

Abstract
Silica particle–associated inflammation is implicated in the genesis of several pulmonary diseases, including silicosis and lung cancer. In this study we investigated the role of phosphatidylcholine-specific phospholipase C (PC-PLC) in silica-stimulated induction of TNF-α and IL-1β and how PC-PLC activity is regulated by silica in a rat alveolar macrophage model. We demonstrated that inhibition of PC-PLC, which was achieved with tricychodecan-9-yl-xanthate (D609), blocked the silica-stimulated induction of TNF-α and IL-1β in alveolar macrophage, suggesting that PC-PLC is involved in the silica-associated inflammatory response. PC-PLC activity was increased significantly by silica exposure, and this could be inhibited by MnTBAP, which catalyzes both the dismutation of O2.- to O2 and H2O2 and the dismutation of H2O2 to O2 and H2O, revealing that PC-PLC activity is regulated in a redox-dependent manner. This is further confirmed by the finding that PC-PLC activity was increased by exogenous H2O2. The intracellular calcium chelator BAPTA blocked the H2O2-increased PC-PLC activity, while the calcium ionophore, A23187, enhanced PC-PLC activity. The data indicate that PC-PLC plays critical roles in the silica-associated inflammatory response and that PC-PLC is regulated through redox- and calcium-dependent manners in alveolar macrophages.
Keywords: phosphatidylcholine-specific phospholipase C, redox signaling, calcium, cytokine, hydrogen peroxide
CLINICAL RELEVANCE
This study demonstrates that silica-induced production of hydrogen peroxide is essential to the activation of phosphatidylcholine specific phospholipase C in the mechanism of silica-induced cytokine induction in alveolar macrophages.
Silica pollution is both an occupational and environmental health concern. Exposure to silica particle has been associated with silicosis, lung cancers, and other pulmonary disorders (1). Although the underlying molecular mechanism of how silica causes these diseases is still elusive, it appears that inflammation caused by silica is one of the most common features and plays a critical role in the initiation and development of the pathogenesis of silica-associated diseases (1). The silica-associated inflammatory response is triggered by cytokines released from alveolar macrophages. For example, TNF-α (2, 3)and IL-1β (4, 5), two cytokines that play important roles in the inflammatory response, are increased after silica particle exposure in alveolar macrophages (6). These inflammatory cytokines can in turn recruit more inflammatory cells into the alveolus and propagate the inflammatory response and injuries (7). It is now clear that the production of cytokines such as TNF-α and IL-β upon exposure to silica is dependent on NF-κB signaling pathways in alveolar macrophages (3, 8). The upstream signaling event of how silica induces cytokines expression, however, remains unclear. Recently, it is found that reactive oxygen species (ROS) plays critical roles in silica-caused inflammation response (9, 10). Crystalline silica is a stimulator of the respiratory burst in phagocytes beginning with superoxide (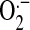 ) that can then result in production of hydrogen peroxide (H2O2), hydroxyl radical (·OH), and peroxynitrite (ONOO−) (10). The surface of fresh ground silica particles also carries siloxyl radicals (Si· or SiO·), which generate ·OH in aqueous medium (11), but that is extremely short lived and would result in either production of H2O2 or oxidation of whatever was immediately adjacent to the site of ·OH generation. On the other hand, the ROS, particularly H2O2, can also function as second messengers in signaling (12, 13). H2O2 can also participate in the activation of NF-κB (14, 15) and the expression of inflammatory cytokines including TNF-α and IL-1β (10). Nonetheless, the pathways through which silica signals in alveolar macrophages are not entirely resolved.
) that can then result in production of hydrogen peroxide (H2O2), hydroxyl radical (·OH), and peroxynitrite (ONOO−) (10). The surface of fresh ground silica particles also carries siloxyl radicals (Si· or SiO·), which generate ·OH in aqueous medium (11), but that is extremely short lived and would result in either production of H2O2 or oxidation of whatever was immediately adjacent to the site of ·OH generation. On the other hand, the ROS, particularly H2O2, can also function as second messengers in signaling (12, 13). H2O2 can also participate in the activation of NF-κB (14, 15) and the expression of inflammatory cytokines including TNF-α and IL-1β (10). Nonetheless, the pathways through which silica signals in alveolar macrophages are not entirely resolved.
Recent studies demonstrated that phosphatidylcholine-specific phospholipase C (PC-PLC) may be involved in the activation of NF-κB signaling pathway and the release of inflammatory molecules (16, 17). PC-PLC hydrolyzes phosphatidylcholine to phosphorycholine and diacylglycerol (DAG), an activator of most protein kinase C (PKC) isoforms. It has been reported that PC-PLC plays significant roles in several signal transduction mechanisms, including MAPK and NF-κB signaling pathways (18, 19). PC-PLC activity can be increased by many stimulators, including growth factors (20), LPS (16), cytokines (21, 22), and others (23, 24). We examined whether PC-PLC plays a role in the silica-induced inflammatory response in alveolar macrophages. Here we present findings that silica increased PC-PLC activity in a calcium- and redox-dependent manner, and that PC-PLC was involved in silica-induced expression of TNF-α and IL-1β in rat alveolar macrophages.
