

Abstract
The liquid layer lining the pulmonary alveolar wall critically determines the lung's immune defense against inhaled pathogens, because it provides a liquid milieu in the air-filled alveolus for dispersal of immune cells and defensive surfactant proteins. However, mechanisms underlying formation of the liquid are unknown. We achieved visualization of the alveolar wall liquid (AWL) in situ in mouse lungs by means of optical-sectioning microscopy. Continuous liquid secretion was present in alveoli of wild-type (WT) mice under baseline conditions. This secretion was blocked by inhibitors of the cystic fibrosis transmembrane regulator (CFTR). The secretion was absent in Cftr−/− mice, and it was blocked when chloride was depleted from the perfusate of WT mice, providing the first evidence that CFTR-dependent chloride secretion causes AWL formation. Injected microparticles demonstrated flow of the AWL. The flow was blocked by CFTR inhibition and was absent in Cftr−/− mice. We conclude that CFTR-dependent liquid secretion is present in alveoli of the adult mouse. Defective alveolar secretion might impair alveolar immune defense and promote alveolar disease.
Keywords: lung, mouse, CFTR, alveolar secretion, optical-sectioning microscopy
CLINICAL RELEVANCE
Our studies of the liquid dynamics of the alveolar wall liquid by real-time microscopy indicate that chloride secretion causes alveolar wall liquid formation, a mechanism previously unknown. This will extend the understanding of innate immune defense.
The thin liquid layer lining the alveolar wall of the lung constitutes an important component of the systemic defense against inhaled pathogens. The epithelial liquid establishes a liquid phase adjacent to the alveolar epithelium, enabling secreted phospholipids and proteins to maintain alveolar patency (1) and to establish alveolar immune defense (2). However, mechanisms underlying formation of the alveolar wall liquid (AWL) remain unidentified. In fact, the bulk of the evidence favors the view that in the adult lung, active mechanisms in the alveolar wall remove, rather than secrete, alveolar liquid (3).
The difficulty is that studies of AWL have employed conventional morphologic approaches that are likely to alter physiologic liquid conditions in the alveolus (4, 5). To some extent, the liquid morphology was preserved by the application of low-temperature electron microscopy that revealed continuity of the AWL (6). However, understanding of liquid formation and flow in the alveolar wall continues to be hampered by the lack of direct, real-time methods.
Here, we address these issues through the first direct visualization of AWL by means of optical-sectioning microscopy of the mouse lung. Liquid secreted in proximal airways might drain into alveoli to form AWL. Subsequently, the wall liquid might be absorbed across the alveolar wall, as suggested by data supporting liquid absorption properties of isolated alveolar cells (7, 8). Alternatively, the wall liquid might originate in the alveolus itself and then flow proximally to the small airways. We considered that a distinction between these possibilities would be evident in the time-dependent dilution of a soluble fluorescent marker that is freely dispersed in the AWL.
MATERIALS AND METHODS
Reagents and Solutions
Reagents and fluorophores.
Fluorescein isothiocyanate (FITC)-dextran (MW 20 kD and 4 kD), ouabain, glibenclamide, amiloride, bumetanide, and terbutaline were purchased from Sigma-Aldrich (St. Louis, MO). All fluorophores were purchased from Molecular Probes (Eugene, OR). Fluorescent latex beads were purchased from Polyscience (Niles, IL). Cystic fibrosis transmembrane regulator (CFTR)inh-172 was a generous gift from Dr. Michael Matthay (UCSF, San Francisco, CA).
Solutions.
HEPES.
All solutions were prepared in HEPES buffer (Na+150 mM, K+ 5 mM, Ca2+ 1 mM, Mg2+ 1 mM, HEPES 20 mM, glucose 10 mM, dextran-70 4%, and fetal bovine serum 1%). Osmolarity of all solutions was quantified (Micro-osmometer 210; Fiske Associates, Norwood, MA) and set at 320 mosm—that is, isosmolar with mouse plasma (9). pH was set at 7.4 (φ300 pH meter, Beckman Instruments, Fullerton, CA) unless otherwise indicated. Inclusion of 4% dextran-70 (70 kD) in HEPES buffer maintains colloid osmotic pressure (COP) of 20 cm H2O (10).
Cl--modified HEPES.
As indicated, in some experiments we used Cl- depleted HEPES buffer that we prepared by replacing Cl- with gluconate, resulting in a Cl- concentration < 0.1 mM. We prepared HEPES buffer at different Cl- concentrations by mixing Cl--containing and Cl--depleted buffers. For all solutions, we quantified Cl- concentration by means of a Cl- electrode (9417B, Orion; Thermo Electron Corporation, Waltham, MA). All determinations of Cl- concentration were performed against standard calibration procedures.
Gel Chromatography
To establish adequacy of fluorophore binding we subjected FITC-dextran to gel chromatography. FITC-dextran in HEPES buffer (100 μl) was added to a gel filtration column (Sephadex G-25; Sigma-Aldrich) consisting of void volume, 3 ml and molecular exclusion > 5 kD. We sequentially collected samples (200 μl) eluted off the column, transferred the samples to a 96-well plate, and quantified sample fluorescence in an imaging station (4000MM; Eastman Kodak, Rochester, NY). All of the fluorescence occurred in the void volume (Figure 1). Further FITC given as free dye eluted in fractions appearing considerably later than the void volume fractions (Figure 1). Hence, FITC was tightly complexed to dextran, thereby ruling out the possibility that free fluorophore dissociated from the complex under experimental conditions.
Figure 1.

Gel chromatography. Fraction number, numerical order of samples collected off a Sephadex column (molecular exclusion, > 5 kD). Chromatography responses for FITC (red dots) and FITC-dextran (black dots) are indicated. For FITC-dextran (20 kD) void volume samples were collected in the first 15 fractions. Fraction volumes were 200 μl. Replicated three times each.
