

Abstract
Sinorhizobium meliloti is a gram-negative soil bacterium found either in free-living form or as a nitrogen-fixing endosymbiont of leguminous plants such as Medicago sativa (alfalfa). S. meliloti synthesizes an unusual sulfate-modified form of lipopolysaccharide (LPS). A recent study reported the identification of a gene, lpsS, which encodes an LPS sulfotransferase activity in S. meliloti. Mutants bearing a disrupted version of lpsS exhibit an altered symbiosis, in that they elicit more nodules than wild type. However, under free-living conditions, the lpsS mutant displayed no change in LPS sulfation. These data suggest that the expression of lpsS is differentially regulated, such that it is transcriptionally repressed during free-living conditions but upregulated during symbiosis. Here, I show that the expression of lpsS is upregulated in strains that constitutively express the symbiotic regulator SyrA. SyrA is a small protein that lacks an apparent DNA binding domain and is predicted to be located in the cytoplasmic membrane yet is sufficient to upregulate lpsS transcription. Furthermore, SyrA can mediate the transcriptional upregulation of exo genes involved in the biosynthesis of the symbiotic exopolysaccharide succinoglycan. The SyrA-mediated transcriptional upregulation of lpsS and exo transcription is blocked in mutants harboring a mutation in chvI, which encodes the response regulator of a conserved two-component system. Thus, SyrA likely acts indirectly to promote transcriptional upregulation of lpsS and exo genes through a mechanism that requires the ExoS/ChvI two-component system.
When nitrogen is limiting, leguminous plants enter into symbioses with members of the genera Rhizobium, Bradyrhizobium, Mesorhizobium, Azorhizobium, and Sinorhizobium (collectively called rhizobia) resulting in the formation of nodules. Within the nodule, differentiated cytoplasmic rhizobia called bacteroids reduce molecular dinitrogen to ammonia. Bacterial colonization of the nodule requires morphological alteration of epidermal cells called root hairs, resulting in the formation of a curled structure referred to as a shepherd's crook. Shepherd's crook formation is followed developmentally by the formation of an infection thread, a tubular ingrowth of the root hair that penetrates the plant. The infection thread is occupied by the bacteria, allowing their entry into the plant interior. Finally, the rhizobia are released from the infection thread into the plant cytoplasm, where they differentiate into nitrogen-fixing bacteroids (7, 8, 24, 30, 54, 63).
Symbiosis between rhizobia and legumes is dependent on bacterial synthesis of Nod factor, a lipochitooligosaccharide composed of β-(1,4)-linked N-acetylglucosamine residues and N-acylated at the nonreducing end (13, 14, 18, 23). The Nod factor produced by S. meliloti carries a 16:2 N-acyl group and 6-O-acetyl group at the nonreducing end of the molecule. It also carries a 6-O-sulfate modification at the reducing end (34), which is essential for its biological activity. Nod factor biosynthesis is dependent upon nod genes, the transcription of which is upregulated during interaction with the plant host. Upregulation of nod gene transcription requires three LysR family members: NodD1, NodD2, and NodD3. NodD1 and NodD2 activate transcription of nod genes in response to plant-derived compounds such as luteolin (49) and betaines such as trigonelline and stachydrine (50). NodD3 does not appear to require a coinducer for transcriptional activation (31, 37, 46). Transcription of nodD3 is dependent on SyrM, a LysR family transcriptional activator (37, 60). Interestingly, NodD3 can activate the transcription of syrM (60). Thus, these proteins are proposed to participate in a self-amplifying loop (60).
Colonization of alfalfa nodules by S. meliloti also requires biosynthesis of succinoglycan, an acidic exopolysaccharide. Succinoglycan biosynthesis is dependent on the products of exo genes, which are transcriptionally upregulated in the exoR::Tn5 and exoS::Tn5 mutants. The exoS gene encodes the sensor kinase of a two-component system, with chvI encoding its cognate response regulator (10, 17). The exoR gene encodes a negative regulator of exo gene transcription (17, 52). Succinoglycan biosynthesis is also increased in strains that overexpress syrA, which encodes a small (9,002 Da), basic protein (pI 9.07) (1). SyrA has been reported to upregulate succinoglycan biosynthesis in a posttranscriptional manner (1). Like nodD3, syrA transcription is regulated by SyrM. Thus, SyrA serves to link the biosynthesis of the two critical symbiotic polysaccharides in S. meliloti.
Cell surface polysaccharides such as such as lipopolysaccharide (LPS) and capsular polysaccharide (K-antigen) are also required for optimum symbiosis. In S. meliloti, the LPS undergoes an unusual covalent modification by sulfate (9, 29). Although common in mammalian cells, sulfated carbohydrates appear to be rare in bacteria, having only been reported in S. meliloti (9), Mycobacterium (45, 55), Mesorhizobium loti (62), and Pseudoalteromonas (56) to date. The physiological function of these sulfated molecules remains obscure, although mutants of S. meliloti and M. loti with decreased polysaccharide sulfation exhibit alterations in symbiosis (11, 62; D. H. Keating, G. R. O. Campbell, and G. C. Walker, submitted for publication). A recent publication reported the identification of a gene, lpsS, which encodes an LPS sulfotransferase activity in S. meliloti (11). Mutants bearing disrupted forms of lpsS produce nearly equivalent amounts of sulfated LPS as wild-type cells under free-living conditions. However, the lpsS mutant showed an altered symbiosis with alfalfa, eliciting the formation of nitrogen-fixing nodules at a greater rate than wild type (11).