MATERIALS AND METHODS
Reagents and Materials
Unless otherwise noted, chemicals were from Sigma (St. Louis, MO) and were at least analytical grade. BAPTA-AM was from Invitrogen (Molecular Probes, Eugene, OR). D609 was from BIOMOL International, L.P. (Plymouth Meeting, PA). [3H]choline was purchased from Amersham Biosciences (Pittsburgh, PA). MnTBAP was kindly provided by Dr. Brian J. Day of the National Jewish Medical and Research Center (Denver, CO). TRIzol reagent was from Life Technologies (Grand Island, NY). DNA-free was from Ambion (Austin, TX). TaqMan reverse transcription reagent and SYBR Green PCR Master Mix were from Applied Biosystems (Foster City, CA). All chemicals used were at least analytical grade.
Silica Preparation
Ground silica was obtained from UNIMIN (Belvidere, IL). Four hundred grams of crushed silica was heated at 450°C for 52 h and then refluxed in 4N HNO3 for 4 h to destroy organic contaminants, including LPS, and remove surface metals such as Fe and Al. The material was then rinsed with Milli-Q water (prepared with Millipore Milli-Q water system; Millipore, Billerica, MA) several times and refluxed again. Next, the silica sample was suspended in NaOH solution (pH 9.3), stirred for 1 h, and allowed to settle for 30 min. The supernatant was then collected and centrifuged at 15,000 × g for 10 min to collect < 2 μm particles. The silica was resuspended in Milli-Q water, and rinsed seven more times. After the eighth rinsing and centrifugation, the sample was resuspended in 0.1% HCl, swirled for 4 h, and collected by centrifugation. The sample was rinsed five times with Milli-Q water, collected by centrifugation, and dried.
Cell Culture
The NR8383 rat AM cell line (25) was a generous gift from Dr. G. H. Zhang, University of Texas Health Science Center at San Antonio. Cells were cultured in F-12K medium (Life Technologies, Grand Island, NY), supplemented with 15% heat-inactivated fetal bovine serum (Omega Sci., Tarzana, CA), 100 U/ml penicillin, and 100 μg/ml streptomycin in 5% CO2 at 37°C.
Determination of PC-PLC Activity
PC-PLC activity was determined using the method described by Zamorano and coworkers (22). Briefly, NR Cells were cultured for 48 h in the presence of [methyl-3H]choline chloride (1.0 μCi/ml). Medium was replaced with label-free Krebs Ringer Phosphate (KRP: 1 M PBS, 1.3 mM CaCl2, 1 mM MgSO4, 5 mM glucose, 1 mM HEPES, pH 7.4) 2 h before treatment. After treatment as indicated, cells were collected and the cell pellet was rinsed with 1× PBS before being resuspended in CH3OH/CHCL3/H2O (2.5:1.25:1) to separate aqueous and organic phases. The water-soluble fraction containing choline metabolites was loaded on a LK6D silica gel Thin Layer Chromatography (TLC) plate (Whatman International Ltd, Maidstone, UK) in a solvent system of methanol/0.5%NaCl/28%NH3 (50:50:1, vol/vol). Unlabeled phophorylcholine was used as positive control. Spots were visualized by staining with iodine vapor and the bands matched to that of the unlabeled phophorylcholine were scraped off. The radioactivity of each band was measured with a LS6500 Multi-Purpose Scintillation counter (Beckman Coulter, Fullerton, CA). The radioactivity of phosphorylcholine, the product of PC-PLC, reflects the corresponding PC-PLC activity.
Cytokine mRNA Assay
NR Cells were cultured for 24 h before treatment. After treatment, cells were collected and washed with PBS. Total RNA was extracted using TRIzol reagent and treated with DNA-free reagent according to the manufacturers' protocols. DNA-free RNA samples were reverse transcribed using the TaqMan reverse transcription system (Applied Biosystems) and real-time PCRs were run with a Cepheid 1.2 real-time PCR machine. Briefly, 5 μl of reverse transcription reaction product was added to reaction tubes containing 12.5 μl SYBR Green PCR Master Mix and primer pair specific for cytokine mRNA; the total PCR sample was 25 μl. GAPDH was used as internal control. The primers were as follows: TNF-α, sense 5′-CCAGGAGAAAGTCAGCCTCCT-3′, antisense 5′-TCATACCAGGGCTTGAGCTCA-3′; IL-1β, sense 5′-CACCTCTCAAGCAGAGCACAG-3′, antisense 5′-GGGTTCCATGGTGAAGTCAAC-3′; GAPDH, sense 5′-ACCCCCAATGTATCCGTTGT-3, antisense 5′-TACTCCTTGGAGGCCATGT-3.