Animals
C57BL/6 mice were purchased from Jackson Laboratories. Cftr−/− (Cftrtm1Unc) mice were purchased from the Case Western Reserve University animal facility. All mice were > 5 wk old (18–28 g), and the experiments were performed in accordance with NIH guidelines and approved by the committee for institutional use and care of animals at St. Luke's-Roosevelt Hospital Center.
Lung Methods
Isolated lung preparation.
Our methods for establishing the isolated perfused lung have been described (11). Briefly, after induction of anesthesia (2% halothane, pentobarbital 2.5 mg/kg, intraperitoneally), heparin (25,000 U/Kg) was injected into the left ventricle. The lungs were removed from the thorax through a sternotomy, then pump-perfused through cannulae inserted in the pulmonary artery and the left atrium. Except where stated otherwise, the perfusion solution was HEPES buffer prepared as described above (Reagents and Solutions), and baseline perfusion at 37°C was set at constant pressures of 10, 3, and 5 cm H2O (Powerlab; ADIinstruments, Colorado Springs, CO) in the pulmonary artery, left atrium, and airway, respectively, to establish flow of 0.5–1 ml/min. In lung ventilation experiments, we perfused the lungs with heparinized, pooled whole blood obtained from two mice. Then, we mechanically ventilated the lungs (Harvard Apparatus, Holliston, MA) at a tidal volume of 7 ml/kg and respiratory frequency of 60 breaths/min.
Microscopy.
Lungs were positioned under either a confocal (LSM 510; Zeiss, Thornwood, NY) or a two-photon (Radiance 2100 MP; Bio-Rad, Hercules, CA) microscope. We imaged each alveolus as a stack of 2- to 5-μm-thick optical sections taken serially from the pleural aspect of the alveolus down to a depth of up to 40 μm.
Alveolar micropuncture and microinfusion.
We have previously described these methods (12). Briefly, we prepared beveled glass micropipettes of tip diameter of 6–8 μm. We backfilled each micropipette with the infusion solution, and then, using a micropipette manipulator (Narishige, East Meadow, NY), we micropunctured single alveoli and then microinfused the solution through the micropipette. We determined microinfused volume by quantifying the volume of liquid displaced in the micropipette shank. Knowing the inner diameter of the shank (0.8 mm), we calculated the volume displaced by applying the cylinder formula to the shank length emptied by microinfusion. Determined thus, our estimated microinfusion rates were ∼ 10 nl/min. After microinfusion, the micropipette was withdrawn and images were acquired from nonmicropunctured alveoli lying 1–2 alveolar diameters from the micropuncture site. Dye leakage from the micropuncture site that causes diffuse fluorescence staining of the subpleural interstitium was not evident, indicating that all microinfusions were intra-alveolar.
Microinfused solutions.
For all alveolar microinfusions, the vehicle was HEPES buffer (see above in Reagents and Solutions) with pH established at 6.5 to approximate the reported AWL pH (13). Durations of intra-alveolar microinfusions were as follows: 10 s each for FITC-dextran (3 mg/ml) and fluorescent beads, and 10 min each for calcein red (10 μM), and for the inhibitors glibenclamide (0.1 mM), CFTRinh-172 (20 μM), amiloride (10 μM or 2 mM), and terbutaline (2 μM). For each inhibitor, we followed with microinfusion of FITC-dextran (20 kD) containing the inhibitor at the same concentration. In some experiments we added ouabain (1 mM) and bumetanide (10 μM) to the perfusate 20 min before FITC-dextran microinfusion. As stated, in some experiments we used FITC alone, or FITC-dextran of smaller molecular size (4 kD).
Imaging.
The lung surface was stabilized with a coverslip and subpleural alveoli were imaged with a ×40, 0.8 NA water immersion objective (Zeiss). Fluorescence excitation and detection wavelengths for FITC were 488 and 505–530, and for calcein red they were 543 and > 560 nm. Excitation for two-photon was 800 nm; emission was collected through appropriate filters. Imaging was begun 5 min after the microinfusion of FITC-dextran. For time-dependent determinations of AWL fluorescence, images (5 μm optical thickness) were obtained every 2 min as z-stacks across the vertical alveolar diameter. Fluorescence intensity was determined by image-analysis software (Zeiss) at a resolution of 512 × 512 pixels.
AWL width.
To quantify AWL width by means of the length quantification tool (LQT) of the imaging software (Zeiss), we obtained images at resolution of 1,024 × 1,024 pixels. Since dimensional assessments might be affected by optical errors arising from the alveolar curvature, we calibrated the LQT output against known diameters of fluorescent beads of different sizes (0.4–2 μm; Polysciences). We microinfused the beads into alveoli. Next, we obtained LQT determinations of bead diameter both in 2-μm-thick optical sections of alveoli, as well as in vitro by placing beads on a coverslip. Intra-alveolar bead diameter was identical to the diameter determined in vitro (Figures 2A and 2B), indicating that intra-alveolar bead images were free of detectable optical distortion and that LQT determinations were obtained at a resolution of 0.3 μm.
Figure 2.

Calibration of length quantification by optical sectioning microscopy. A shows magnified region of an alveolar wall (AE). The image was taken as a 2-μm optical section 15 μm below the visceral pleura. Pseudocolors show fluorescence of beads (green) of different sizes, and of calcein red–loaded alveolar epithelial cells (red). (B) Diameters for three nominal bead sizes are plotted for data quantified in the alveolus against that in vitro. Mean ± SE, n = 10 for each group. Line drawn by linear regression (P < 0.05).
pH.
AWL pH was determined by the BCECF method (14). Intra-alveolar BCECF (3 μM), injected by alveolar micropuncture and microinfusion, was excited alternatingly at 458 and 488 nm. The corresponding emissions were collected through a 505-nm lowpass filter and the 488/458 ratio determined. For pH determination, the ratio was read against calibration curves relating BCECF fluorescence to pH. Calibration curves were generated in vitro for pH 3–11.
