

Abstract
The airway epithelium provides a protective barrier against inhaled environmental toxins and microorganisms, and epithelial injury initiates a number of processes to restore its barrier integrity, including activation of matrix metalloproteinases such as MMP-9 (92-kD gelatinase B). Airway epithelial cells continuously produce nitric oxide (NO), which has been linked to cell migration and MMP-9 regulation in several cell types, but the importance of epithelial NO in mediating airway epithelial repair or MMP-9 activation is unknown. Using primary or immortalized human bronchial epithelial cells, we demonstrate that low concentrations of NO promote epithelial cell migration and wound repair in an in vitro wound assay, which was associated with increased localized expression and activation of MMP-9. In addition, in HBE1 cells that were stably transfected with inducible NOS (NOS2), to mimic constitutive epithelial NOS2 expression in vivo, NOS inhibition decreased epithelial wound repair and MMP-9 expression. The stimulatory effects of NO on epithelial wound repair and MMP-9 expression were dependent on cGMP-mediated pathways and were inhibited by 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ), an inhibitor of soluble guanylyl cyclase. Inhibition of cGMP-dependent protein kinase (PKG) attenuated NO-mediated epithelial wound closure, but did not affect MMP-9 expression. However, pharmacologic MMP inhibition and siRNA knockdown of MMP-9 expression demonstrated the contribution of MMP-9 to NO-mediated wound closure. Overall, our results demonstrate that NOS2-derived NO contributes to airway epithelial repair by both PKG-dependent and -independent mechanisms, and involves NO-dependent expression and activation of MMP-9.
Keywords: cell migration, cGMP, guanylyl cyclase, DT-2
CLINICAL RELEVANCE
This article discusses mechanisms by which airway epithelial-derived nitric oxide (NO) facilitates cell migration and epithelial repair following injury, and this study contributes to our overall understanding of epithelial NO biology.
The airway epithelium serves a primary role as a protective barrier against inhaled environmental toxins and microorganisms. Conditions of epithelial injury are associated with activation of a wound repair response within the epithelium, involving a complex sequence of events promoting epithelial cell migration and proliferation. Among these events are the induction and activation of several members of the matrix metalloproteinase (MMP) family, which comprise membrane-associated or secreted neutral endopeptidases that are involved in extracellular matrix degradation and remodeling during wound repair, inflammation, angiogenesis, tumor invasion, and metastasis (1, 2). One of these MMPs, MMP-9 (92-kD gelatinase B), is typically activated during injurious conditions, and is produced by different cell types, including macrophages and neutrophils, as well as the airway epithelium, and plays various roles in growth, development, inflammation, and remodeling (1–3). Studies with MMP-9–deficient mice have shown diverse roles of MMP-9 in lung injury and in models of allergic airway diseases (3–5). Expression and activation of MMP-9 is subject to tight regulation at different levels. The transcriptional regulation of MMP-9 is complex and depends on the activation of mitogen-activated protein kinase (MAPK) and NF-κB signaling pathways, in addition to other regulatory elements (6, 7). Furthermore, the mechanisms that control MMP-9 activation after it has been secreted are still incompletely understood (8). Although MMP-9 is normally expressed at low levels within airway epithelial cells, it can be induced by proinflammatory cytokines such as TNF-α, and appears to be an important factor that regulates epithelial cell migration and wound repair after injury (9–11).
The respiratory epithelium continuously produces nitric oxide (NO) largely due to constitutive expression of inducible nitric oxide synthase (NOS2) (12). Although NOS2 is typically induced under conditions of infection or inflammation, constitutive NOS2 expression within the airway epithelial may serve functions in innate host defense (12), or regulation of epithelial ion transport (13, 14). Another suggested function of epithelial NOS2 is to promote wound repair (15, 16), which may be related to the ability of NO to promote cell migration in various cell types (17–19). However, the importance for NOS2 in airway epithelial repair is currently unknown.
Given the various effects of NO on cell signaling and gene expression (20, 21), epithelial NOS2 could serve to regulate epithelial expression of genes involved in inflammatory-immune processes or epithelial repair and remodeling. Indeed, the potential for regulation of MMP-9 expression and activation by NOS2-derived NO has received considerable interest in previous studies (22, 23). Although several studies have suggested that NO inhibits cytokine-mediated MMP-9 expression in mesangial cells or vascular smooth muscle cells (22, 24), others have reported contrasting observations in vascular smooth muscle cells and chrondocytes (25). We have previously demonstrated that cytokine-induced MMP-9 expression in tracheobronchial epithelial cells is suppressed by exogenous high concentrations of NO and S-nitrosothiols (26), but it is unclear whether this is relevant to MMP-9 regulation by endogenous NOS activity under normal physiologic conditions.
The present studies were designed to investigate the involvement of epithelial NOS2 in airway epithelial cell migration and repair in an in vitro wound model, using an exogenous NO donor (DETA NONOate) and stable transfection with NOS2 in an attempt to mimic continuous NO production by airway epithelial cells in vivo. Overall, our results demonstrate that low, physiologically relevant concentrations of NO promote airway epithelial cell migration and wound repair, and that this involves cGMP-dependent expression and activation of MMP-9.
MATERIALS AND METHODS
Cell Culture and Treatments
Experiments were performed with primary normal human bronchial epithelial cells (NHBE) obtained from Cambrex Bio Science (Walkersville, MD) between passage numbers 3 and 5, a papilloma virus–immortalized human bronchial epithelial cell line (HBE1) (kindly provided by Drs. R. Wu [University of California, Davis, CA] and J. Yankaskas [University of North Carolina, Chapel Hill, NC]), and HBE1 cells that were stably transfected with NOS2 (26) and will be referred to as HBE1-NOS2 cells. Because HBE1 cells do not express detectable NOS2, we generated HBE1-NOS2 cells in an attempt to mimic constitutive airway epithelial NOS2 expression in vivo (27). Cells were grown and maintained as previously described (33) and experiments were performed in 6-, 12-, or 24-well plates (Corning, Corning, NY), or in 8-well chamber slides (Nunc, Rochester, NY). After changing the medium or after cell injury (see next section), cells were treated with the slow-releasing NO-donor, diethylenetriamine NONOate (DETA NONOate 10–500 μM; Cayman Chemical, Ann Arbor, MI; t1/2 = 20 h at 37°C), human TNF-α (100 ng/ml; Sigma, St Louis, MO), or 8-bromoguanosine 3′5′-cyclic monophosphate (8-Br-cGMP 10 μM; Sigma) for up to 24 h. Cells were pretreated, where indicated, for 30 min with the soluble guanylyl cylase inhibitor 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ 10 μM; Sigma), endogenous NOS activity was blocked using NG-monomethyl-L-arginine (l-NMMA 1 mM; Alexis, San Diego, CA), or the specific NOS2 inhibitor, 1400W (100 μM; Alexis). Where indicated, the PKG inhibitors KT5823 (10 μM; Sigma) and DT-2 (10 μM) (28), or the MMP inhibitor (MMP Inhibitor II 1 μM; Calbiochem, La Jolla, CA) were added 30 min before experimentation. None of the agents used significantly affected cell morphology or viability under these conditions.