Statistical Analysis
We used the comparative ΔΔCt method for the relative mRNA quantitation. The relative quantification of target, normalized to an internal control (GAPDH), and an untreated sample is given by relative quantification = 2−ΔΔCt ΔΔCt being defined as the difference of mean ΔCt(HNE treated sample) and mean ΔCt(untreated sample) and ΔCt as the difference in mean Ct (cytokines) and Ct (GAPDH) as the internal control. Threshold cycles (Ct) were selected in the line in which all samples were in logarithmic phase.
All data were expressed as the mean ± SE. Sigma Stat software was used for statistical analysis, and statistical significance was accepted when P < 0.05. The Tukey test was used for comparison of mRNA level, and one-way ANOVA was used for comparison of PC-PLC activity.
RESULTS
Involvement of PC-PLC in Silica-Induced Production of Cytokines in Alveolar Macrophages
Cytokine induction after silica particle exposure in alveolar macrophages has been well established (6). PC-PLC has been reported to be involved in NF-κB activation and cytokine production (16, 17). To study whether PC-PLC is involved in the silica-caused inflammatory response, we investigated the effect of D609, a purportedly specific inhibitor of PC-PLC, which we verified inhibited the catalytic activity, on the release of TNF-α and IL-1β, two critical cytokines involved in silica-associated inflammation. As shown in Figure 1, exposure of rat alveolar macrophages (NR8383 cells) to 50 μg/ml of silica particles increased the expression of both TNF-α (Figure 1A) and IL-β (Figure 1B) at the mRNA level, significantly. Pretreatment with 40 μM D609 for 10 min abrogated silica-stimulated cytokine expression, suggesting that PC-PLC was required for silica-stimulated induction of inflammatory cytokines.
Figure 1.


PC-PLC is involved in silica-induced production of cytokines at RNA level in alveolar macrophages. (A) TNF-α; (B) IL-1β. Rat alveolar macrophages (NR8383 cells) were treated with 50 μg/ml silica particle for 3 h, the total RNA was extracted, and then the relative mRNA levels of TNF-α and IL-1β were determined with real time RT-PCR assay. To confirm the involvement of PC-PLC in cytokine expression, cells were pretreated with 40 μM D609 for 10 min before silica treatment. n = 3, *P < 0.01.
Silica Increases PC-PLC Activity in a Redox-Dependent Manner
PC-PLC activity can be increased by many reagents, but no prior data were available on the effect of silica exposure on PC-PLC activity. To determine if silica activates PC-PLC in alveolar macrophages, we analyzed the production of phosphorycholine by using a [3H]choline and TCL method. Phosphorycholine is a hydrolysis product of phosphatidylcholne catalyzed by PC-PLC and its amount (shown as phosphory[3H]choline in TCL assay) reflects the activity of PC-PLC. As expected, 50 μg/ml of silica increased PC-PLC activity significantly in the alveolar macrophages (Figure 2).
Figure 2.

Silica increases PC-PLC activity. [3H]choline-labeled NR8383 cells were treated with 50 μg/ml silica for 60 min, the [3H]phosphorylcholine was extracted and separated with the TLC method as described in Materials and Methods, and then the radioactivity was counted with a scintillation counter. To confirm the involvement of PC-PLC, cells were pretreated with 40 μM D609 for 10 min before silica treatment. n = 3, *P < 0.01.
Little is known about how PC-PLC activity is regulated in alveolar macrophages. Based on the evidence that (1) PC-PLC is involved in H2O2-primed alveolar macrophage stimulation (26) and (2) silica exposure is related to the generation of ROS (10), we hypothesize that PC-PLC activity is regulated in a redox-dependent manner. To test this, we first investigated whether an ROS scavenger could block silica-increased PC-PLC activity. Data in Figure 3 demonstrated that pretreatment with 100 μM MnTBAP for 10 min before exposure to 50 μg/ml of silica particles abrogated the silica-increased PC-PLC activity. MnTBAP possesses both superoxide dismutase (SOD) and catalase activity, and can thus eliminate ROS (27). MnTBAP is also small enough to easily get into the interface of silica and the cell, which is important as illustrated by previous studies in which enzymes have been shown to be incapable of doing this unless added at very high concentrations (28). The data here revealed that ROS was involved in PC-PLC activation by silica.
Figure 3.

MnTBAP blocked silica-increased PC-PLC activity. [3H]choline-labeled cells were pretreated with 100 mM MnTBAP for 10 min, after which cells were incubated with or without 50 μg/ml silica for an additional 60 min. Next the [3H]phosphorylcholine was extracted and separated by TLC, and the radioactivity was counted with a scintillation counter. n = 3, *P < 0.01.
To further confirm the hypothesis, we treated cells with exogenous H2O2 and then determined the PC-PLC activity. Fifty micromoles of exogenous H2O2 increased the production of [3H]phosphorycholine significantly in 1 h, and this increase was completely blocked by D609 (Figure 4), suggesting that H2O2 can stimulate PC-PLC in alveolar macrophages.
Figure 4.