Statistical Analysis
Results are expressed as mean ± SE. ANOVA (Newman-Keuls test) was used to determine statistically significant differences between groups. P < 0.05 was considered significant.
RESULTS
AWL Fluorescence Dynamics
Alveolar microinfusion of FITC-dextran.
To load fluorophore in the AWL, we perfused lungs with fluorescence-free buffer. Next, we micropunctured an alveolus and gave a 10-s infusion of FITC-dextran (20 kD) to fill 6–8 alveoli. As we reported previously (12), the infused liquid drained spontaneously from the alveoli, rapidly re-establishing the air-filled alveolar lumen and revealing AWL fluorescence as a thin juxta-epithelial layer (Figure 3A).
Figure 3.

Confocal microscopy of fluorescent AWL tracers introduced by alveolar microinfusion. Images are of 2-μm optical sections of single mouse alveoli at a depth of 20 μm below the visceral pleura. The alveolar lumen (ALV), a septum (thin arrow), and an alveolar corner (double arrows) are marked. (A) Alveolar images show protocol of fluorophore loading by alveolar microinfusion. Left, red fluorescence indicates intracellular fluorescence of calcein red in alveolar epithelium before microinfusion. Middle, image taken 5 min after alveolar microinfusion of FITC-dextran shows green fluorescence along the alveolar epithelial wall, indicating fluorescence loading in the AWL. Note thicker AWL width at alveolar corner. AWL fluorescence is also evident in an adjacent alveolus (thick arrow). Right, image taken 25 min after FITC-dextran loading shows diminished green fluorescence both along the alveolar epithelial wall and at the alveolar corner. (B) Time-dependent changes of AWL fluorescence. For FITC-dextran (black) and FITC-dextran in the presence of ouabain (red), lines were drawn by exponential regression of mean AWL fluorescence against time. P < 0.05. Mean ± SE; n = 5 each group.
We quantified AWL fluorescence in single optical slices of the alveolus taken at a fixed depth below the pleura. We also determined integrated fluorescence of an image stack comprising a fixed number of optical sections taken across the alveolar diameter in the z-direction from the pleura. By both quantifications, AWL fluorescence decayed exponentially (Figure 3B) (not shown for integrated fluorescence). Similar decays were obtained after intra-alveolar microinfusion of FITC alone, or FITC-dextran containing dextran of a smaller molecular weight (4 kD) (not shown), indicating that molecular size was not a determining factor in the fluorescence decay.
To prevent tissue fluid accumulation at the experimental site, we maintained the colloid osmotic pressure (COP) of the perfusion buffer at 20 cm H2O, which approximates the COP for mouse plasma. Since we did not detect loss of image resolution and fluorescence intensity, which denote tissue fluid accumulation (our unpublished findings), we interpret that significant edema formation did not occur at the imaged site.
Increased macromolecular permeability of the alveolar membrane could cause loss of FITC-dextran from the alveolar space. To determine the trans-alveolar macromolecular permeability, we intravascularly perfused lungs with HEPES buffer containing fluorescent dextran (FITC-dextran, 20 kD) for 60 min, followed by perfusion with fluorescence-free buffer. Fluorescent dextran did not appear in the alveolar lumen (Figure 4A). Hence, FITC-dextran was not transported across the alveolar wall. This result indicates that there was no increase of macromolecular permeability of the alveolar epithelial membrane and that as similar to in vivo conditions, the alveolar barrier effectively blocked passive macromolecular transport.
Figure 4.


Methodological considerations. Images are of 2-μm optical sections of a single mouse alveolus at a depth of 20 μm below the visceral pleura. Lumens of the alveolus (ALV) and capillaries (*), a septum (arrow), and an alveolar corner (double arrows) are marked. (A) Red pseudocolor shows calcein fluorescence in capillary endothelial cells. Images were taken during sequential vascular perfusion with nonfluorescent buffer (left), FITC-dextran (green fluorescence, middle), then buffer again (right). The images were taken in each perfusion period at the indicated times after start of perfusion. Note: vascular infusions of FITC-dextran, then buffer, leave no residual alveolar fluorescence (right). (B) Red pseudocolor shows calcein fluorescence in alveolar epithelium. Left image was taken immediately after FITC-dextran (green) microfusion. Right image was taken 20 min later, after intra-alveolar buffer microinfusion. (C) Effect of varying image acquisition rate on time-dependent changes of AWL fluorescence. Data are from imaging series obtained at the indicated intervals between successive image acquisitions. Lines were drawn by exponential regression of mean AWL fluorescence against time. P < 0.05. Mean ± SE; n = 3 each group. (D) Plot from single experiment shows AWL pH during AWL fluorescence decay. Lungs were inflated with indicated concentrations of CO2. Note: pH decreases only in the presence of high CO2. Repeated four times.
In FITC-dextran–loaded alveoli, alveolar microinfusion of buffer at the end of the 20-min imaging period completely washed out AWL fluorescence and revealed no trans-epithelial or intra-cellular FITC fluorescence (Figure 4B). Hence, FITC-dextran was not transported across the alveolar wall or taken up by the alveolar epithelium.
Since fluorescence decay might result from fluorophore photobleaching by the excitation light, in separate experiments we determined that AWL fluorescence decays under identical imaging conditions, but at relatively fast and slow image acquisition rates. Fluorescence decays were identical for all acquisition rates (Figure 4C), indicating that the fluorescence decrease was not attributable to photobleaching.
Since pH could potentially modify FITC fluorescence, we detected pH by including the membrane-impermeable form of the pH-sensitive dye, BCECF in the AWL. During the AWL fluorescence transient, pH remained constant (Figure 4D). For positive control, we determined that AWL acidification was detectable by inflating the lung with CO2 gas (Figure 4D). These findings rule out pH as a factor in causing the AWL fluorescence decay.
Ion transport.