In Vitro Wound Repair Assay
To investigate the effects of NO on epithelial cell migration, we used a common in vitro wound assay, in which confluent cell monolayers are mechanically wounded by creating a linear scratch. Although wound repair in such in vitro injury models is a complex process involving both cell migration and proliferation, the initial phase of wound repair involves primarily cell migration (29, 30). After introduction of a linear wound of ∼ 0.5 mm width using a sterile P-200 pipette tip, cell monolayers were washed with media to remove cell debris, fresh media was added to each well, and appropriate reagents were administered. Closure of linear wounds was followed for 24 h, using an IX70 inverted microscope (Olympus, Center Valley, PA) using UltraView v4 software (Perkin Elmer Life Sciences, Wellesley, MA). Wound closure was expressed as a percentage of the initial wound area, quantitated using NIH ImageJ software.
Cell Migration
For more quantitative analysis of epithelial cell migration, cells were seeded at 5.0 × 104 cells/well in 8.0 μm polycarbonate membranes (Nunc), and the ability of cells to migrate into these membranes was followed. Twenty-four hours after cell treatments, nonmigrated cells were removed with a cotton swab, and cells that migrated through the membrane pores were fixed with 4% paraformaldehyde (PFA), stained with 0.5% crystal violet in 20% methanol, and extracted in 0.2 M sodium acetate (pH 4.5) for quantitation by absorbance at 562 nm using a Biotek Synergy HT microplate reader (Winooski, VT).
Analysis of MMP-9 by Gelatin Zymography
Secretion of MMP-9 in the culture media was analyzed by gelatin zymography, as described previously (26). Gelatinolytic bands were scanned and digitized for quantitation of band intensity using NIH ImageJ software.
RT-PCR Analysis of MMP-9 Expression
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA) according to the manufucturer's protocol. Reverse transcription (RT) was performed using 2–5 μg of total RNA and PCR reactions were performed using a GeneAmp PCR System 9,700 (Applied Biosystems, Foster City, CA), as detailed previously (26). PCR products were resolved by 1% agarose gel electrophoresis and visualized by ethidium bromide staining. The expression of MMP-9 and uPA was normalized to that of GAPDH by band densitometry analysis using NIH ImageJ software.
MMP-9 Reporter Assay
To determine the transcriptional activation of MMP-9 by NO, cells were transiently transfected with the pGL3/MMP-9/Luc-670 construct (generously provided by Dr. D. Boyd, University of Texas, Houston, TX) (31). Briefly, 1 μg of construct was incubated with the transfection reagent (Lipofectamine and Plus Reagent; Invitrogen) at room temperature, and 100 μl of the transfection mixture was added per well and incubated for 24 h at 37°C. After appropriate treatments, luciferase activity was determined in cell lysates, according to the manufacturer's procedures (Promega, Madison, WI).
Measurement of NO and Metabolites
Generation of NO into the medium was monitored directly using an amiNO-FLAT Nitric Oxide Sensor (Harvard Apparatus, Holliston, MA). Alternatively, total NO production was determined by measuring accumulation of the metabolites nitrite and nitrate in the culture media. Briefly, 50 μl sample was mixed with 40 μL NADPH (1 mM; Sigma) and 40 μl nitrate reductase (160 mU/ml; Sigma) and incubated for 1 h in the dark at room temperature to convert nitrate to nitrite. Nitrite was subsequently measured by addition of 900 μl Griess reagent, and resulting absorbance at 543 nm was measured after 10 min, and compared with external standards of nitrate/nitrite that were processed similarly.
Determination of Cellular cGMP
Cellular cGMP was determined by a RIA following manufacturer's protocol (Amersham Biosciences, Piscataway, NJ). After treatments, cells were extracted in cold 0.5 M TCA (Fisher, Fair Lawn, NJ) for 30 min at 4°C, and the protein precipitate was removed by centrifugation at 6,000 × g for 10 min. The supernatant was extracted three times with 5 vols of water-saturated diethyl ether to remove TCA, and analyzed by RIA, using the acetylation protocol (range 2–128 fmol/sample).
Immunofluorescence Analysis of MMP-9 and uPA
Cells were grown to confluence on coverslips or 8-well chamber slides (Nunc) and wounded as previously described. After addition of test reagents and incubation for 24 h, cells were rinsed twice with PBS, fixed in 4% PFA/PBS for 15 min, and blocked in 2% drymilk/PBS/0.1% triton X for 45 min. Cells were probed with an MMP-9 mouse monoclonal antibody (Oncogene, Cambridge, MA) or urokinase plasminogen activator (uPA) rabbit polyclonal antibody (Santa Cruz, Santa Cruz, CA) in 2% BSA/PBS/triton X for 1 h at 37°C, washed twice with PBS, and then incubated with a secondary antibody (Alexa-Fluor 568 goat anti-mouse; Molecular Probes Inc., Eugene, OR) in 2% BSA/PBS for 1 h at 37°C. After two washes with PBS, nuclei were stained with Sytox green (1:10,000 in PBS) for 5 min. After final washing, slides were imaged using an Olympus BX50 confocal laser scanning microscope, with Lasersharp 2,000 software (Bio-Rad Laboratories, Hercules, CA).
Inhibition of MMP-9 Expression Using siRNA
To address the direct role of MMP-9 in epithelial wound closure, MMP-9 expression was silenced using pre-designed MMP-9 siRNA (Santa Cruz), following manufacturer's guidelines. Briefly, 10 μM MMP-9 siRNA or scramble control was mixed with the siRNA transfection reagent and cells were treated for 5–7 h, followed by the addition of fresh media. This siRNA treatment was repeated the next day to maximize MMP-9 knockdown, and cells were used in experiments 24 h after the second transfection. Cells were treated with DETA NONOate (10 μM) and MMP-9 expression was monitored by RT-PCR and immunofluorescence staining.