The difference between the free-living and symbiotic phenotypes could be explained by repressed lpsS expression under laboratory conditions and upregulated expression during symbiosis. Here, I report that transcription of lpsS (as well as exo genes involved in biosynthesis of succinoglycan) is increased in strains that constitutively express the symbiotic regulator SyrA. Surprisingly, the ability of SyrA to mediate transcriptional upregulation of lpsS and exo genes is blocked in mutants that affect the ExoS/ChvI two-component system.
MATERIALS AND METHODS
Bacterial strains and media.
All strains used are derivatives of S. meliloti Rm1021 (41) and are described in Table 1. All strains were grown in LB (12), tryptone yeast extract (TY) (3), or M9 (38) medium with antibiotic concentrations as previously described (48).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristic(s)a | Reference or source |
|---|---|---|
| S. meliloti strains | ||
| Rm1021 | Strr SU47 | 41 |
| Rm8002 | Rm1021; phoA mutant; Strr | 36 |
| DKR153 | lpsS::pDW33 | 11 |
| DKR192 | Rm1021/pTE3 | This study |
| DKR381 | Rm1021/pTE3::syrA | This study |
| DKR340 | lpsS::pDW33/pTE3 | This study |
| DKR341 | lpsS::pDW33/pRmJT5 | This study |
| DKR360 | lpsS::pDW33/pTE3::syrM | This study |
| DKR361 | lpsS::pDW33/pTE3::syrA | This study |
| DKR475 | lpsS::pDW33/pMS03/TE3 | This study |
| DKR477 | lpsS::pDW33/pMS03::nodPQ/pTE3 | This study |
| DKR476 | lpsS::pDW33/pMS03/pTE3::syrA | This study |
| DKR478 | lpsS::pDW33/pMS::nodPQ/pTE3::syrA | This study |
| DKR395 | nodD3::pVO155 | This study |
| DKR400 | lpsS::pDW33 nodD3::pVO155/pTE3 | This study |
| DKR402 | lpsS::pDW33 nodD3::pVO155/pTE3::syrA | This study |
| DKR405 | syrM::pVO155 | This study |
| DKR408 | lpsS::pDW33 syrM::pVO155/pTE3 | This study |
| DKR409 | lpsS::pDW33 syrM::pVO155/pTE3::syrA | This study |
| DKR396 | lpsS::pVO155 | This study |
| DKR398 | lpsS::pVO155/pTE3 | This study |
| DKR399 | lpsS::pVO155/pTE3::syrA | This study |
| EC69 | chvI::pDW33 encoding K214T mutation | D. H. Wells, E. J. Chen, and S. R. Long |
| DKR403 | lpsS::pVO155 chvI::pDW33 (K214T)/pTE3 | This study |
| DKR404 | lpsS::pVO155 chvI::pDW33 (K214T)/pTE3::syrA | This study |
| DKR373 | lpsS::pDW33 exoR::Tn5 | 28 |
| DKR375 | lpsS::pDW33 exoS::Tn5 | 28 |
| DKR497 | lpsS::pDW33 exoS::Tn5/pTE3 | This study |
| DKR498 | lpsS::pDW33 exoS::Tn5/pTE3::syrA | This study |
| DKR494 | exoS::Tn5/pMS03 | This study |
| DKR495 | exoS::Tn5/pMS03::nodPQ | This study |
| DKR342 | exoY::pVO155/pTE3 | This study |
| DKR385 | exoY::pVO155/pTE3::syrA | This study |
| DKR411 | exoY::pVO155 chvI::pDW33 (K214T)/pTE3 | This study |
| DKR412 | exoY::pVO155 chvI::pDW33 (K214T)/pTE3::syrA | This study |
| DW223 | exoH::pVO155 | 64 |
| DKR420 | exoH::pVO155/pTE3 | This study |
| DKR421 | exoH::pVO155/pTE3::syrA | This study |
| DKR451 | Rm8002/pDKR451 syrA1-31::phoA24-C terminus | This study |
| DKR482 | Rm8002/pDKR451 syrAfull-length::phoA24-C terminus | This study |
| DKR457 | Rm8002/pTE3 | This study |
| DKR458 | Rm8002/pTE3::syrA | This study |
| Plasmids | ||
| pTE3 | pLAFR harboring serovar Typhimurium trp promoter | 19 |
| pRF771 | pLAFR harboring serovar Typhimurium trp promoter and an improved multicloning site | 64 |
| pVO155 | Insertional activation plasmid Nmr | 48 |
| pDW33 | Insertional activation plasmid Hygr | 11 |
| pMS03 | Broad-host-range vector high-copy-number derivative of pMB393 containing the trp promoter from serovar Typhimurium | 62 |
| pRmJT5 | pLAFR harboring 20-kb fragment with host specific nod genes | 61 |
| pS73 | pTE3::syrM | 61 |
| pMB89 | pTE3::syrA | 1 |
| pDKR395 | pVO155::nodD3 | This study |
| pDKR396 | pVO155::lpsS | This study |
| pDKR405 | pVO155::syrM | This study |
| pDKR451 | pRF771::syrA1-31::phoA24-C terminus | This study |
| pDKR482 | pRF771::syrAfull-length::phoA24-C terminus | This study |
| pDKR453 | pCR2.1::syrA1-31 | This study |
| pDKR454 | pCR2.1::phoA24-C terminus | This study |
| pDKR452 | pCR2.1::syrAfull-length | This study |
| pGTO101 | pMS03::nodPQ | 62 |
All strains are derived from strain S. meliloti Rm1021.