To determine the role of active mechanisms underlying the fluorescence decay, we treated alveoli with pharmacologic modifiers of ion transport. Ouabain, the Na+-K+-ATPase blocker, completely inhibited the AWL fluorescence decay (Figures 3B and 5A), indicating the involvement of active mechanisms. This inhibition, in which AWL fluorescence remained stable for up to 20 min, further indicates that our imaging methods did not cause photobleaching. Terbutaline, the Na+-K+-ATPase agonist (15) and activator of apical Na+ channels (16), increased AWL fluorescence (Figure 5A), indicating that AWL absorption could be induced. Given at two different concentrations (17, 18), amiloride, the apical Na+ channel inhibitor (16), had no effect on AWL fluorescence (Figure 5A). However, amiloride blocked terbutaline-induced absorption (Figure 5A), indicating that alveolar liquid absorption was absent under resting conditions, but that it could be agonist induced.
Figure 5.


Effect of inhibitors on alveolar wall liquid (AWL) dynamics. UN, untreated; OB, ouabain (1 mM); TB, terbutaline (2 μM); AL, amiloride (low: 10 μM, high: 2 mM); GB, glibenclamide (0.1 mM); IH, CFTRinh-172 (20 μM); BU, bumetanide (10 μM); WT, wild-type mouse; KO, Cftr−/− mouse. Group bars show data obtained in the first 10 min of imaging. Mean ± SE. n = 4 for each group. (A, D) Data show effects of inhibitors. *P < 0.05 compared with untreated WT. †P < 0.05 compared with untreated KO. (B, C) Data show effects of vascular perfusions with buffer solutions containing Cl- at the indicated concentrations (Cl-vasc). P < 0.05 compared with Cl-vasc of 110.
Vascular Cl-.
To determine the role of Cl- in the AWL fluorescence decay, we perfused lungs with HEPES buffer containing different Cl- contents (see “Cl--modified HEPES” under Reagents and Solutions in Materials and Methods). Reducing Cl- concentration below physiologic levels in the vascular perfusion progressively decreased AWL fluorescence decay (Figures 5B and 5C). Importantly, Cl--depleted perfusion, or perfusion with the Na+-K+- Cl- cotransport inhibitor, bumetanide (19) in Cl--containing perfusion, each completely blocked the fluorescence decay (Figure 5C). These findings indicated that basolateral Cl- determined AWL fluorescence decay.
CFTR.
Since CFTR is a Cl- channel (20), we considered the CFTR role in the AWL fluorescence decay. We perfused the lungs with HEPES buffer. Evidence for CFTR involvement was obtained in that the fluorescence decay was completely inhibited in Cftr−/− mice (Figure 5D). Further, the CFTR inhibitors, glibenclamide (21) and CFTRinh-172 (22), each inhibited the fluorescence decay in wild-type (WT) mice (Figure 5D). In Cftr−/− mice, amiloride had no effect on AWL fluorescence (Figure 5D), indicating absence of underlying Na+ absorption, while terbutaline increased AWL fluorescence (Figure 5D). These findings indicate that, as with WT mice, basal absorption was absent in mice expressing defective CFTR, although absorption could be pharmacologically induced.
AWL Width
To quantify AWL width, we used high magnification to view ∼ 10-μm segments of the alveolar wall in each image (Figure 6A). Over flat parts of the wall, we placed the LQT in vertical orientation to the air–liquid interface and determined fluorescence thickness at 2-μm intervals along the alveolar wall (Figure 6A). At alveolar corners, we determined the maximum AWL width (Figure 6A).
Figure 6.



Width of alveolar wall liquid (AWL). UN, untreated; TB, terbutaline (2 μM); AL, amiloride (low: 10 μM, high: 2 mM); IH, CFTRinh-172 (20 μM); WT, wild-type mouse; KO, Cftr−/− mouse. (A) Images of a single alveolus (left), taken in a 2-μm optical section at a depth of 20 μm below the pleura, are shown at high magnification (rectangles) for indicated locations at the alveolar septum (AS) and an alveolar corner (AC). Red pseudocolor shows intracellular fluorescence of calcein red in alveolar epithelium (AE). Green pseudocolor shows FITC-dextran in the AWL (AWL). White lines indicate placement of the line quantification tool for AWL width determinations. At the alveolar corner, we determined the maximum AWL width. (B) Plots from single experiments show time course of AWL width changes determined in alveolar corners, for the conditions indicated. (C) Group data show changes in AWL width changes determined in alveolar corners, 10 min after beginning of alveolar imaging. Mean ± SE. n = 4 for each group. *P < 0.05 compared with untreated. (D) Images show a single alveolus of a Cftr−/− mouse at time points after FITC-dextran loading by alveolar microinfusion. The dotted line shows the alveolar margin. Optical sections of 5 μm thickness were taken at 20 μm below the pleura. Uneven AWL distribution is indicated by fluorescence accumulations at corners (arrows), but absence of fluorescence in the marked segment of the alveolar epithelial wall (stars). Note that AWL fluorescence is unchanged between the two images (arrows). (E) Bars show frequency of distribution of AWL width. Widths were determined at 2-μm intervals along the alveolar perimeter (60–80 determinations). NF, no fluorescence; Septae, flat regions of alveolar wall; corners, septal junctions. Mean ± SE, n = 3 alveoli, *P < 0.05 compared with WT. Solid bars, WT; shaded bars, KO; open bars, WT + IH.
We restricted time-dependent determinations of AWL width to alveolar corners, since width changes were better determined here than in the thinner liquid layer over the flat sections of the alveolar wall. Our determinations indicated that AWL width remained unchanged during AWL fluorescence decay (Figures 6B and 6C), thereby ruling out the presence of liquid inflow from the bronchiole. Consistent with their effects on AWL fluorescence decay (Figure 5A), terbutaline decreased AWL width (Figures 6B and 6C), while ouabain and amiloride each had no effect on the width (Figure 6C). These considerations indicate that the AWL fluorescence decay resulted from dilution of the microinfused FITC-dextran by liquid secreted by alveolar cells.