Gelatinase Activity by In Situ Zymography
HBE1 cells were grown to confluence in 8-well chamber slides, wounded with a P-200 tip, and subjected to treatment with DETA NONOate (10 μM) in the absence or presence of the MMP-9 siRNA transfection reagent (10 μM). As a positive control, cells were incubated with the MMP activator p-chloromercuribenzoic acid (p-CMB, 200 μM; Sigma) for 2 h. After treatments, cells were overlayed with DQ-Gelatin (100 μg/ml; Molecular Probes/Invitrogen) for 2 h at 37°C, washed with PBS, stained for nuclei using 4′,6-diamidino-2-phenylindole (DAPI), fixed with 4% PFA, and analyzed using fluorescence microscopy.
Analysis of proMMP-9 Activation
Recombinant human proMMP-9 was kindly provided by Dr. T. Akaike (Kumamoto University, Japan). ProMMP-9 (200 ng/ml) was incubated with either DETA NONOate (10 μM), the MMP activator p-CMB (200 μM), or the S-nitrosothiol, S-nitrosocysteine (SNOC; 200 μM) prepared as indicated (23) in the presence of the fluorogenic substrate DQ-Gelatin (50 μg/ml; Molecular Probes). MMP activation was monitored by increased fluorescence (485/515 nm) up to 8 h.
RESULTS
NO Promotes Epithelial Cell Migration and Wound Repair
We explored the role of NO in two in vitro assays of epithelial cell migration and wound repair using either NHBE or HBE1 cells. As illustrated in Figure 1A (panels a-f) and summarized in Figure 1B, low concentrations of the slow releasing NO-donor DETA NONOate (10 μM) promoted wound repair in a linear wound assay in NHBE or HBE1 cells. In addition, wound repair in HBE1 cells that were stably transfected with NOS2 was markedly decreased in the presence of the NOS inhibitor l-NMMA (1 mM) (Figures 1A, panel h-i, and 1B), as well as using the NOS2-specific inhibitor, 1400W (100 μM) (data not shown). Using different concentrations of DETA NONOate (10–500 μM), the effect of NO on wound closure was found to be dose-dependent and in fact inhibited by higher DETA NONOate concentrations (500 μM; Figure 1C). Analysis of NO or its metabolites indicated that rates of NO production from 10 μM DETA NONOate and HBE1-NOS2 cells were comparable and in the low nM range (Table 1). Nitrite/nitrate accumulation in HBE1-NOS2 media was linear over time, indicating continuous production of low levels of NO, which were below the detection limit of our NO electrode, presumably because cells actively metabolize NO. Indeed, measured NO levels from DETA NONOate were consistently lower in the presence of HBE1 cells.
Figure 1.

Airway epithelial cell migration and wound repair is enhanced by NO. (A) Confluent NHBE and HBE1 cells (panels a and d) were wounded using a p-200 pipet tip and either untreated (panels b and e) or treated with DETA NONOate (10 μM) for 24 h (panels c and f). Confluent HBE1-NOS2 cells (panel g) were similarly injured and wound closure was monitored in the absence (panel h) or presence of the NOS inhibitor, l-NMMA, added 30 min before wounding (panel i). (B) Analysis of % wound closure using NIH ImageJ software to quantitate wound areas. (C) Analysis of % wound closure in NHBE (solid bars) and HBE1 (open bars) cells using varying doses of DETA NONOate (10–500 μM). (D) Cell migration was examined using 0.8-μm pore-size polycarbonate transwell membranes. NHBE1 (solid bars) and HBE1 (open bars) cells were either treated with l-NMMA or DETA NONOate (10 μM) and HBE1-NOS2 cells (shaded bars) were treated with l-NMMA where values are compared with HBE1 CTL. Data represent mean ± SE from at least three separate experiments.
TABLE 1.
PRODUCTION OF NITRIC OXIDE AND ITS METABOLITES BY DETA NONOATE OR HBE1-NOS2 CELLS
| Cell Type | Treatment |
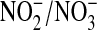 (μM) (μM) |
NO (nM)* |
|---|---|---|---|
| DETA NONOate (μM) | |||
| HBE1 | 0 | 0.4 ± 0.1 | < 10 |
| 10 | 14.1 ± 0.7 | 17.5 ± 1.7 | |
| 100 | 67.5 ± 3.8 | N.D. | |
| 500 | 389.1 ± 6.7 | 258.4 ± 8.0 | |
| l-NMMA (mM) | |||
| HBE1-NOS2 | 0 | 20.8 ± 1.3 | < 10 |
| 1 | 2.5 ± 0.9 | N.D. |
Definition of abbreviations: DETA NONOate, diethylenetriaamine NONOate; l-NMMA, NG-monomethyl-l-arginine; NO, nitric oxide.
Accumulation of the NO metabolites  and
and 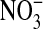 were measured in conditioned media 24 h after cell treatment with DETA NONOate (10, 100, 500 μM) or l-NMMA (1 mM), using Griess reagent after reduction of
were measured in conditioned media 24 h after cell treatment with DETA NONOate (10, 100, 500 μM) or l-NMMA (1 mM), using Griess reagent after reduction of 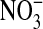 with nitrate reductase. Peak levels of NO concentrations within the media were determined using an aminoFLAT Nitric Oxide Sensor (Innovative Instruments, Inc., Tampa, FL). Listed values represent mean ± SEM of 5-8 measurements for
with nitrate reductase. Peak levels of NO concentrations within the media were determined using an aminoFLAT Nitric Oxide Sensor (Innovative Instruments, Inc., Tampa, FL). Listed values represent mean ± SEM of 5-8 measurements for 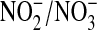 and mean ± SEM of 3 separate measurements for NO.
and mean ± SEM of 3 separate measurements for NO.
NO measurements from DETA NONOate were performed in the absence of HBE1 cells, and were consistently lower when HBE1 were present.
Epithelial cell migration was examined more directly using 0.8-μm pore size polycarbonate transwells (Figure 1D). Cell migration of NHBE or HBE1 cells was enhanced by low exogenous NO (DETA NONOate; 10 μM). Similarly, HBE1-NOS2 cells migrated faster compared with control HBE1 cells, and this was reduced in the presence of l-NMMA (Figure 1C). As shown, l-NMMA had no significant effect on wound repair or cell migration in NHBE or HBE1 cells. These results indicate that constitutive NOS2 activity and NO production by airway epithelial cells, as seen in vivo, promotes their ability to migrate and repair wounds.