Strain construction.
Plasmids were introduced into S. meliloti by triparental mating as described previously (16). Strain DKR396 was constructed by introduction of the plasmid pDKR396 (which harbors an internal fragment of lpsS) into Rm1021 and selection for neomycin-resistant colonies. pDKR396 cannot replicate within S. meliloti; thus, neomycin-resistant colonies arise from recombination events that integrate the plasmid into the genome at the lpsS locus, disrupting the lpsS gene. The insertion events were then confirmed by PCR. Strains DKR395 and DKR405 were constructed in the same manner. Strains DKR400 and DKR402 were constructed by transduction of the nodD3::pVO155 from strain DKR395 into DKR340 and DKR361, respectively. Strains DKR408 and DKR409 were constructed by transduction of syrM::pVO155 from strain DKR405 into strains DKR340 into DKR361, respectively. Strains DKR403 and DKR404 were constructed by transduction of the chvI::pDW33 (K214T) allele from strain EC69 to strains DKR398 and DKR399, respectively. Strains DKR411 and DKR412 were constructed by transduction of the chvI::pDW33 (K214T) allele from strain EC69 to strains DKR342 and DKR385, respectively. Strains DKR413 and DKR414 were constructed by transduction of the chvI::pDW33 (K214T) allele from strain EC69 to strains DKR420 and DKR421, respectively.
Plasmid construction.
Plasmid pDKR396 was constructed by amplifying an internal fragment of lpsS from Rm1021 chromosomal DNA using the primers 5′-AGGTCGACGGAAGGGATTTCATTCA-3′ and 5′-TCGGATCCGCGGCGAGCTCCTCGTA-3′. This fragment was then cloned into plasmid pCR2.1 (Invitrogen), and its presence was verified by colony PCR and restriction enzyme digestion. The lpsS-containing fragment was then isolated from the pCR2.1 plasmid by restriction enzyme digestion with BamH1 and Sal1 and ligated into pVO155 (48), digested with the same enzymes.
Plasmid pDKR395 was constructed by amplifying nodD3 via PCR using the primers 5′-AGGTCGACGAGCGCGTGGCTCGGGA-3′ and 5′-TGGGATCCAACATGCCCCATCGACA-3′. The fragments were then cloned into plasmid pVO155 by the method described for plasmid pDKR396.
Plasmid pDKR405 was constructed using the primers 5′-ACGCGAGTCGACAGATGATGAACCT-3′ and 5′-AAGGATCCGAGCGGAGCGGCGCCCA-3′ to amplify syrM. The fragments were then cloned into plasmid pVO155 by the method described for plasmid pDKR396.
Plasmid pDKR452 was constructed using primers 5′-CTATCGATCAGTTGGACGCTGCCGA-3′ and 5′-GACATATGATTGCGGTTCTCGCTGA-3′ to amplify the syrA open reading frame. The PCR product was then cloned into pCR2.1. The fragment was then cloned into plasmid pCR2.1 and verified by colony PCR and restriction enzyme digestion.
Plasmid pDKR453 was constructed using primers 5′-CTATCGATCAGTTGGACGCTGCCGA-3′ and 5′-GCGTCGTAACCATATGGCCGGGGCAGGGCT-3′ to amplify an N-terminal portion of the syrA open reading frame (residues 1 to 31). The PCR product was then cloned by the method described for plasmid pDKR452.
Plasmid pDKR454 was constructed using primers 5′-CACATATGCCTGTTCTGGAAAACCGGGCTG-3′ and 5′-CGGTACCGAAAATTCACTGCCGGGC-3′ to amplify the C-terminal portion of the phoA open reading frame (residues 24 to the C terminus [phoA24-C terminus]). The PCR product was then cloned by the method described for plasmid pDKR452.
Plasmid pDKR451 was constructed by ligating the Xba1/Nde1 fragment from plasmid pDKR453 with the BamH1/Nde1 fragment from plasmid pDKR454. The fragment was then ligated into plasmid pRF771 (64) digested with BamH1 and Xba1 and verified by colony PCR and restriction enzyme digestion.