AWL distributions were markedly different between WT and Cftr−/− mice (Figures 6D and 6E). In WT mice, although AWL widths were different at septal and corner regions of the alveolus (Figure 6E), AWL distribution was continuous and present at all regions of the alveolar perimeter (Figure 3A). By contrast, in Cftr−/− mice and WT mice treated with CFTRinh-172, the distribution was nonuniform in that a substantial proportion of the alveolar perimeter contained no detectable AWL (Figures 6D and 6E). Moreover, AWL width was markedly diminished over septae, although the corner widths remained similar to WT (Figure 6E).
AWL Flow
In alveoli microinfused with fluorescent beads (diameter 0.8 μm) (Figure 7A), optical sections taken at different levels indicated that the beads floated on the AWL surface (Figure 7A), and that they traveled finite distances in a direction deep to the pleural surface (Figure 7A). Displacements of single mobile beads occurred linearly with time in WT mice (Figure 7B), indicating that the beads were convectively removed from the plane of the optical slice. By contrast, bead mobility was completely blocked in glibenclamide-treated alveoli and it was absent in Cftr−/− mice (Figure 7B), indicating that the bead mobility was due to CFTR-dependent secretion and not to nonspecific effects such as those due to gravity. These findings affirmed the presence of liquid flow in the alveolar wall that was predominantly driven by epithelial secretion.
Figure 7.

Convective flow in the AWL. (A) Composite of sequential x-y images taken in a 2-μm optical section at indicated depth and time. ALV, alveolar lumen. Calcein-red fluorescence of alveolar epithelium (AE) is shown in red pseudocolor; green pseudocolor indicates FITC-dextran in the AWL. Microinfused bead (blue, diameter 0.8 μm) on the AWL surface is separated from the alveolar epithelium (double-headed arrow). (B) Distance–time relation of single-bead movement in wild-type (WT), Cftr−/− (KO), and glibenclamide-treated wild-type (WT+GB) alveoli. Lines drawn by linear regression with coefficient of determination of 0.99 (P < 0.05). *P < 0.05 compared with WT. Mean ± SE, and n = 5 for each point.
Methodological Considerations
In several groups of experiments we ruled out interference from methodological errors (see Figure E1 in the online supplement). AWL fluorescence decay was not affected by changing capillary pressure from 3 to 7 cm H2O, which changes filtration rate (23), or by changing AWL pH to 7.4 (Figure E1). To determine the effects of mechanical ventilation under blood-perfused conditions, we ventilated the lungs as described above (see Isolated lung preparation in Materials and Methods). In these experiments, we stopped ventilation for 5 min to load the FITC-dextran in the AWL. Then, we determined AWL fluorescence at 5, 15, and 25 min, stopping ventilation for 1–2 min at each time point to obtain images. The rate of AWL fluorescence decay was not different between constantly inflated and ventilated lungs, thereby ruling out the constant inflation condition as a source of error (Figure E1).
DISCUSSION
To visualize the AWL in intact alveoli, we loaded fluorophore by direct alveolar microinfusion of FITC-dextran. FITC fluorescence revealed the AWL as a thin layer of juxta-epithelial fluorescence that progressively decayed. The fluorescence decay for FITC-dextran was determined by vascular Cl- and it was ouabain inhibitable, pointing to active Cl- transport as the critical mechanism underlying the loss of AWL fluorescence. Tracer particles instilled in the AWL moved vectorially in a direction deep to the optical section, reflecting presence of secretory flow. However, the final destination of the beads remains unclear. The presence of active secretory mechanisms was also supported by the findings that the CFTR inhibitors, glibenclamide and CFTRinh-172, blocked both the fluorescence decay and the particle flow. Importantly, neither fluorescence decay nor particle flow was evident in the Cftr−/− mouse. These findings taken together indicate for the first time that the alveolar wall secretes liquid and that the secretion is driven by CFTR-dependent Cl- transport.
The presence of Cl- transport in isolated type 2 cells and the adult alveolus has been reported (24, 25). Thus, in isolated type 2 cells γ-aminobutyric increases Cl- efflux (24). Jiang and coworkers showed that cAMP-stimulated CFTR partially drives vascular-alveolar Cl- transport in mouse lung (25). To avoid nonspecific effects of Cl--depleted alveolar conditions (26, 27), we maintained Cl- concentration in the microinfused intra-alveolar liquid in the physiologic range (28). Our findings indicate that vascular Cl- concentration related directly to the AWL secretion rate. The Cl- role in this secretion was further supported in that AWL fluorescence decay was completely blocked by the Cl- transport inhibitor, bumetanide. Taking our findings together with those of Jiang and colleagues, we interpret that basolateral-apical Cl- transport determines AWL secretion.
Ion transport kinetics underlying apical Cl- secretion remain inadequately understood (29). However, it is generally proposed that in Cl- secreting epithelia, as for example in airway gland epithelia, the basolateral Na+-K+-ATPase establishes a Na+ gradient that drives Cl- uptake (30) by the basolateral Na+-K+- Cl- cotransporter, raising the intracellular Cl- potential and thereby increasing the Cl- electrochemical gradient across the cell membrane (31). CFTR-regulated Cl- channnels in the apical membrane facilitate apical Cl- secretion that may be entirely passive (31). To maintain electroneutrality co-vectorial Na+ transport occurs through transcellular or paracellular pathways. Water follows in response to the osmotic gradient. Na+ re-entry in the cell is facilitated by either apical Na+ channels or basolateral Na+-exchangers (29). This mechanism is supported by data demonstrating that inhibition of the Na+-K+-ATPase with ouabain (32), the basolateral Na+-K+- Cl- cotransporter with bumetanide (33), and CFTR with glibenclamide (34) each completely abolished Cl- secretion in tracheal epithelium. We suggest that a similar mechanism underlies alveolar Cl- secretion (Figure 8), since each of these inhibitors blocked the AWL fluorescence decay. Amiloride had no effect on the AWL fluorescence decay, indicating that no detectable eNAC dependent apical Na+ absorption was present and suggesting that alternative pathways of Na+ re-entry might exist. Further evaluation of these ion transport mechanisms is required in the context of the intact alveolus.
