z., Xiao X., Van Nostrand E., Burge C.B., Xiao X., Van Nostrand E., Burge C.B., Van Nostrand E., Burge C.B., Burge C.B. General and specific functions of exonic splicing silencers in splicing control. Mol. Cell. 2006;23:61–70. doi: 10.1016/j.molcel.2006.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.I., Reed R.B., Grabowski P.J., Artzt K., Reed R.B., Grabowski P.J., Artzt K., Grabowski P.J., Artzt K., Artzt K. Function of quaking in myelination: regulation of alternative splicing. Proc. Natl. Acad. Sci. 2002;99:4233–4238. doi: 10.1073/pnas.072090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G., Hoon S., Venkatesh B., Burge C.B., Hoon S., Venkatesh B., Burge C.B., Venkatesh B., Burge C.B., Burge C.B. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc. Natl. Acad. Sci. 2004;101:15700–15705. doi: 10.1073/pnas.0404901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G.W., Van Nostrand E., Holste D., Poggio T., Burge C.B., Van Nostrand E., Holste D., Poggio T., Burge C.B., Holste D., Poggio T., Burge C.B., Poggio T., Burge C.B., Burge C.B. Identification and analysis of alternative splicing events conserved in human and mouse. Proc. Natl. Acad. Sci. 2005;102:2850–2855. doi: 10.1073/pnas.0409742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.H., Chasin L.A., Chasin L.A. Computational definition of sequence motifs governing constitutive exon splicing. Genes & Dev. 2004;18:1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.H., Heller K.A., Hefter I., Leslie C.S., Chasin L.A., Heller K.A., Hefter I., Leslie C.S., Chasin L.A., Hefter I., Leslie C.S., Chasin L.A., Leslie C.S., Chasin L.A., Chasin L.A. Sequence information for the splicing of human pre-mRNA identified by support vector machine classification. Genome Res. 2003;13:2637–2650. doi: 10.1101/gr.1679003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.H., Leslie C.S., Chasin L.A., Leslie C.S., Chasin L.A., Chasin L.A. Computational searches for splicing signals. Methods. 2005;37:292–305. doi: 10.1016/j.ymeth.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Zheng Z.M. Regulation of alternative RNA splicing by exon definition and exon sequences in viral and mammalian gene expression. J. Biomed. Sci. 2004;11:278–294. doi: 10.1159/000077096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.L., Baraniak A.P., Lou H., Baraniak A.P., Lou H., Lou H. Role for Fox-1/Fox-2 in mediating the neuronal pathway of calcitonin/calcitonin gene-related peptide alternative RNA processing. Mol. Cell. Biol. 2007;27:830–841. doi: 10.1128/MCB.01015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Hasman R.A., Young K.M., Kedersha N.L., Lou H., Hasman R.A., Young K.M., Kedersha N.L., Lou H., Young K.M., Kedersha N.L., Lou H., Kedersha N.L., Lou H., Lou H. U1 snRNP-dependent function of TIAR in the regulation of alternative RNA processing of the human calcitonin/CGRP pre-mRNA. Mol. Cell. Biol. 2003;23:5959–5971. doi: 10.1128/MCB.23.17.5959-5971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Hasman R.A., Barron V.A., Luo G., Lou H., Hasman R.A., Barron V.A., Luo G., Lou H., Barron V.A., Luo G., Lou H., Luo G., Lou H., Lou H. A nuclear function of Hu proteins as neuron-specific alternative RNA processing regulators. Mol. Biol. Cell. 2006;17:5105–5114. doi: 10.1091/mbc.E06-02-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]