Plasmid pDKR482 was constructed by ligating the Xba1/Nde1 fragment from plasmid pDKR452 with the BamH1/Nde1 fragment from plasmid pDKR454 and cloning into pRF771 in the manner described for plasmid pDR451.
Preparation of extracts for LPS analysis.
Extracts were prepared according to Reuhs et al. (53), as modified by Cronan et al. (11). The pellet was resuspended in 50 μl of sample loading buffer, and the polysaccharides were fractionated by Tris-Tricine-polyacrylamide gel electrophoresis (PAGE) as described previously (47). The polysaccharides were then visualized by silver staining (Bio-Rad).
In vivo labeling of LPS.
Wild-type and lpsS mutants were cultured in TY medium containing 5 μCi of Na235SO4 (ICN) as described previously (11). The LPS was then extracted as described above and fractionated by Tris-Tricine-PAGE (47). The PAGE gel was then silver stained (Bio-Rad) to determine the relative amount of extracted LPS and dried, and the incorporated 35SO4 was visualized by autoradiography and quantified by phosphorimaging (Amersham Pharmacia).
Preparation of cell surface protein extracts.
Extracts were prepared as described previously (29). The resulting pellet was resuspended in 100 μl of buffer A (0.05 M Na2HPO4, 0.005 M EDTA; pH 7), and protein concentration was determined by a modified Bradford assay (Bio-Rad).
In vitro cell surface sulfation assay.
In vitro LPS sulfation was assayed as described previously (29). A total of 0.25 to 1 μg of a particulate extract was combined with 1 μl of S. meliloti LPS (which was added as a sulfate acceptor), 5 μCi of 35SO4-labeled PAPS (3′-phosphoadenosine-5′-phosphosulfate) prepared as described previously (20, 35, 58), and 2 μl of 5× buffer B (50 mM Tris-HCl [pH 8], 30 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, 10% glycerol) in a total reaction volume of 10 μl. The mixture was then allowed to incubate for 30 min at 30°C, and the reaction was stopped by incubation for 2 min at 95°C. The samples were then heated at 95°C for 5 min in sodium dodecyl sulfate sample buffer and fractionated on a 12.5% sodium dodecyl sulfate-PAGE gel. The gel was dried, and the incorporation of 35SO4 into LPS in the particulate fraction was measured using a phosphorimager (Amersham Pharmacia).
PAPS analysis by thin-layer chromatography.
PAPS was analyzed as described previously (4), as modified by Townsend et al. (62). Briefly, wild-type strains harboring vector control, multicopy syrA, or multicopy nodPQ, were cultured in 1 ml of TY medium with Na235SO4 to saturation. The cells were centrifuged at 8,000 × g, resuspended in 1 ml of water, and centrifuged again at 8,000 × g. The resulting cell mass was resuspended in 0.2 ml of water, and 20 μl of 11 N formic acid was added. The mixture was mixed by vortexing, incubated on ice for 30 min, and centrifuged at 8,000 × g for 10 min. Fifty microliters of each supernatant was then spotted on a polyethyleneimine (PEI)-cellulose thin-layer chromatography (TLC) plate (Baker), the plate was immersed in methanol, and allowed to dry before being placed in a TLC chamber containing 100 ml of 0.9 M LiCl2. After the solvent front reached the top of the TLC plate, the plate was again immersed in methanol for 2 min and allowed to dry. The 35SO4 incorporation was visualized by autoradiography and quantified by phosphorimaging.
Reverse transcription (RT)-PCR assay.
S. meliloti strains were cultured to stationary phase in LB medium (optical density at 600 nm [OD600] of 2.5). The cells were then washed with LB and frozen at −20°C. RNA was extracted from the pellets via modified phenol-chloroform extraction (Trizol). Dilutions of the RNA were then used as templates for the synthesis of cDNA (First-Strand cDNA synthesis kit; Fermentas). Two microliters of cDNA from each dilution was used to amplify DNA via PCR. The DNA was then fractionated on a 1% agarose gel, and the DNA was detected with ethidium bromide staining, followed by fluorescent imaging (Typhoon-Amersham).
Alkaline phosphatase assay.
Plasmids containing syrA-phoA translational fusions were introduced into strain Rm8002 (which exhibits greatly reduced alkaline phosphatase activity [36]) by triparental mating. The plasmid-bearing strains were then streaked onto LB plates containing 60 μg/ml 5-bromo-4-chloro-3-indolyl phosphate. The plates were grown for 5 days and then photographed.
β-Glucuronidase assay.
Cells were grown to stationary phase (OD600of 2.5) and then harvested. β-Glucuronidase activity was assayed under free-living conditions according to Jefferson et al. (27).
RESULTS
Expression of the lpsS gene is upregulated by plasmid pRmJT5, which encodes the symbiotic regulators SyrM, NodD3, and SyrA.