Bolus H2O2 increases PC-PLC activity. [3H]choline labeled cells were pretreated with 40 μM D609 for 30 min, and then were incubated with/without 50 μM H2O2 for an additional 15 min. [3H]phosphorylcholine was extracted and separated by TLC. The radioactivity was counted with a scintillation counter. n = 4, *P < 0.01.
Activation of PC-PLC Is Calcium-Dependent
It is now well established that ROS are involved in the activation of various signaling pathways via several mechanisms including protein thiol modification and changes in intracellular calcium concentration ([Ca2+]i) (12). It has been known for many years that hydroperoxides are capable of elevating [Ca2+]i through a variety of mechanisms, including influx from the media, release from the endoplasmic reticulum, and release of annexin VI from membranes (29). Previous reports suggested that calcium influx was likely involved in PC-PLC activation (30, 31). Thus, we determined if calcium elevation is also involved in PC-PLC activation by H2O2. Pretreatment for 30 min with 5 mM BAPTA-AM (an agent that is hydrolyzed to produce the intracellular calcium chelator, BAPTA), which buffers hydroperoxide-induced increases in [Ca2+]i in alveolar macrophages (32), inhibited H2O2-increased PC-PLC activity (Figure 5A). In contrast, when cells were pretreated for 5 min with 250 nM A23187, a calcium ionophore that allows entry of calcium, which was 1.3 mM in the medium, the H2O2-induced PC-PLC activation was significantly enhanced (Figure 5B). Thus, our data suggest that elevation of [Ca2+]i is involved in H2O2-increased PC-PLC activation in alveolar macrophages.
Figure 5.


PC-PLC activity is regulated in a calcium-dependent manner. (A) BAPTA inhibits H2O2-increased PC-PLC activity. [3H]choline-labeled cells were pretreated with 5 mM BAPTA, a calcium chelator, for 30 min, and then were incubated with or without 50 μM H2O2 for an additional 15 min. n = 3, *P < 0.01. (B) A23187 increases PC-PLC activity. [3H]choline-labeled cells were treated with 250 nM of A23187, a calcium ionophore, for 5 min, and the [3H]phosphorylcholine was extracted and separated by TLC. Radioactivity of the phosphorycholine band was then counted with a scintillation counter. n = 3, *P < 0.01.
DISCUSSION
Inflammation underlies the pathogenesis of silica-induced lung diseases, though the signaling pathways through which silica particle stimulates the production of cytokines remains only partially resolved. In this study we showed that the induction of both TNF-α and IL-1β upon silica exposure of alveolar macrophages is through PC-PLC. We also demonstrated that PC-PLC activity was regulated via redox- and calcium-dependent signaling. Our data suggest that PC-PLC may be an upstream molecule involved in the inflammation response in alveolar macrophages.
Several studies have demonstrated that PC-PLC plays a role in cell signaling. PC-PLC has been shown to be involved in cell growth (33, 34), differentiation (35), and apoptosis (36). Schutze and colleagues first reported that PC-PLC was involved in the activation of NF-κB signaling (37), and this was confirmed by others (21, 38). Later, the requirement of PC-PLC activity for the induction of cytokines, such as TNF-α (39), IL-1α (40), and Il-1β (41), was reported. Our finding that PC-PLC is critical for the induction of TNF-α and IL-1β by silica further confirms the role of PC-PLC in cytokine regulation. The hydrolysis of phosphatidylcholine by PC-PLC generates phosphorycholine and. DAG. The latter is a well-known second messenger that is involved in several signaling pathways, including activation of most PKC isoforms, while little is known about the role, if any, of phosphorycholine in cell signaling.
An alternative potential mechanism of how PC-PLC is involved in NF-κB signaling or TNF-α and IL-1β regulation is through its involvement in signaling leading to the generation of H2O2. Although we have shown that H2O2 can activate PC-PLC, we and others have found that PC-PLC may play a critical role in regulation of respiratory burst, the major source of stimulated H2O2 production by macrophages (26). In other words, PC-PLC activation appears to be both upstream and downstream of H2O2 generation. Once produced, H2O2 can activate NF-κB signaling (14, 42), which is involved in TNF-α and IL-1β regulation.
Many reagents have been reported to increase PC-PLC activity, including growth factors such as insulin (20); cytokines such as TNF-α (21, 43), IFN-γ (44), and IL-4 (22); LPS (16); prostaglandin F2α (23); endothelin-1 (24); and others (17, 45, 46). In this study we found that PC-PLC was also activated by silica in alveolar macrophages (Figure 2). How PC-PLC is activated upon stimulation is currently not well characterized. Since many PC-PLC activators have specific receptors on cell membrane, it is assumed that PC-PLC is activated through receptor-mediated signaling pathways. It has been reported that PKC (20) and Ras (47) can affect PC-PLC activity. In contrast, Murthy and Makhlouf found that PC-PLC was not PKC sensitive (30). Our data suggest that PC-PLC activity is regulated in a redox-dependent manner in alveolar macrophages, as shown by the inhibition of silica-stimulated PC-PLC activity by MnTBAP, an SOD and catalase mimic (Figure 3), and by the finding that bolus H2O2 increased PC-PLC activity (Figure 4). Here the source of ROS upon the exposure of silica may be either from siloxyl radicals (Si· or SiO·) on the surface of silica particles (11), and/or from the respiratory burst initiated by phagocytosis of the particles. Many PC-PLC stimulators, if not all, seem to use ROS as a signaling messenger. For example, TNF-α (48), LPS (49, 50), and insulin (51) have been found to activate signaling by generating ROS. Thus, our results suggesting that PC-PLC activation by silica is regulated via a redox-dependent manner is an important (although perhaps not entirely surprising) finding.