NO Enhances MMP-9 Expression in Airway Epithelial Cells
To address whether the influence of NO-mediated cell migration is related to changes in MMP-9 expression and activation in airway epithelial cells, we determined the ability of NO to alter MMP-9 expression in NHBE or HBE1 cells. Contrary to previous observations with S-nitrosothiols or high concentrations of NO donor compounds (26), low-nM NO concentrations generated from DETA NONOate (10 μM) significantly enhanced MMP-9 mRNA expression (Figure 2A). Enhanced MMP-9 protein expression by NO in HBE1 cells was also observed, as determined by gelatin zymography (Figure 2B). Consistent with these findings, MMP-9 mRNA expression and protein secretion by HBE1-NOS2 cells was decreased in the presence of L-NMMA (Figures 2A and 2B), illustrating similar effects of endogenously produced NO. Dose–response studies showed that higher concentrations of DETA NONOate (100–500 μM) did not enhance MMP-9 expression but were inhibitory (Figure 2C), comparable to dose-dependent effects on wound closure. Similar effects of NO were also observed under conditions in which MMP-9 expression was stimulated with TNF-α (data not shown). Basal levels of MMP-9 expression in HBE1-NOS2 cells were not significantly higher than in untransfected HBE1 cells, and NOS2 inhibition appeared to reduce MMP-9 expression to levels below those in HBE1 cells. This indicates that basal MMP-9 expression in HBE1-NOS2 cells was perhaps lowered by factors independent of NOS2. However, the selective effect of NOS inhibition in HBE1-NOS2 cells compared with HBE1 cells clearly demonstrates a contribution of NOS2 to MMP-9 expression.
Figure 2.

NO induces MMP-9 expression in airway epithelial cells. (A) NHBE (solid bars), HBE1 (open bars), or HBE1-NOS2 (shaded bars) cells were grown to 80–90% confluence and treated with either DETA NONOate (10 μM) or l-NMMA for 24 h, after which MMP-9 mRNA was analyzed by semiquantitative RT-PCR and quantitated using densitometry in relation to GADPH expression. (B) Accumulation of secreted MMP-9 protein in the media of HBE1 and HBE1-NOS2 cells was analyzed by gelatin zymography, after 24 h incubation in the absence or presence of DETA NONOate (10 μM) or l-NMMA. (C) Analysis of MMP-9 mRNA expression in HBE1 cells using varying doses of DETA NONOate (10–500 μM). (D) Subconfluent HBE1 (open bars) or HBE1-NOS2 cells (shaded bars) were transiently transfected with a luciferase reporter construct containing the −670 bp MMP-9 promoter sequence, and incubated with either DETA NONOate (10 μM) or l-NMMA for 24 h, after which luciferase activity was measured from cell extracts. Data represent mean ± SE from at least three separate experiments.
To determine whether NO regulates MMP-9 expression at the transcriptional level, we performed experiments in HBE1 or HBE1-NOS2 cells after transfection with a luciferase reporter construct containing the −670 bp MMP-9 promoter sequence, which includes binding sites for the transcription factors NF-κB and activator protein-1 (31). As shown in Figure 2D, HBE1 cell exposure to DETA NONOate (10 μM) increased luciferase expression, whereas NOS inhibition reduced luciferase reporter activity in HBE1-NOS2 cells, indicating the contribution from endogenous NOS2.
NO-Mediated Epithelial Wound Repair and MMP-9 Expression Involve cGMP-Dependent Pathways
One cellular target involved in NO-mediated signaling is the heme center of soluble guanylyl cyclase (sGC), activation of which induces the production of cGMP. Therefore, we examined the involvement of sGC and cGMP in NO-mediated epithelial cell migration and wound repair in HBE1 cells. As illustrated in Figure 3A, the ability of DETA NONOate (10 μM) to enhance wound repair was almost completely prevented after pretreatment with the selective sGC inhibitor ODQ (10 μM), whereas ODQ did not affect wound repair in control cells. Moreover, inhibition of NOS activity reduced wound closure in injured HBE1-NOS2 cells, but did not affect wound closure of control HBE1 cells. Analysis of cellular cGMP illustrated increases upon HBE1 cell exposure to NO (DETA NONOate; 10 μM), and elevated cGMP levels in HBE1-NOS2 cells compared with nontransfected HBE1 cells (Figure 3B). As expected, these increases in cGMP were prevented by pretreatment with ODQ and l-NMMA in HBE1-NOS2 cells.
Figure 3.

Epithelial wound repair and MMP-9 expression are regulated by cGMP-dependent mechanisms. (A) HBE1 (open bars) or HBE1-NOS2 cells (shaded bars) were either untreated or pretreated with ODQ or l-NMMA 30 min before treatment with DETA NONOate (10 μM) for 24 h to examine epithelial wound closure. (B) Cellular levels of cGMP in HBE1 (open bars) or HBE1-NOS2 (shaded bars) cells were determined by RIA, after cell treatment with DETA NONOate, l-NMMA, or ODQ. (C) Analysis of MMP-9 mRNA by semiquantitative RT-PCR in HBE1 cells (open bars) or HBE1-NOS2 (shaded bars) after 24 h incubation with DETA NONOate (10 μM) in the absence or presence of ODQ, was added 30 min before DETA NONOate. Top: representative RT-PCR analysis of MMP-9 and GAPDH expression. Bottom: MMP-9 expression was normalized to GAPDH expression. (D) HBE1 cells were incubated in the absence or presence of the cGMP analog 8-Bromo-cGMP for 24 h, and % wound closure and MMP-9 expression was examined. HBE1 cells were pretreated with PKG inhibitor, KT5823, or DT-2 for 30 min before wounding and then incubated in the absence or presence of DETA NONOate (10 μM) for 24 h, before (E) analysis of % wound closure and (F) MMP-9 mRNA expression. Data represent mean ± SE from at least three separate experiments.
Pretreatment with ODQ also completely prevented DETA NONOate-mediated stimulation of MMP-9 expression in HBE1 cells (Figure 3C) and markedly decreased MMP-9 expression in HBE1-NOS2 cells in contrast to normal HBE1 cells, indicating that NOS2-derived NO promotes MMP-9 expression via sGC-mediated cGMP production. Consistent with the effects of NO, addition of the stable cGMP analog, 8-bromo-cGMP (10 μM) resulted in enhanced epithelial wound repair as well as increased MMP-9 RNA expression (Figure 3D).