Mutants bearing disrupted forms of lpsS elicit increased numbers of nitrogen-fixing nodules on the plant host alfalfa, compared to wild-type S. meliloti (11). However, under free-living conditions, lpsS::pDW33 mutants display no change in growth rate, LPS structure, or LPS sulfation compared to wild type (11). I hypothesized that the difference in observed phenotypes between free-living growth and growth in planta could result from differential expression of lpsS, such that it is transcribed at a low level under free-living conditions but transcriptionally upregulated during symbiosis. To test this hypothesis, I utilized a previously constructed lpsS::pDW33 mutant, which results in a transcriptional fusion of lpsS to uidA (11) encoding β-glucuronidase. Using this transcriptional fusion, I examined lpsS transcription under laboratory conditions in strains that constitutively express known symbiotic regulatory genes. The regulation of many symbiotic genes in S. meliloti involves three transcriptional activators: NodD1, which promotes transcription of nod genes in the presence of the flavonoid luteolin (49); NodD2, which promotes transcription in response to betaines such as trigonelline and stachydrine (50); and NodD3/SyrM, which upregulates nod genes (and other symbiotically relevant genes) during symbiosis (2, 60).
A previous report detected no changes in lpsS transcription in cells cultured in the presence of luteolin (11); therefore, I examined the effect of NodD3/SyrM on expression of lpsS. The genes nodD3 and syrM are expressed at a very low level during free-living growth (60) but are upregulated during symbiosis. While the regulatory mechanism of nodD3 and syrM in planta is only incompletely understood, the expression levels of nodD3 and syrM are known to be increased when placed on the low-copy-number plasmid pRmJT5 (61), which contains nodD3, syrM, and syrA, as well as several other host-specific nod genes. Introduction of pRmJT5 increased transcription of the lpsS::uidA fusion by 2.8-fold compared to plasmid pTE3 (Fig. 1A), which I employed as a vector control. Therefore, the expression of the lpsS::uidA fusion is upregulated by a gene or genes present on plasmid pRmJT5.
FIG. 1.
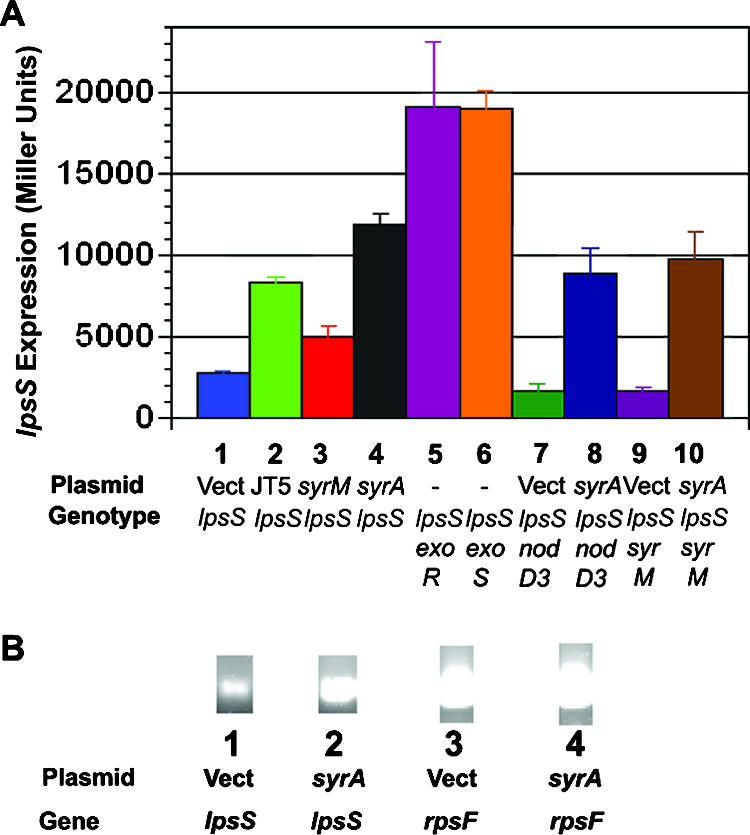
lpsS expression is affected by plasmids overexpressing SyrA. (A) Expression of lpsS::uidA transcriptional fusions. Plasmids harboring S. meliloti host-specific nod genes were introduced into the lpsS::pDW33 insertion (which results in a transcriptional fusion of lpsS to the uidA gene). In addition, the exoR::Tn5, exoS::Tn5, syrM::pVO155, and nodD3::pVO155 regulatory mutations were introduced into lpsS::pDW33 by transduction. The strains were then grown to stationary phase (OD600 of 2.5) and assayed for β-glucuronidase activity as described in Materials and Methods. Activity is in Miller units. Error bars represent standard deviations of experiments carried out in triplicate. Strains lacking the lpsS::pDW33 showed background levels of β-glucuronidase activity (≤50 Miller units). Lane 1, lpsS::pDW33/pTE3 (vector); lane 2, lpsS::pDW33/pRmJT5 (which contains a 20-kb fragment of pSymA with host-specific nod genes); lane 3, lpsS::pDW33/pTE3::syrM; lane 4 lpsS::pDW33/pTE3::syrA; lane 5, lpsS::pDW33 exoR::Tn5; lane 6, lpsS::pDW33 exoS::Tn5; lane 7, lpsS::pDW33 nodD3::pVO155/pTE3; lane 8, lpsS::pDW33 nodD3::pVO155/pTE3::syrA; lane 9, lpsS::pDW33 syrM::pVO155/pTE3; lane 10, lpsS::pDW33 syrM::pVO155/pTE3::syrA. (B) Measurement of lpsS expression by RT-PCR. S. meliloti strains were cultured in LB medium. RNA was extracted from the pellets and cDNA was prepared and used as template for DNA amplification by PCR as described in Materials and Methods. Lane 1, cDNA prepared from wild type/pTE3, amplified with lpsS-specific primers; lane 2, cDNA prepared from wild type/pTE3::syrA, amplified with lpsS-specific primers; lane 3, cDNA prepared from wild type/pTE3, amplified with rpsF-specific primers; lane 4, cDNA prepared from wild type/pTE3::syrA, amplified with rpsF-specific primers.