In this study we also demonstrated that the intracellular calcium chelator blocked PC-PLC activation (Figure 5A), while the calcium ionophore enhanced the PC-PLC activation (Figure 5B), suggesting that calcium elevation is an upstream event for PC-PLC activation. This result is consistent with findings from other research groups (30, 31), who found that calcium chelators could inhibit PC-PLC activity. On the other hand, Nofer and coworkers reported that PC-PLC could facilitate the thapsigargin-induced loss of intracellular calcium, and the authors suggested that there existed a physiologic feedback mechanism activated by calcium influx that acted through consecutive activation of PC-PLC to limit the rise of [Ca2+]i (52). It is currently not clear how silica elevated the [Ca2+]i. Several studies suggest that elevated [Ca2+]i is from influx from the extracellular environment (53, 54). However, Chen and colleagues observed that chelation of extracellular calcium had no effect on silica-elevated [Ca2+]i and concluded that silica increased [Ca2+]i via mobilization of intracellular calcium pool (55). It seems that ROS is involved in silica-elevated [Ca2+]i elevation, since antioxidant treatment blocked the [Ca2+]i increase (56). Indeed, several studies (including studies from our lab) have reported the elevation of [Ca2+]i after ROS stimulation (32, 57). The exact mechanisms by which silica increases [Ca2+]i and by which calcium activates PC-PLC remain to be determined.
Based on the numerous reports of the involvement of PC-PLC in inflammatory responses, further studies are warranted, including the mechanisms by which PC-PLC is activated by various agents and how PC-PLC interacts with other signaling pathways. It must be mentioned here that currently the major tool to identify the involvement of PC-PLC in pathophysiologic processes in the mammalian system remains the use of its pharmacologic inhibitor, D609. Although the DNA sequence of PC-PLC from bacteria is established (58), the gene for PC-PLC has not been identified in any eukaryote. Indeed, there is no statistically significant homology between bacterial PC-PLCs and that of any mammalian protein. As such, reports that antibodies to bacterial PC-PLC react with the mammalian enzyme must be viewed with skepticism. Until mammalian PC-PLC can be isolated or cloned, D609 and the activity assay for PC-PLC remain the only tools for investigation of this important enzyme.
Cytokines, such as IL-1β and TNF-α, released from alveolar macrophages after silica particle exposure play critical roles in silica-associated pulmonary pathogenesis. As shown by the present study and others (4, 5), both IL-1β and TNF-α were increased by silica exposure. Interleukin-1β has been implicated in the deposition of collagen and pulmonary fibrosis elicited by silica and bleomycin (59). TNF-α is also required for the development of silica-induced fibrosis and silicosis is absent in TNF-α knockout mice (60). We showed that PC-PLC is involved in TNF-α and IL-β induction by silica. Furthermore, we demonstrated that PC-PLC regulation is redox- and calcium-dependent. Our findings about the role of PC-PLC in silica-induced cytokine expression and the regulatory mechanism of PC-PLC should provide new insight into the inflammatory response.
Acknowledgments
The authors thank Drs. Virginia Ramon and Peggy O'Day for the silica particles.
This work was supported by NIH grant HL37556 (H.J.F.) and funds from the University of California, Merced.