We next explored the involvement of downstream mechanisms in cGMP signaling in regulating airway epithelial wound repair and MMP-9 expression, using two structurally distinct inhibitors of PKG, the small molecule KT5823 (10 μM) and the peptide-based inhibitor DT-2 (10 μM) (28) (Figure 3E). As shown, both PKG inhibitors significantly reduced wound repair, both in unstimulated HBE1 cells and after exposure to DETA NONOate (10 μM), consistent with the importance of PKG-mediated pathways in cell migration (19, 32). However, neither PKG inhibitor significantly affected basal or NO-stimulated expression of MMP-9 (Figure 3F), suggesting that NO-mediated MMP-9 expression in HBE1 occurs by a PKG-independent mechanism.
NO Regulates Epithelial Wound Repair: Direct Involvement of MMP-9
Consistent with earlier reports (9, 11), injury to HBE1 cells resulted in increased cell-associated MMP-9 expression, primarily at or near the wound edge (Figure 4A). Moreover, cell-associated MMP-9 expression was markedly enhanced after cell stimulation with either TNF-α (100 ng/ml) (Figure 4A, panels a and b) or DETA NONOate (10 μM) (Figure 4A, panels a and c). Consistent with the stimulatory effects of exogenous NO, inhibition of endogenous NOS activity markedly reduced MMP-9 levels at or near the wound edge of injured HBE1-NOS2 cells (Figure 4A, panels d and e). To explore the involvement of MMP-9 in epithelial wound closure in the linear wound assay, we tested the effects of a pharmacologic MMP inhibitor, MMP inhibitor II, on TNF-α or NO-mediated epithelial wound closure. Indeed, pretreatment with the MMP inhibitor in both cases markedly inhibited the extent of wound closure (Figure 4B). Similarly, NO-stimulated epithelial cell migration in a transwell assay was largely prevented in the presence of the MMP inhibitor (Figure 4C).
Figure 4.

NO-induced MMP-9 expression regulates airway epithelial migration and wound repair. (A) HBE1 cells in chamber slides were wounded and incubated in the absence (a) or presence of TNF-α (100 ng/ml) (b) or DETA NONOate (10 μM) (c), for 24 h. Similarly, HBE1-NOS2 cells were wounded and incubated in the absence (d) or presence (e) of l-NMMA. After incubations, cell monolayers were fixed with 4% PFA and MMP-9 expression was analyzed by immunofluorescence (red). Nuclear staining (green) is shown for contrast. Arrows indicate direction of migration. (B) Quantitative analysis of HBE1 cell wound closure after cell treatment with either TNF-α (100 ng/ml) or DETA NONOate (10 μM) in the absence or presence of an MMP inhibitor (1 μM; 30 min preincubation) for 24 h. Wound areas were measured using NIH ImageJ software, and results are shown. (C) HBE1 cell migration through 8.0-μm polycarbonate transwell membranes was quantitated 24 h after exposure to DETA NONOate (10 μM) in the absence or presence of MMP inhibitor I (1 μM; 30 min preincubation). (D) HBE1 cells were transfected with either MMP-9 targeted siRNA or scrambled siRNA sequence (neg CTL) before DETA NONOate treatment (10 μM), and effect on MMP-9 expression was examined after 24 h by RT-PCR. (E) Untransfected or siRNA transfected HBE1 monolayers were wounded, and epithelial wound closure in the absence or presence of DETA NONOate (10 μM) was determined using NIH ImageJ software. Data represent mean ± SE from at least three separate experiments.
Although the results above suggest a contribution of MMP-9 in NO-mediated epithelial migration and wound repair, involvement of other MMPs cannot be ruled out due to the lack of specificity of the MMP inhibitor. To more specifically address the involvement of MMP-9 in NO-mediated epithelial wound closure, we used a siRNA approach to silence MMP-9 expression in HBE1 cells. As illustrated in Figure 4D, transfection with MMP-9–targeted siRNA reduced MMP-9 expression in unstimulated HBE1 cells, and markedly reduced NO-stimulated expression of MMP-9. Control transfections with a scrambled siRNA sequence (neg CTL) had no effect. Similar findings were obtained upon analysis of MMP-9 protein (data not shown). MMP-9 gene silencing by siRNA was found to reduce airway epithelial wound closure in untreated HBE1 cells, and markedly decreased NO-stimulated wound closure to a rate that was similar to that seen in untransfected control cells (Figure 4E). Since siRNA strategies can potentially increase cellular interferon production (33), we verified whether MMP-9 siRNA caused inadvertent induction of NOS2 expression, but observed no detectable NOS2 expression in HBE1 cells transfected with either MMP-9 or control siRNA (not shown). Together, these results demonstrate that the stimulatory effects of NO on airway epithelial cell migrations and wound repair involve the contribution of MMP-9.
Effects of NO on MMP-9 Activation
After secretion, MMP-9 is subject to various extracellular or cell-associated factors that regulate its activation (8). Hence, in addition to controlling MMP-9 expression, NO could potentially also directly or indirectly modulate MMP-9 activation. To visualize cell-associated MMP-9 activity in wounded HBE cells, we used an in situ zymography approach using a fluorescent gelatin substrate (DQ-Gelatin). As illustrated in Figure 5A, localized gelatinase activity could be observed at or near the wound edge of HBE1 cells after linear wounding (panel a). Prior transfection of HBE1 cells with either scrambled MMP-9 siRNA (panel b) or MMP-9 siRNA (panel c) indicated that this gelatinase activity is largely due to MMP-9. Chemical MMP activation with p-CMB (positive CTL) caused a more global increase in gelatinase activity (panel d). Consistent with results in Figure 4A, cell treatment with either TNF-α (100 ng/ml) or DETA NONOate (10 μM) markedly enhanced gelatinase activity at the wound edge and in migrating cells (Figure 5A, panels e and g), which was markedly inhibited by MMP-9 siRNA (Figure 5A, panels f and h), demonstrating that the observed gelatinase activity originates from MMP-9 and that NO enhances MMP-9 activation.
Figure 5.

NO indirectly regulates gelatinase activity at the epithelial wound edge. (A) In situ zymography analysis of gelatinase activity in wounded HBE1 cells. Confluent HBE1 cells were wounded and incubated as indicated in Materials and Methods in the absence (a), or presence of either scrambled siRNA (b), MMP-9 siRNA sequence (c), or the MMP activator p-CMB (d). In addition, untransfected cells (e and g) or MMP-9 siRNA transfected cells (f and h) were incubated in the presence of TNF-α (100 ng/ml) (e and f) or DETA NONOate (10 μM) (g and h). Arrows indicate direction of migration. (B) Recombinant human proMMP-9 was incubated with DETA NONOate (10 μM; X symbols), the nitrosating agent S-nitrosocysteine (200 μM; diamonds), or the MMP activator p-CMB (200 μM; circles), and activation was followed using fluorogenic MMP-9 substrate (DQ-Gelatin). Squares, control. (C) Analysis of uPA RNA in NHBE cells and protein expression in HBE1 cells after wounding and incubation in the absence or presence of DETA NONOate (10 μM). Left: representative RT-PCR analysis of uPA expression. Right: Analysis of uPA protein by immunohistochemistry. Arrows indicate direction of migration. Data represent mean ± SE from at least two separate experiments.