Upregulation of expression of lpsS is dependent upon the symbiotic regulator syrA.
Having shown that plasmid pRmJT5 increases the expression of lpsS::uidA transcriptional fusion, I then sought to determine whether SyrM and/or NodD3 was responsible for this transcriptional upregulation. I utilized plasmid pS73 (61), a derivative of plasmid pTE3 containing the syrM gene placed downstream of the Salmonella enterica serovar Typhimurium trp promoter (which leads to constitutive expression in S. meliloti [19]). I found that multicopy syrM resulted in a 1.7-fold increase in lpsS::uidA transcription, compared to the vector control (Fig. 1A). Due to the presence of the strong trp promoter, plasmid pS73 would be expected to produce SyrM at a higher level than plasmid pRmJT5, suggesting that SyrM might be operating in an indirect manner to promote expression of lpsS. SyrM has been reported to directly activate transcription of only two genes: nodD3 and syrA (1, 46). NodD3 upregulates transcription by binding to a conserved 5′ region of nod genes called a nod box. Since the lpsS gene does not contain an apparent nod box, it seemed unlikely that NodD3 was responsible for the increase in lpsS transcription. Plasmid pRmJT5 also contains the gene syrA, which encodes a protein that leads to increased biosynthesis of the exopolysaccharide succinoglycan (1). I introduced the plasmid pMB89 (1) that places syrA under control of the trp promoter and measured expression of the lpsS::uidA fusion. Expression of the lpsS::uidA fusion was upregulated fourfold in the presence of multicopy syrA (Fig. 1A) compared to the vector control. Analysis of steady-state RNA levels by RT-PCR also showed an increase in lpsS transcription in the presence of multicopy syrA (Fig. 1B). Thus, overexpression of syrA results in an increase in lpsS transcription.
lpsS-dependent sulfotransferase activity is increased in SyrA overexpressing backgrounds.
Transcription of lpsS is elevated in the presence of multicopy syrA, which would be expected to increase the steady-state levels of LpsS. Antisera directed against LpsS was not available; thus, LpsS protein levels could not be measured directly. However, increased steady-state levels of LpsS protein would be expected to result in an increase in LPS sulfotransferase activity. Thus, I measured LPS sulfotransferase activity in extracts derived from cells overexpressing nodD3, syrM, and syrA. Extracts from strains bearing either plasmid pRmJT5 or overproducing SyrM did not exhibit a significant increase in LPS sulfotransferase activity (Fig. 2A). The reason for the lack of an increase in LPS sulfotransferase activity in the strain containing pRmJT5 is not known but may result from an increase in intracellular PAPS in the extract (which competes with the sulfate donor in the in vitro assay). However, extracts prepared from strains overexpressing syrA showed a twofold increase in sulfotransferase activity (Fig. 2A). Mutants that lack a functional copy of lpsS but overexpress syrA showed only 20% of the sulfotransferase activity observed in wild type (Fig. 2A), demonstrating that the increase in LPS sulfotransferase activity is LpsS dependent. Therefore, multicopy syrA upregulates the transcription of the lpsS gene and results in increased LpsS activity.
FIG. 2.