Originally Published in Press as DOI: 10.1165/rcmb.2006-0297OC on December 7, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Ding M, Chen F, Shi X, Yucesoy B, Mossman B, Vallyathan V. Diseases caused by silica: mechanisms of injury and disease development. Int Immunopharmacol 2002;2:173–182. [DOI] [PubMed] [Google Scholar]
- 2.Oghiso Y, Kubota Y. Enhanced interleukin 1 production by alveolar macrophages and increase in Ia-positive lung cells in silica-exposed rats. Microbiol Immunol 1986;30:1189–1198. [DOI] [PubMed] [Google Scholar]
- 3.Rojanasakul Y, Ye J, Chen F, Wang L, Cheng N, Castranova V, Vallyathan V, Shi X. Dependence of NF-kappaB activation and free radical generation on silica-induced TNF-alpha production in macrophages. Mol Cell Biochem 1999;200:119–125. [DOI] [PubMed] [Google Scholar]
- 4.Davis GS, Pfeiffer LM, Hemenway DR. Persistent overexpression of interleukin-1beta and tumor necrosis factor-alpha in murine silicosis. J Environ Pathol Toxicol Oncol 1998;17:99–114. [PubMed] [Google Scholar]
- 5.Srivastava KD, Rom WN, Jagirdar J, Yie TA, Gordon T, Tchou-Wong KM. Crucial role of interleukin-1beta and nitric oxide synthase in silica-induced inflammation and apoptosis in mice. Am J Respir Crit Care Med 2002;165:527–533. [DOI] [PubMed] [Google Scholar]
- 6.Porter DW, Ye J, Ma J, Barger M, Robinson VA, Ramsey D, McLaurin J, Khan A, Landsittel D, Teass A, et al. Time course of pulmonary response of rats to inhalation of crystalline silica: NF-kappa B activation, inflammation, cytokine production, and damage. Inhal Toxicol 2002;14:349–367. [DOI] [PubMed] [Google Scholar]
- 7.Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med 1998;157:1666–1680. [DOI] [PubMed] [Google Scholar]
- 8.Chen F, Shi X. NF-kappaB, a pivotal transcription factor in silica-induced diseases. Mol Cell Biochem 2002;234–235:169–176. [PubMed] [Google Scholar]
- 9.Rimal B, Greenberg AK, Rom WN. Basic pathogenetic mechanisms in silicosis: current understanding. Curr Opin Pulm Med 2005;11:169–173. [DOI] [PubMed] [Google Scholar]
- 10.Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med 2003;34:1507–1516. [DOI] [PubMed] [Google Scholar]
- 11.Vallyathan V, Shi XL, Dalal NS, Irr W, Castranova V. Generation of free radicals from freshly fractured silica dust. Potential role in acute silica-induced lung injury. Am Rev Respir Dis 1988;138:1213–1219. [DOI] [PubMed] [Google Scholar]
- 12.Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med 2002;166:S4–S8. [DOI] [PubMed] [Google Scholar]
- 13.Giron-Calle J, Forman HJ. Phospholipase D and priming of the respiratory burst by H(2)O(2) in NR8383 alveolar macrophages. Am J Respir Cell Mol Biol 2000;23:748–754. [DOI] [PubMed] [Google Scholar]
- 14.Kaul N, Choi J, Forman HJ. Transmembrane redox signaling activates NF-kappaB in macrophages. Free Radic Biol Med 1998;24:202–207. [DOI] [PubMed] [Google Scholar]
- 15.Kang JL, Go YH, Hur KC, Castranova V. Silica-induced nuclear factor-kappaB activation: involvement of reactive oxygen species and protein tyrosine kinase activation. J Toxicol Environ Health A 2000;60:27–46. [DOI] [PubMed] [Google Scholar]
- 16.Carter AB, Monick MM, Hunninghake GW. Lipopolysaccharide-induced NF-kappaB activation and cytokine release in human alveolar macrophages is PKC-independent and TK- and PC-PLC-dependent. Am J Respir Cell Mol Biol 1998;18:384–391. [DOI] [PubMed] [Google Scholar]
- 17.Wu HH, Hsieh WS, Yang YY, Tsai MC. Lipoteichoic acid induces prostaglandin E(2) release and cyclooxygenase-2 synthesis in rat cortical neuronal cells: involvement of PKCepsilon and ERK activation. Life Sci 2006;79:272–280. [DOI] [PubMed] [Google Scholar]
- 18.Exton JH. Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta 1994;1212:26–42. [DOI] [PubMed] [Google Scholar]
- 19.van Dijk M, Muriana FJ, van Der Hoeven PC, de Widt J, Schaap D, Moolenaar WH, van Blitterswijk WJ. Diacylglycerol generated by exogenous phospholipase C activates the mitogen-activated protein kinase pathway independent of Ras- and phorbol ester-sensitive protein kinase C: dependence on protein kinase C-zeta. Biochem J 1997;323:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donchenko V, Zannetti A, Baldini PM. Insulin-stimulated hydrolysis of phosphatidylcholine by phospholipase C and phospholipase D in cultured rat hepatocytes. Biochim Biophys Acta 1994;1222:492–500. [DOI] [PubMed] [Google Scholar]