Figure 8.

Mechanisms of AWL secretion. AEC, alveolar epithelial cell; BL, baso-lateral; AP, apical. Arrows indicate ion movement. Numbers are sequence of proposed events, namely the basolateral Na+-K+-ATPase establishes an outward Na+ gradient across the cell membrane (1) that drives the basolateral Na+-K+-Cl- cotransporter (2), increasing cytosolic Cl-. Apical CFTR facilitate outward Cl- transport (3) down the Cl- potential gradient. Outward Na+ transport via transcellular (4) or paracellular (5) pathways maintains apical electroneutrality. Water follows passively (5).
Our findings support Macklin's long-standing proposal (35), and Bastacky and coworkers' more recent electronmicroscopic evidence (6), that a thin layer of secreted liquid covers the alveolar wall. However, a large literature indicates that alveoli absorb liquid. A detailed consideration of this literature is outside the present scope. However, as well reviewed recently (3), in alveolar type 2 epithelial cells the presence of an appropriate Na+ potential gradient (36), apical Na+ channels, and amiloride-inhibitable Na+ transport (37), provide evidence in favor of apical-basolateral Na+-driven liquid transport. The presence of such liquid transport across type 2 cells has been directly demonstrated (8) and more recently, attributed to nucleotide-gated (38, 39) and CFTR-dependent, Cl--driven (27, 40) mechanisms. Recent evidence indicates that alveolar type 1 epithelial cells might also support active liquid transport (41).
Despite this evidence, we point out that no report provides direct data showing liquid absorption across the intact alveolar membrane. We might have failed to detect absorption across type 2 cells in the present experiments. This possibility is indicated in that consistent with reported data (3), terbutaline stimulated amiloride-inhibitable absorption, as reflected in increase of AWL fluorescence. Further investigation is required to determine whether this terbutaline-induced effect uncovered absorptive flux across type 2 cells in the present model.
Several methodological issues are relevant to our interpretations. Potential concerns are that the absence of ventilation and blood flow and changes in alveolar pH might vitiate physiologic conditions in the AWL. We addressed these issues through several experiments in which we varied conditions both in the lung as well as in the microinfused solution. The fluorescence decay was unaffected by hemodynamics, ventilation, or pH within a range that might be applicable to conditions in vivo. These considerations rule out the possibility that procedural factors induced alveolar secretion. Nevertheless, the relevance of our findings to in vivo mechanisms must be interpreted with caution.
As an alternative to our present interpretation of the fluorescence decay, we considered that FITC-dextran might have been removed by epithelial uptake. Such an uptake should be revealed by intra- or paracellular fluorescence of the alveolar epithelial lining. However, in alveoli given buffer washout after the usual period of fluorescence decay, no residual fluorescence was evident in the alveolar lining, thereby ruling out epithelial uptake as a significant mechanism. Varying the rate of fluorescence excitation did not affect the data, thereby ruling out photobleaching as a cause of the fluorescence decay. We also considered that the fluorescence decay might be due to the dilutional effect of liquid inflow from the terminal bronchiole. Since such an inflow could cause time-dependent increases of AWL width, we quantified the width in magnified images of the alveolar wall. However, AWL width remained unchanged during the fluorescence decay, thereby ruling out the presence of liquid inflow from the bronchiole. These considerations make it unlikely that a mechanism other than alveolar secretion caused the fluorescence decay.
Our dimensional estimates of the AWL width (Figure 6E) are somewhat different from those obtained by Bastacky and colleagues by low-temperature electronmicroscopy (6). To achieve rapid freezing, Bastacky and coworkers fixed lungs under hyperinflated conditions that attenuate tissue thickness. Under these conditions, AWL width averaged 0.14 μm over flat septae, but 0.89 μm at alveolar corners. Although we confirmed that widths differ markedly between septae and corners, our width estimates were higher probably because we held the lungs at a lower inflation pressure that approximated end-expiratory conditions. However, we agree with Bastacky and colleagues that in WT mice, the AWL is distributed as a continuous layer and that the alveolus lacks the liquid-free zones that some postulate (42).
Although our findings indicate that net flux is secretory in the alveolus, the fate of the secretion remains unclear. Presently, the AWL secretion rate for the entire alveolus (JAWL) remains unknown. Based on our findings, we calculate JAWL of 150 × 10−3 ml/h for the mouse lung (see Appendix). This estimate might be in error to the extent that our assumptions of alveolar geometry and uniform secretion rate are incorrect. However, given this caution, we note that the reported fluid absorption rate in the human distal bronchiole is 23 × 10−3 ml/h/cm2 (43). The ratio of JAWL to this bronchiolar absorption rate gives a surface area of ∼ 7 cm2 for the distal bronchiole, across which the entire alveolar secretion is potentially cleared. For mouse, this bronchial surface area amounts to ∼ 1% of the lung's total surface area. Hence, it is possible that reabsorption rates of distal bronchi are sufficient to clear alveolar secretion.
In conclusion, our findings are the first evidence that CFTR-regulated chloride secretion occurs in the wall of the adult alveolus and that this secretion establishes bulk flow of the AWL. Moreover, several questions remain. The specific alveolar sites of AWL formation and the sites to which the liquid flows and is possibly absorbed remain uncertain. Thus, the extent to which liquid absorption occurs across type 2 cells in conjunction with AWL secretion requires consideration. Further studies are needed to address these and other questions, especially with regard to the possible disease relevance of Cl--dependent secretion in the adult alveolus.
Acknowledgments
Dr. Shonit Das (Lung Biology Laboratory, Columbia University, SLRHC, New York, NY) provided the gel chromatography data.