Increased LpsS activity in strains overexpressing SyrA. Plasmids harboring S. meliloti host-specific nod genes were introduced into Rm1021 (wild type). In addition, the exoR::Tn5 and exoS::Tn5 regulatory mutations were introduced into Rm1021 by transduction. (A) LPS sulfotransferase activity. The strains were grown to saturation (OD600 of 2.5) and extracts were assayed for LPS sulfotransferase activity as described in Materials and Methods. LPS sulfotransferase activity represents sulfate incorporated into LPS (as measured by phosphorimaging)/mg of protein. Lane 1, Rm1021 (wild type)/pTE3 (vector); lane 2, Rm1021/pRmJT5 (which contains a 20-kb fragment of pSymA with host-specific nod genes); lane 3 Rm1021/pTE3::syrM; lane 4, Rm1021/pTE3::syrA; lane 5, exoR::Tn5; lane 6, exoS::Tn5; lane 7, chvI(K214T)/pTE3; lane 8, chvI(K214T)/pTE3::syrA; lane 9, lpsS::pDW33/pTE3; lane 10, lpsS::pDW33/pTE3::syrA. Error bars represent standard deviations of experiments carried out in triplicate. (B) LPS sulfation in strains overexpressing SyrA. Strains were grown to saturation (OD600 of 2.5) in the presence of Na235SO4 (ICN). Cell surface polysaccharides were then extracted and fractionated by Tris-Tricine-PAGE, and the incorporation of sulfate was measured by phosphorimaging as described in Materials and Methods. Lane 1, Rm1021 (wild type)/pTE3 (vector); lane 2, Rm1021/pRmJT5 (which contains a 20-kb fragment of pSymA with host-specific nod genes); lane 3, Rm1021/pTE3::syrM; lane 4, Rm1021/pTE3::syrA; lane 5, exoR::Tn5; lane 6, exoS::Tn5. (C) Measurement of PAPS biosynthesis. Strains were cultured in the presence of 35SO4, and PAPS and APS (adenosine-5′-phosphosulfate, a derivative of PAPS) was recovered by formic acid extraction as described in Materials and Methods. The formic acid extracts were then subjected to fractionation on PEI-cellulose, and the radioactive material was detected by phosphorimaging. Migration of PAPS and APS was determined by comparison to labeled standards (not shown). The asterisk signifies a high mobility spot that did not comigrate with any of the standards. Lane 1, Rm1021 (wild type) containing pTE3 (Vect) and pMS03 (Vect); lane 2, Rm1021 containing pTE3::syrA and pMS03; lane 3, Rm1021 containing pTE3 and pMS03::nodPQ; lane 4, Rm1021 containing pTE3::syrA and pMS03::nodPQ. (D) Overexpression of nodPQ results in a SyrA-dependent increase in LPS sulfation. Either pMS03 (vector control) or pMS03 containing nodPQ from M. loti was introduced into wild-type strains harboring either pTE3 or pTE3::syrA. The incorporation of sulfate was then measured as described in panel B. Lane 1, Rm1021 (wild type), containing pTE3 (Vect) and pMS03 (Vect); lane 2, Rm1021 containing pTE3::syrA and pMS03; lane 3, Rm1021 containing pTE3 and pMS03::nodPQ; lane 4, Rm1021 containing pTE3::syrA and pMS03::nodPQ; lane 5, exoS::Tn5 containing pMS03; lane 6, exoS::Tn5 containing pMS03::nodPQ; lane 7, lpsS::pDW33 containing pTE3 and pMS03; lane 8, lpsS::pDW33 containing pTE3::syrA and pMS03; lane 9, lpsS::pDW33 containing pTE3 and pMS03::nodPQ; lane 10, lpsS::pDW33 containing pTE3::syrA and pMS03::nodPQ. Error bars represent standard deviations of experiments carried out in triplicate.
Although overexpression of syrA resulted in a measurable increase in LpsS activity, it was unclear whether this would contribute to a change in overall LPS sulfation. Thus, I measured the incorporation of radiolabeled sulfate into LPS in vivo in the presence and absence of pRmJT5 or multicopy plasmids bearing syrM and syrA (Fig. 2B). Plasmid pRmJT5 resulted in a 2.5-fold increase in LPS sulfation compared to the vector control, but overexpression of syrM or syrA did not result in a measurable increase in LPS sulfation (Fig. 2B). A similar result was observed previously in strains with multicopy forms of lpsS (11). These strains showed increased LPS sulfotransferase activity but did not display an increase in LPS sulfation. However, strains harboring multicopy forms of lpsS did show increased LPS sulfation compared to vector controls when luteolin was added to the culture. Addition of luteolin also led to increased LPS sulfation in strains overexpressing syrA, compared to vector control strains (my unpublished results). Luteolin is known to induce the transcription of several classes of genes including nodPQ. The nodPQ genes catalyze the synthesis of PAPS, the activated form of sulfate used by LpsS and other sulfotransferases. Plasmid pRmJT5 encodes the nodPQ genes, as well as the regulators syrM, syrA, and nodD3. NodD3 can activate transcription of both the plasmid and genomic copies of nodPQ and would be expected to result in elevated biosynthesis of PAPS (60). Thus, it seemed possible that the differences in LPS sulfation between strains harboring multicopy syrA and pRmJT5 might result from differences in intracellular PAPS concentration. I measured the PAPS biosynthesis of a strain carrying pRmJT5 and observed an increase in the intracellular PAPS concentration with respect to strains carrying vector alone (my unpublished results). Therefore, I hypothesized that plasmid pRmJT5 led to an increase in LPS sulfation for two reasons: the ability to increase lpsS expression and the ability to increase PAPS concentration resulting from upregulation of nodPQ transcription. Conversely, overexpression of syrA increased lpsS expression, leading to elevated levels of LpsS-dependent sulfotransferase activity, but did not increase LPS sulfation because of limiting PAPS. To test whether LPS sulfation was limited by the internal PAPS concentration in strains overexpressing syrA, I utilized a plasmid (62) that places the nodPQ genes from M. loti under control of the trp promoter in the plasmid pMS03. PAPS was undetectable in strains harboring vector alone, suggesting that PAPS is limiting for LPS sulfation in S. meliloti. However, overexpression of nodPQ from this plasmid in a wild-type background resulted in an increase in PAPS as measured by TLC on PEI-cellulose (Fig. 2C) and a 1.5-fold increase in LPS sulfation (Fig. 2D). However, when nodPQ and syrA were both overexpressed, I observed a 2.3-fold increase in LPS sulfation (Fig. 2D). Therefore, syrA is capable of increasing LPS sulfation, provided that sufficient PAPS is present in the cell.