- 21.Chen C, Chou C, Sun Y, Huang W. Tumor necrosis factor alpha-induced activation of downstream NF-kappaB site of the promoter mediates epithelial ICAM-1 expression and monocyte adhesion. Involvement of PKCalpha, tyrosine kinase, and IKK2, but not MAPKs, pathway. Cell Signal 2001;13:543–553. [DOI] [PubMed] [Google Scholar]
- 22.Zamorano J, Rivas MD, Garcia-Trinidad A, Qu CK, Keegan AD. Phosphatidylcholine-specific phospholipase C activity is necessary for the activation of STAT6. J Immunol 2003;171:4203–4209. [DOI] [PubMed] [Google Scholar]
- 23.Sakai T, Sugiyama T, Banno Y, Kato Y, Nozawa Y. Involvement of phosphatidylcholine hydrolysis by phospholipase C in prostaglandin F2alpha-induced 1,2-diacylglycerol formation in osteoblast-like MC3T3–E1 cells. J Bone Miner Metab 2004;22:198–206. [DOI] [PubMed] [Google Scholar]
- 24.Liu GL, Shaw L, Heagerty AM, Ohanian V, Ohanian J. Endothelin-1 stimulates hydrolysis of phosphatidylcholine by phospholipases C and D in intact rat mesenteric arteries. J Vasc Res 1999;36:35–46. [DOI] [PubMed] [Google Scholar]
- 25.Helmke RJ, Boyd RL, German VF, Mangos JA. From growth factor dependence to growth factor responsiveness: the genesis of an alveolar macrophage cell line. In Vitro Cell Dev Biol 1987;23:567–574. [DOI] [PubMed] [Google Scholar]
- 26.Giron-Calle J, Srivatsa K, Forman HJ. Priming of alveolar macrophage respiratory burst by H(2)O(2) is prevented by phosphatidylcholine-specific phospholipase C inhibitor Tricyclodecan-9-yl-xanthate (D609). J Pharmacol Exp Ther 2002;301:87–94. [DOI] [PubMed] [Google Scholar]
- 27.Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys 1997;347:256–262. [DOI] [PubMed] [Google Scholar]
- 28.Ryer-Powder JE, Forman HJ. Adhering lung macrophages produce superoxide demonstrated with desferal-Mn(IV). Free Radic Biol Med 1989;6:513–518. [DOI] [PubMed] [Google Scholar]
- 29.Hoyal CR, Thomas AP, Forman HJ. Hydroperoxide-induced increases in intracellular calcium due to annexin VI translocation and inactivation of plasma membrane Ca2+-ATPase. J Biol Chem 1996;271:29205–29210. [DOI] [PubMed] [Google Scholar]
- 30.Murthy KS, Makhlouf GM. Agonist-mediated activation of phosphatidylcholine-specific phospholipase C and D in intestinal smooth muscle. Mol Pharmacol 1995;48:293–304. [PubMed] [Google Scholar]
- 31.McGehee DS, Aldersberg M, Liu KP, Hsuing S, Heath MJ, Tamir H. Mechanism of extracellular Ca2+ receptor-stimulated hormone release from sheep thyroid parafollicular cells. J Physiol 1997;502:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoyal CR, Gozal E, Zhou H, Foldenauer K, Forman HJ. Modulation of the rat alveolar macrophage respiratory burst by hydroperoxides is calcium dependent. Arch Biochem Biophys 1996;326:166–171. [DOI] [PubMed] [Google Scholar]
- 33.Plo I, Lautier D, Levade T, Sekouri H, Jaffrezou JP, Laurent G, Bettaieb A. Phosphatidylcholine-specific phospholipase C and phospholipase D are respectively implicated in mitogen-activated protein kinase and nuclear factor kappaB activation in tumour-necrosis-factor-alpha-treated immature acute-myeloid-leukaemia cells. Biochem J 2000;351:459–467. [PMC free article] [PubMed] [Google Scholar]
- 34.van Dijk MC, Hilkmann H, van Blitterswijk WJ. Platelet-derived growth factor activation of mitogen-activated protein kinase depends on the sequential activation of phosphatidylcholine-specific phospholipase C, protein kinase C-zeta and Raf-1. Biochem J 1997;325:303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X, Lu H, Zhou L, Huang Y, Chen H. Changes of phosphatidylcholine-specific phospholipase C in hepatocarcinogenesis and in the proliferation and differentiation of rat liver cancer cells. Cell Biol Int 1997;21:375–381. [DOI] [PubMed] [Google Scholar]
- 36.Du C, Zhao Q, Araki S, Zhang S, Miao J. Apoptosis mediated by phosphatidylcholine-specific phospholipase C is associated with cAMP, p53 level, and cell-cycle distribution in vascular endothelial cells. Endothelium 2003;10:141–147. [DOI] [PubMed] [Google Scholar]
- 37.Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Kronke M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 1992; 71:765–776. [DOI] [PubMed]
- 38.Krunkosky TM, Fischer BM, Martin LD, Jones N, Akley NJ, Adler KB. Effects of TNF-alpha on expression of ICAM-1 in human airway epithelial cells in vitro: signaling pathways controlling surface and gene expression. Am J Respir Cell Mol Biol 2000;22:685–692. [DOI] [PubMed] [Google Scholar]
- 39.Zhang F, Zhao G, Dong Z. Phosphatidylcholine-specific phospholipase C regulates activation of RAW264.7 macrophage-like cells by lipopeptide JBT3002. J Leukoc Biol 2001;69:1060–1066. [PubMed] [Google Scholar]