Appendix
Estimation of AWL secretion rate (JAWL)
In AWL loaded with FITC-dextran, we assumed that (1) fluorescence was proportional to dextran concentration, (2) the fluorescence decay indicated tracer dilution by alveolar water secretion, (3) AWL volume (VAWL) was constant (as reflected in constant AWL width), and (4) all secreted liquid exited the alveolus.
Thus, at any given time, the dextran mass (M) in the AWL can be determined as:
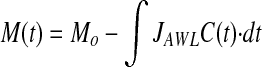 |
(1) |
, where Mo is mass at time t = 0, and C is dextran concentration (M/VAWL).
Equation 1 states that the current mass equals the initial mass minus that which has left the alveolus by convection. The mass that has left by convection is the integral of the product of the flow rate out of the alveolus and the time-dependant dextran concentration of the AWL. Dividing each term by VAWL, and recognizing that JAWL is constant, Equation 1 becomes:
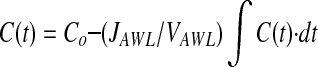 |
(2) |
Substituting fluorescence intensity (F(t), grey levels) for concentration yields:
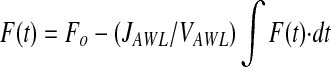 |
(3) |
,where Fo is the initial fluorescence intensity. Equation 3 solves as the first order exponential
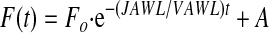 |
(4) |
,where A is a constant.
From the exponential decay of the fluorescence data in Figure 3B we calculate JAWL/VAWL as 0.036 min-1 (R2 = 0.98). Assuming spherical geometry for the alveolus, an average alveolar diameter of 40 μm and an average AWL width (W) of 1.2 μm
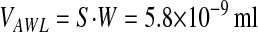 |
(5) |
,where S is the surface area of the single alveolus (5 × 10−5 cm2)
From Equations 4 and 5,
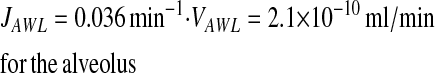 |
or
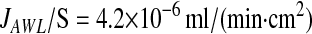 |
.Assuming total alveolar surface area for mouse to be 600 cm2 (44), we estimate:
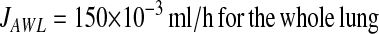 |
.
The studies reported here were supported by NIH grants HL36024, HL64896, and HL78645 (J.B.); HL-75503 (K.P.); and HL080878 (C.E.P).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2006-0347OC on February 8, 2007
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Clements JA. Lung surfactant: a personal perspective. Annu Rev Physiol 1997;59:1–21. [DOI] [PubMed] [Google Scholar]
- 2.Wright JR. Immunomodulatory functions of surfactant. Physiol Rev 1997;77:931–962. [DOI] [PubMed] [Google Scholar]
- 3.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev 2002;82:569–600. [DOI] [PubMed] [Google Scholar]
- 4.Gil J, Weibel ER. Improvements in demonstration of lining layer of lung alveoli by electron microscopy. Respir Physiol 1969;8:13–36. [DOI] [PubMed] [Google Scholar]
- 5.Bachofen H, Schurch S. Alveolar surface forces and lung architecture. Comp Biochem Physiol A Mol Integr Physiol 2001;129:183–193. [DOI] [PubMed] [Google Scholar]
- 6.Bastacky J, Lee CY, Goerke J, Koushafar H, Yager D, Kenaga L, Speed TP, Chen Y, Clements JA. Alveolar lining layer is thin and continuous: low-temperature scanning electron microscopy of rat lung. J Appl Physiol 1995;79:1615–1628. [DOI] [PubMed] [Google Scholar]
- 7.Fang X, Song Y, Zemans R, Hirsch J, Matthay MA. Fluid transport across cultured rat alveolar epithelial cells: a novel in vitro system. Am J Physiol Lung Cell Mol Physiol 2004;287:L104–L110. [DOI] [PubMed] [Google Scholar]
- 8.Mason RJ, Williams MC, Widdicombe JH, Sanders MJ, Misfeldt DS, Berry LC Jr. Transepithelial transport by pulmonary alveolar type II cells in primary culture. Proc. Natl. Acad. Sci. USA 1982;79:6033–6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vallon V, Verkman AS, Schnermann J. Luminal hypotonicity in proximal tubules of aquaporin-1-knockout mice. Am J Physiol Renal Physiol 2000;278:F1030–F1033. [DOI] [PubMed] [Google Scholar]
- 10.Ragette R, Fu C, Bhattacharya J. Barrier effects of hyperosmolar signaling in microvascular endothelium of rat lung. J Clin Invest 1997;100: 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ying X, Minamiya Y, Fu C, Bhattacharya J. Ca2+ waves in lung capillary endothelium. Circ Res 1996;79:898–908. [DOI] [PubMed] [Google Scholar]
- 12.Wang PM, Ashino Y, Ichimura H, Bhattacharya J. Rapid alveolar liquid removal by a novel convective mechanism. Am J Physiol Lung Cell Mol Physiol 2001;281:L1327–L1334. [DOI] [PubMed] [Google Scholar]
- 13.Nielson DW, Goerke J, Clements JA. Alveolar subphase pH in the lungs of anesthetized rabbits. Proc Natl Acad Sci USA 1981;78:7119–7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayaraman S, Song Y, Vetrivel L, Shankar L, Verkman AS. Noninvasive in vivo fluorescence measurement of airway-surface liquid depth, salt concentration, and pH. J Clin Invest 2001;107:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki S, Zuege D, Berthiaume Y. Sodium-independent modulation of Na(+)-K(+)-ATPase activity by beta-adrenergic agonist in alveolar type II cells. Am J Physiol 1995;268:L983–L990. [DOI] [PubMed] [Google Scholar]
- 16.Matalon S, O'Brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol 1999;61:627–661. [DOI] [PubMed] [Google Scholar]
- 17.Hardiman KM, Lindsey JR, Matalon S. Lack of amiloride-sensitive transport across alveolar and respiratory epithelium of iNOS(−/−) mice in vivo. Am J Physiol Lung Cell Mol Physiol 2001;281:L722–L731. [DOI] [PubMed] [Google Scholar]