One potential mechanism by which NO may promote MMP-9 activation is by direct S-nitrosylation of the cysteine switch in the proMMP-9 (23). To test for this possibility, we incubated recombinant human proMMP-9 with DETA NONOate (10 μM) or the known MMP activator, p-CMB (200 μM), and followed activation using the fluorogenic MMP-9 substrate (DQ-Gelatin). While p-CMB was found to activate proMMP-9, as expected, exogenous NO (DETA NONOate; 10 μM) failed to enhance proMMP-9 activation (Figure 4B). In contrast, S-nitrosocysteine (200 μM) caused mild MMP-9 activation, consistent with earlier findings (23). These findings suggest that, under our experimental conditions, NO was not directly responsible for MMP-9 activation in HBE1 cells. To address a potential indirect mechanism by which NO promotes MMP-9 activation, we examined the effects of NO on expression of urokinase plasminogen activator (uPA), which has been previously demonstrated to be capable of activating MMP-9 (34). Indeed, treatment of either NHBE cells (Figure 5C) or HBE1 cells (not shown) with DETA NONOate (10 μM) resulted in increased uPA mRNA expression and enhanced uPA immunoreactivity in migrating cells after linear injury, revealing one potential mechanism by which NO can promote MMP-9 activation.
DISCUSSION
The major finding of the present study is that low nM concentrations of NO, either added exogenously in the form of DETA NONOate (10 μM) or produced endogenously in HBE1 cells that were stably transfected with NOS2, promote airway epithelial cell migration and wound repair after epithelial injury. These findings illustrate a potential functional role for constitutive NOS2 activity within the airway epithelium in vivo (27). In addition to proposed roles in innate host defense and regulation of inflammatory processes (12, 35), airway epithelial NOS2 may also serve to facilitate epithelial repair processes and preserve epithelial barrier function after injurious insults (36). Analogous roles for NOS2-derived NO have previously been indicated in cutaneous wound healing and intestinal epithelial repair (15, 16).
A second major conclusion from our studies is that NO-mediated epithelial wound repair is at least partly mediated by MMP-9, based on NO-dependent presence of cell-associated MMP-9 and gelatinase activity at the wound edge, and on inhibitory effects of pharmacologic MMP inhibition and siRNA knockdown of MMP-9. Although MMP-9 may be present at very low levels in resting airway epithelial cells in vivo, its expression has been shown to increase rapidly during early stages of epithelial wound repair, and previous studies have indicated a contributing role of MMP-9 in airway epithelial cell migration and wound repair (3, 9, 11). Hence, the ability of physiologically relevant concentrations of NO to enhance MMP-9 expression and activation may reflect an important mechanism by which epithelial NO contributes to epithelial repair mechanisms under conditions of airway injury. Although several other studies have reported that NO is capable of enhancing MMP-9 expression or activation (23, 25), we and others have previously demonstrated that high concentrations of NO and S-nitrosothiols, potentially reflecting conditions of airway inflammation in which NO production is elevated, inhibit MMP-9 expression (22). Indeed, exposure of HBE1 cells to higher concentrations of DETA NONOate (500 μM), which generates near μM concentrations of NO, were found to inhibit epithelial cell migration as well as MMP-9 expression. Hence, the beneficial effects of physiologic levels of NO on epithelial MMP-9 expression and cell migration may be overruled during inflammation by inhibitory effects due to elevated NO production.
Extensive studies over the past decade have revealed many diverse mechanisms by which NO can affect cell function or regulate gene expression (20, 21). Indeed, NO has been demonstrated to be capable of regulating MMP-9 expression and activation at various levels, by influencing its transcriptional activation, mRNA stability, as well as proMMP activation (22, 23, 25). Our results indicate that NO acts largely by enhancing MMP-9 gene transcription by a cGMP-dependent signaling mechanism, analogous to MMP regulation in endothelial cells (25, 37). Indeed, various components of the NO-dependent cGMP signaling pathway, including cGMP-specific protein kinases (PKG) and phosphodiesterases, are present within airway epithelial cells, and have been linked to NO-dependent regulation of ciliary function (38). Although NOS2 has typically not been associated with cGMP-mediated pathways, our results demonstrate that continuous NO production by constitutive NOS2 activity results in elevated production of cGMP in airway epithelial cells, and promotes epithelial cell migration and MMP-9 expression by cGMP-dependent pathways. Previous studies have shown that NO can promote cell migration by cGMP-mediated activation of PKG (18, 19). Accordingly, our studies indicated that pharmacologic inhibitors of PKG (KT5823 and DT-2) attenuated NO-mediated airway epithelial cell migration. However, the same inhibitors failed to inhibit MMP-9 expression under these conditions, even though NO-mediated MMP-9 expression depends on cGMP. Given the observed importance of MMP-9 in airway epithelial cell migration and wound repair, it follows that effects of NO on cell migration are mediated by both PKG-dependent and -independent cGMP-related mechanisms. These latter mechanisms might involve the activation of cyclic nucleotide-gated ion channels, known to be expressed in airway epithelial cells (39). Future studies will be needed to further elucidate the downstream targets for cGMP that participate in NO-mediated airway epithelial cell MMP-9 regulation and/or migration.
In addition to regulating MMP-9 expression, NO could also directly or indirectly contribute to MMP-9 activation, by oxidative or nitrosative mechanisms (23, 40). However, our results in Figure 4B demonstrate that NO, at concentrations that promote cell migration and MMP-9 expression, is incapable of directly activating proMMP-9. Moreover, NO-stimulated wound closure in HBE1 cells was unaffected in the presence of 100 U/ml superoxide dismutase (SOD) (not shown), arguing against involvement of superoxide anion–dependent peroxynitrite formation. Finally, the fact that NO-dependent epithelial cell migration was completely prevented by sGC inhibition further argues against an oxidative mechanism in MMP-9 activation in our studies.