- 40.Liu MT, Huang HM, Jeng KC, Ou SC, Kuo JS. Induction of cytokine genes and IL-1alpha by chemical hypoxia in PC12 cells. Life Sci 2000;67:2147–2157. [DOI] [PubMed]
- 41.Holian A, Kelley K, Hamilton RF Jr. Mechanisms associated with human alveolar macrophage stimulation by particulates. Environ Health Perspect 1994;102:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 2006;72:1493–1505. [DOI] [PubMed] [Google Scholar]
- 43.Lin CC, Hsiao LD, Chien CS, Lee CW, Hsieh JT, Yang CM. Tumor necrosis factor-alpha-induced cyclooxygenase-2 expression in human tracheal smooth muscle cells: involvement of p42/p44 and p38 mitogen-activated protein kinases and nuclear factor-kappaB. Cell Signal 2004;16:597–607. [DOI] [PubMed] [Google Scholar]
- 44.von Knethen A, Brune B. PKC alpha depletion in RAW264.7 macrophages following microbial/IFNgamma stimulation is PC-PLC-mediated. Antioxid Redox Signal 2005;7:1217–1222. [DOI] [PubMed] [Google Scholar]
- 45.Li F, Wu N, Su RB, Zheng JQ, Xu B, Lu XQ, Cong B, Li J. Involvement of phosphatidylcholine-selective phospholipase C in activation of mitogen-activated protein kinase pathways in imidazoline receptor antisera-selected protein. J Cell Biochem 2006;98:1615–1628. [DOI] [PubMed] [Google Scholar]
- 46.Seo JS, Kim MS, Lee SH, Kim KH, Lee HH, Jeong HD, Chung JK. Uronema marinum: identification and biochemical characterization of phosphatidylcholine-hydrolyzing phospholipase C. Exp Parasitol 2005;110:22–29. [DOI] [PubMed] [Google Scholar]
- 47.Cai H, Erhardt P, Szeberenyi J, Diaz-Meco MT, Johansen T, Moscat J, Cooper GM. Hydrolysis of phosphatidylcholine is stimulated by Ras proteins during mitogenic signal transduction. Mol Cell Biol 1992;12:5329–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garg AK, Aggarwal BB. Reactive oxygen intermediates in TNF signaling. Mol Immunol 2002;39:509–517. [DOI] [PubMed] [Google Scholar]
- 49.Sanlioglu S, Williams CM, Samavati L, Butler NS, Wang G, McCray PB Jr, Ritchie TC, Hunninghake GW, Zandi E, Engelhardt JF. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-kappa B. J Biol Chem 2001;276:30188–30198. [DOI] [PubMed] [Google Scholar]
- 50.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol 2004;173:3589–3593. [DOI] [PubMed] [Google Scholar]
- 51.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal 2005;7:1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nofer JR, Tepel M, Walter M, Seedorf U, Assmann G, Zidek W. Phosphatidylcholine-specific phospholipase C regulates thapsigargin-induced calcium influx in human lymphocytes. J Biol Chem 1997;272:32861–32868. [DOI] [PubMed] [Google Scholar]
- 53.Gercken G, Berg I, Dorger M, Schluter T. Mechanisms of particle-induced activation of alveolar macrophages. Toxicol Lett 1996;88:121–129. [DOI] [PubMed] [Google Scholar]
- 54.Tarnok A, Schluter T, Berg I, Gercken G. Silica induces changes in cytosolic free calcium, cytosolic pH, and plasma membrane potential in bovine alveolar macrophages. Anal Cell Pathol 1997;15:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen J, Armstrong LC, Liu SJ, Gerriets JE, Last JA. Silica increases cytosolic free calcium ion concentration of alveolar macrophages in vitro. Toxicol Appl Pharmacol 1991;111:211–220. [DOI] [PubMed] [Google Scholar]
- 56.Kim YK, Jang YY, Han ES, Lee CS. Depressant effect of ambroxol on stimulated functional responses and cell death in rat alveolar macrophages exposed to silica in vitro. J Pharmacol Exp Ther 2002;300:629–637. [DOI] [PubMed] [Google Scholar]
- 57.Hoyal CR, Giron-Calle J, Forman HJ. The alveolar macrophage as a model of calcium signaling in oxidative stress. J Toxicol Environ Health B Crit Rev 1998;1:117–134. [DOI] [PubMed] [Google Scholar]
- 58.Pomerantsev AP, Kalnin KV, Osorio M, Leppla SH. Phosphatidylcholine-specific phospholipase C and sphingomyelinase activities in bacteria of the Bacillus cereus group. Infect Immun 2003;71:6591–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piguet PF, Vesin C, Grau GE, Thompson RC. Interleukin 1 receptor antagonist (IL-1ra) prevents or cures pulmonary fibrosis elicited in mice by bleomycin or silica. Cytokine 1993;5:57–61. [DOI] [PubMed] [Google Scholar]
- 60.Ortiz LA, Lasky J, Lungarella G, Cavarra E, Martorana P, Banks WA, Peschon JJ, Schmidts HL, Brody AR, Friedman M. Upregulation of the p75 but not the p55 TNF-alpha receptor mRNA after silica and bleomycin exposure and protection from lung injury in double receptor knockout mice. Am J Respir Cell Mol Biol 1999;20:825–833. [DOI] [PubMed] [Google Scholar]