- 18.Hardiman KM, McNicholas-Bevensee CM, Fortenberry J, Myles CT, Malik B, Eaton DC, Matalon S. Regulation of amiloride-sensitive Na(+) transport by basal nitric oxide. Am J Respir Cell Mol Biol 2004;30:720–728. [DOI] [PubMed] [Google Scholar]
- 19.Pond BB, Berglund K, Kuner T, Feng G, Augustine GJ, Schwartz-Bloom RD. The chloride transporter Na(+)-K(+)-Cl- cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J Neurosci 2006;26:1396–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 1991;253:202–205. [DOI] [PubMed] [Google Scholar]
- 21.Schultz BD, Singh AK, Devor DC, Bridges RJ. Pharmacology of CFTR chloride channel activity. Physiol Rev 1999;79:S109–S144. [DOI] [PubMed] [Google Scholar]
- 22.Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest 2002;110:1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Safdar Z, Wang P, Ichimura H, Issekutz AC, Quadri S, Bhattacharya J. Hyperosmolarity enhances the lung capillary barrier. J Clin Invest 2003;112:1541–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin N, Kolliputi N, Gou D, Weng T, Liu L. A novel function of ionotropic gamma-aminobutyric acid receptors involving alveolar fluid homeostasis. J Biol Chem 2006;281:36012–36020. [DOI] [PubMed] [Google Scholar]
- 25.Jiang J, Song Y, Bai C, Koller BH, Matthay MA, Verkman AS. Pleural surface fluorescence measurement of Na+ and Cl- transport across the air space-capillary barrier. J Appl Physiol 2003;94:343–352. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen VG, Duvall MD, Baird MS, Matalon S. cAMP activation of chloride and fluid secretion across the rabbit alveolar epithelium. Am J Physiol 1998;275:L1127–L1133. [DOI] [PubMed] [Google Scholar]
- 27.Mutlu GM, Dumasius V, Burhop J, McShane PJ, Meng FJ, Welch L, Dumasius A, Mohebahmadi N, Thakuria G, Hardiman K, et al. Upregulation of alveolar epithelial active Na+ transport is dependent on beta2-adrenergic receptor signaling. Circ Res 2004;94:1091–1100. [DOI] [PubMed] [Google Scholar]
- 28.Nielson DW. Electrolyte composition of pulmonary alveolar subphase in anesthetized rabbits. J Appl Physiol 1986;60:972–979. [DOI] [PubMed] [Google Scholar]
- 29.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev 2000;80:211–276. [DOI] [PubMed] [Google Scholar]
- 30.Ballard ST, Inglis SK. Liquid secretion properties of airway submucosal glands. J Physiol 2004;556:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welsh MJ. Intracellular chloride activities in canine tracheal epithelium. Direct evidence for sodium-coupled intracellular chloride accumulation in a chloride-secreting epithelium. J Clin Invest 1983;71:1392–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Widdicombe JH, Ueki IF, Bruderman I, Nadel JA. The effects of sodium substitution and ouabain on ion transport by dog tracheal epithelium. Am Rev Respir Dis 1979;120:385–392. [DOI] [PubMed] [Google Scholar]
- 33.Gerencser GA, Loughlin GM, Crowder MA. Chloride transport in the ferret trachea. Comp Biochem Physiol A Mol Integr Physiol 1999;122: 93–97. [DOI] [PubMed] [Google Scholar]
- 34.Schreiber R, Murle B, Sun J, Kunzelmann K. Electrolyte transport in the mouse trachea: no evidence for a contribution of luminal K(+) conductance. J Membr Biol 2002;189:143–151. [DOI] [PubMed] [Google Scholar]
- 35.Macklin CC. Pulmonary sumps, dust accumulations, alveolar fluid and lymph vessels. Acta Anat (Basel) 1955;23:1–33. [DOI] [PubMed] [Google Scholar]
- 36.Lazrak A, Matalon S. cAMP-induced changes of apical membrane potentials of confluent H441 monolayers. Am J Physiol Lung Cell Mol Physiol 2003;285:L443–L450. [DOI] [PubMed] [Google Scholar]
- 37.Matalon S, Lazrak A, Jain L, Eaton DC. Invited review: biophysical properties of sodium channels in lung alveolar epithelial cells. J Appl Physiol 2002;93:1852–1859. [DOI] [PubMed] [Google Scholar]
- 38.Junor RW, Benjamin AR, Alexandrou D, Guggino SE, Walters DV. A novel role for cyclic nucleotide-gated cation channels in lung liquid homeostasis in sheep. J Physiol 1999;520:255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norlin A, Lu LN, Guggino SE, Matthay MA, Folkesson HG. Contribution of amiloride-insensitive pathways to alveolar fluid clearance in adult rats. J Appl Physiol 2001;90:1489–1496. [DOI] [PubMed] [Google Scholar]
- 40.Fang X, Fukuda N, Barbry P, Sartori C, Verkman AS, Matthay MA. Novel role for CFTR in fluid absorption from the distal airspaces of the lung. J Gen Physiol 2002;119:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG. Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc. Natl. Acad. Sci. USA 2002;99:1966–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hills BA. An alternative view of the role(s) of surfactant and the alveolar model. J Appl Physiol 1999;87:1567–1583. [DOI] [PubMed] [Google Scholar]
- 43.Blouquit S, Morel H, Hinnrasky J, Naline E, Puchelle E, Chinet T. Characterization of ion and fluid transport in human bronchioles. Am J Respir Cell Mol Biol 2002;27:503–510. [DOI] [PubMed] [Google Scholar]
- 44.Geelhaar A, Weibel ER. Morphometric estimation of pulmonary diffusion capacity. 3. The effect of increased oxygen consumption in Japanese Waltzing mice. Respir Physiol 1971;11:354–366. [DOI] [PubMed] [Google Scholar]