Although we were unable to detect activated (cleaved) MMP-9 in HBE1 culture media after cell wounding and exposure to NO, we observed cell-associated and localized MMP-9 and gelatinase activity that was enhanced in response to NO. This suggests that NO may indirectly contribute to localized MMP-9 activation by increasing its sequestration at the cell surface (e.g., by the hylaluronan receptor CD44) (37). Indeed, epithelial expression of CD44 is up-regulated in migrating epithelial cells (41), and CD44 is known to bind active MMP-9 at the cell surface, where it can activate latent growth factors to promote cell migration (8). However, whether NO is capable of modulating CD44 expression of remains to be established. NO could also indirectly contribute to MMP-9 activation by up-regulating or activating factors that contribute to proteolytic activation of secreted or surface-bound MMP-9. One such factor is the urokinase/plasminogen activator (uPA), which has been linked to MMP-9 activation in response to epithelial injury (34). Indeed, our observation that NO is capable of inducing uPA expression points to a potential indirect mechanism by which NO promotes MMP-9 activation. However, given the highly complex nature of MMP-9 activation and NO-mediated cell signaling and gene expression, we cannot rule out other mechanisms that control MMP-9 activation.
In summary, our findings demonstrate that low, physiologically relevant concentrations of NO promote airway epithelial cell migration and wound repair, which is at least partly due to the increased expression and activation of MMP-9. These studies illustrate a potential functional role of constitutive NOS2 activity within the respiratory epithelium, and provide further insights into the general mechanisms involved in epithelial NO biology.
Acknowledgments
The authors thank Dr. Takaaki Akaike for providing recombinant proMMP-9, and Dr. Douglas Boyd for the MMP-9 reporter construct. They also thank Yvonne Janssen-Heininger for scientific discussions, and C. Wendy Koot and Sharon Cawley for technical assistance.
This work was supported in part by National Institutes of Health grants HL60812 and HL074295 (to A.v.d.V.) and HL68991 (to W.R.D.), and by research grants from the Cystic Fibrosis Foundation (to A.v.d.V.) and the Totman Medical Research Trust (to W.R.D.).
Originally Published in Press as DOI: 10.1165/rcmb.2006-0253SM on September 15, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Parks WC, Shapiro SD. Matrix metalloproteinases in lung biology. Respir Res 2001;2:10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit Rev Biochem Mol Biol 2002;37:375–536. [DOI] [PubMed] [Google Scholar]
- 3.Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol 2003;28:12–24. [DOI] [PubMed] [Google Scholar]
- 4.McMillan SJ, Kearley J, Campbell JD, Zhu XW, Larbi KY, Shipley JM, Senior RM, Nourshargh S, Lloyd CM. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol 2004;172:2586–2594. [DOI] [PubMed] [Google Scholar]
- 5.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest 2002;110:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi T, Kishimoto J, Hattori S, Wachi H, Shinkai H, Burgeson RE. Matrix metalloproteinase 9 expression is coordinately modulated by the KRE-M9 and 12-O-tetradecanoyl-phorbol-13-acetate responsive elements. J Invest Dermatol 2004;122:278–285. [DOI] [PubMed] [Google Scholar]
- 7.Ma Z, Shah RC, Chang MJ, Benveniste EN. Coordination of cell signaling, chromatin remodeling, histone modifications, and regulator recruitment in human matrix metalloproteinase 9 gene transcription. Mol Cell Biol 2004;24:5496–5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fridman R, Toth M, Chvyrkova I, Meroueh SO, Mobashery S. Cell surface association of matrix metalloproteinase-9 (gelatinase B). Cancer Metastasis Rev 2003;22:153–166. [DOI] [PubMed] [Google Scholar]
- 9.Legrand C, Gilles C, Zahm JM, Polette M, Buisson AC, Kaplan H, Birembaut P, Tournier JM. Airway epithelial cell migration dynamics. MMP-9 role in cell-extracellular matrix remodeling. J Cell Biol 1999;146:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohan R, Chintala SK, Jung JC, Villar WV, McCabe F, Russo LA, Lee Y, McCarthy BE, Wollenberg KR, Jester JV, et al. Matrix metalloproteinase gelatinase B (MMP-9) coordinates and effects epithelial regeneration. J Biol Chem 2002;277:2065–2072. [DOI] [PubMed] [Google Scholar]
- 11.Coraux C, Martinella-Catusse C, Nawrocki-Raby B, Hajj R, Burlet H, Escotte S, Laplace V, Birembaut P, Puchelle E. Differential expression of matrix metalloproteinases and interleukin-8 during regeneration of human airway epithelium in vivo. J Pathol 2005;206:160–169. [DOI] [PubMed] [Google Scholar]
- 12.Xu W, Zheng S, Dweik RA, Erzurum SC. Role of epithelial nitric oxide in airway viral infection. Free Radic Biol Med 2006;41:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duszyk M. Regulation of anion secretion by nitric oxide in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2001;281:L450–L457. [DOI] [PubMed] [Google Scholar]
- 14.Hardiman KM, McNicholas-Bevensee CM, Fortenberry J, Myles CT, Malik B, Eaton DC, Matalon S. Regulation of amiloride-sensitive Na(+) transport by basal nitric oxide. Am J Respir Cell Mol Biol 2004;30:720–728. [DOI] [PubMed] [Google Scholar]
- 15.Gookin JL, Rhoads JM, Argenzio RA. Inducible nitric oxide synthase mediates early epithelial repair of porcine ileum. Am J Physiol Gastrointest Liver Physiol 2002;283:G157–G168. [DOI] [PubMed] [Google Scholar]
- 16.Yamasaki K, Edington HD, McClosky C, Tzeng E, Lizonova A, Kovesdi I, Steed DL, Billiar TR. Reversal of impaired wound repair in iNOS-deficient mice by topical adenoviral-mediated iNOS gene transfer. J Clin Invest 1998;101:967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez-Rivera E, Lizarbe TR, Martinez-Moreno M, Lopez-Novoa JM, Rodriguez-Barbero A, Rodrigo J, Fernandez AP, Alvarez-Barrientos A, Lamas S, Zaragoza C. Matrix metalloproteinase 13 mediates nitric oxide activation of endothelial cell migration. Proc Natl Acad Sci USA 2005;102:3685–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jadeski LC, Chakraborty C, Lala PK. Nitric oxide-mediated promotion of mammary tumour cell migration requires sequential activation of nitric oxide synthase, guanylate cyclase and mitogen-activated protein kinase. Int J Cancer 2003;106:496–504. [DOI] [PubMed] [Google Scholar]
- 19.Kawasaki K, Smith RS Jr, Hsieh CM, Sun J, Chao J, Liao JK. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol Cell Biol 2003;23:5726–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bogdan C. Nitric oxide and the regulation of gene expression. Trends Cell Biol 2001;11:66–75. [DOI] [PubMed] [Google Scholar]
- 21.Hemish J, Nakaya N, Mittal V, Enikolopov G. Nitric oxide activates diverse signaling pathways to regulate gene expression. J Biol Chem 2003;278:42321–42329. [DOI] [PubMed] [Google Scholar]
- 22.Akool el S, Kleinert H, Hamada FM, Abdelwahab MH, Forstermann U, Pfeilschifter J, Eberhardt W. Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol Cell Biol 2003;23:4901–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 2002;297:1186–1190. [DOI] [PubMed] [Google Scholar]
- 24.Knipp BS, Ailawadi G, Ford JW, Peterson DA, Eagleton MJ, Roelofs KJ, Hannawa KK, Deogracias MP, Ji B, Logsdon C, et al. Increased MMP-9 expression and activity by aortic smooth muscle cells after nitric oxide synthase inhibition is associated with increased nuclear factor-kappaB and activator protein-1 activity. J Surg Res 2004;116:70–80. [DOI] [PubMed] [Google Scholar]
- 25.Marcet-Palacios M, Graham K, Cass C, Befus AD, Mayers I, Radomski MW. Nitric oxide and cyclic GMP increase the expression of matrix metalloproteinase-9 in vascular smooth muscle. J Pharmacol Exp Ther 2003;307:429–436. [DOI] [PubMed] [Google Scholar]
- 26.Okamoto T, Valacchi G, Gohil K, Akaike T, van der Vliet A. S-nitrosothiols inhibit cytokine-mediated induction of matrix metalloproteinase-9 in airway epithelial cells. Am J Respir Cell Mol Biol 2002;27:463–473. [DOI] [PubMed] [Google Scholar]
- 27.Guo FH, Comhair SA, Zheng S, Dweik RA, Eissa NT, Thomassen MJ, Calhoun W, Erzurum SC. Molecular mechanisms of increased nitric oxide (NO) in asthma: evidence for transcriptional and post-translational regulation of NO synthesis. J Immunol 2000;164:5970–5980. [DOI] [PubMed] [Google Scholar]
- 28.Taylor MS, Okwuchukwuasanya C, Nickl CK, Tegge W, Brayden JE, Dostmann WR. Inhibition of cGMP-dependent protein kinase by the cell-permeable peptide DT-2 reveals a novel mechanism of vasoregulation. Mol Pharmacol 2004;65:1111–1119. [DOI] [PubMed] [Google Scholar]
- 29.White SR, Tse R, Marroquin BA. Stress-activated protein kinases mediate cell migration in human airway epithelial cells. Am J Respir Cell Mol Biol 2005;32:301–310. [DOI] [PubMed] [Google Scholar]
- 30.Zahm JM, Kaplan H, Herard AL, Doriot F, Pierrot D, Somelette P, Puchelle E. Cell migration and proliferation during the in vitro wound repair of the respiratory epithelium. Cell Motil Cytoskeleton 1997;37:33–43. [DOI] [PubMed] [Google Scholar]
- 31.Yan C, Wang H, Aggarwal B, Boyd DD. A novel homologous recombination system to study 92 kDa type IV collagenase transcription demonstrates that the NF-kappaB motif drives the transition from a repressed to an activated state of gene expression. FASEB J 2004;18:540–541. [DOI] [PubMed] [Google Scholar]
- 32.Smolenski A, Poller W, Walter U, Lohmann SM. Regulation of human endothelial cell focal adhesion sites and migration by cGMP-dependent protein kinase I. J Biol Chem 2000;275:25723–25732. [DOI] [PubMed] [Google Scholar]
- 33.Jackson AL, Linsley PS. Noise amidst the silence: off-target effects of siRNAs? Trends Genet 2004;20:521–524. [DOI] [PubMed] [Google Scholar]
- 34.Legrand C, Polette M, Tournier JM, de Bentzmann S, Huet E, Monteau M, Birembaut P. uPA/plasmin system-mediated MMP-9 activation is implicated in bronchial epithelial cell migration. Exp Cell Res 2001;264:326–336. [DOI] [PubMed] [Google Scholar]
- 35.Eriksson U, Egermann U, Bihl MP, Gambazzi F, Tamm M, Holt PG, Bingisser RM. Human bronchial epithelium controls TH2 responses by TH1-induced, nitric oxide-mediated STAT5 dephosphorylation: implications for the pathogenesis of asthma. J Immunol 2005;175:2715–2720. [DOI] [PubMed] [Google Scholar]
- 36.Rose F, Guthmann B, Tenenbaum T, Fink L, Ghofrani A, Weissmann N, Konig P, Ermert L, Dahlem G, Haenze J, et al. Apical, but not basolateral, endotoxin preincubation protects alveolar epithelial cells against hydrogen peroxide-induced loss of barrier function: the role of nitric oxide synthesis. J Immunol 2002;169:1474–1481. [DOI] [PubMed] [Google Scholar]
- 37.Zaragoza C, Soria E, Lopez E, Browning D, Balbin M, Lopez-Otin C, Lamas S. Activation of the mitogen activated protein kinase extracellular signal-regulated kinase 1 and 2 by the nitric oxide-cGMP-cGMP-dependent protein kinase axis regulates the expression of matrix metalloproteinase 13 in vascular endothelial cells. Mol Pharmacol 2002;62:927–935. [DOI] [PubMed] [Google Scholar]
- 38.Li D, Shirakami G, Zhan X, Johns RA. Regulation of ciliary beat frequency by the nitric oxide-cyclic guanosine monophosphate signaling pathway in rat airway epithelial cells. Am J Respir Cell Mol Biol 2000;23:175–181. [DOI] [PubMed] [Google Scholar]
- 39.Xu W, Leung S, Wright J, Guggino SE. Expression of cyclic nucleotide-gated cation channels in airway epithelial cells. J Membr Biol 1999;171:117–126. [DOI] [PubMed] [Google Scholar]
- 40.Castier Y, Brandes RP, Leseche G, Tedgui A, Lehoux S. p47phox-dependent NADPH oxidase regulates flow-induced vascular remodeling. Circ Res 2005;97:533–540. [DOI] [PubMed] [Google Scholar]
- 41.Leir SH, Baker JE, Holgate ST, Lackie PM. Increased CD44 expression in human bronchial epithelial repair after damage or plating at low cell densities. Am J Physiol Lung Cell Mol Physiol 2000;278:L1129–L1137. [DOI] [PubMed] [Google Scholar]