Expression of exopolysaccharide biosynthetic genes is upregulated by the symbiotic regulator syrA.
The surprising finding that lpsS transcription was affected by SyrA led to a reexamination of the role of SyrA in the regulation of symbiotically important polysaccharides. A previous study had shown that overexpression of SyrA resulted in increased production of the symbiotic polysaccharide succinoglycan but did not detect increased expression of transcriptional fusions to the succinoglycan biosynthetic genes exoP and exoF (1). I introduced plasmid pMB89 harboring syrA into previously constructed single-copy exoY::uidA and exoH::uidA transcriptional fusions (64) (Fig. 3A). Expression of the exoY::uidA fusion was increased 2.6-fold in the presence of multicopy syrA, while expression of the exoH::uidA fusion was increased fourfold in the presence of multicopy syrA (Fig. 3A), compared to vector alone. Utilizing RT-PCR, I also demonstrated an upregulation of exoY expression (Fig. 3B). Because the previous analysis of syrA-mediated expression had used a transcriptional fusion to exoF, I also constructed a single-copy exoF::uidA transcriptional fusion and measured the expression of this fusion in the presence and absence of multicopy syrA. The exoF::uidA fusion was upregulated threefold in cells harboring multicopy syrA (Fig. 3A) compared to vector alone. Thus, overexpression of syrA upregulates expression of multiple genes involved in succinoglycan biosynthesis.
FIG. 3.
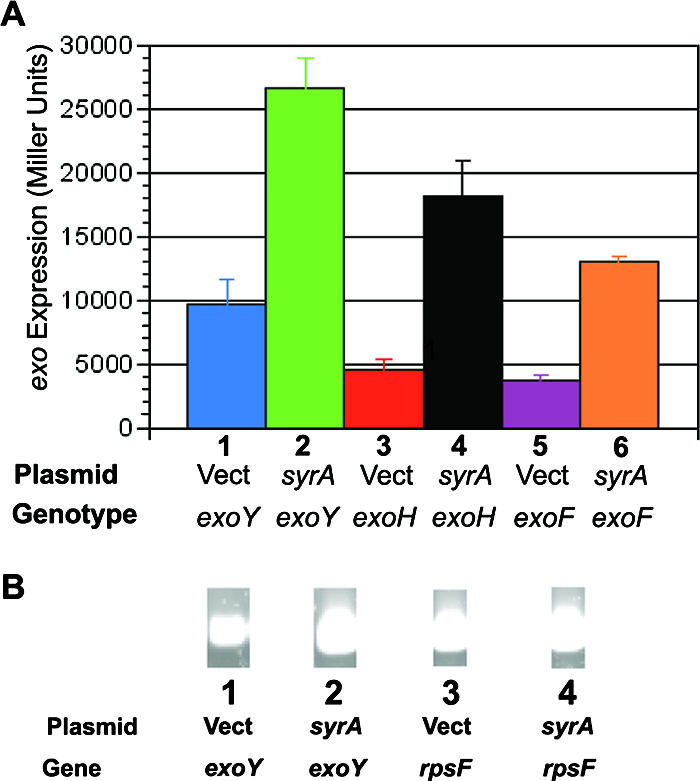
Increased exo gene transcription in strains overexpressing syrA. (A) Expression of exo::uidA transcriptional fusions. Plasmids harboring S. meliloti syrA were introduced into the exoY::pVO155, exoH::pVO155, and exoF::pVO155 mutants (which result in transcriptional fusions to the uidA gene). The strains were then grown to saturation and assayed for β-glucuronidase activity as described in Materials and Methods. Error bars represent standard deviations of experiments carried out in triplicate. Activity is in Miller units. Lane 1, exoY::pVO155/pTE3 (vector); lane 2, exoY::pVO155/pTE3::syrA; lane 3, exoH::pVO155/pTE3; lane 4, exoH::pVO155/pTE3::syrA; lane 5, exoF::pVO155/pTE3; lane 6, exoF::pVO155/pTE3::syrA. (B) Measurement of exoY expression by RT-PCR. Purified RNA was used as template for RT-PCR. Lane 1: cDNA prepared from wild type/pTE3, amplified with exoY-specific primers; lane 2, cDNA prepared from wild type/pTE3::syrA, amplified with exoY-specific primers; lane 3, cDNA prepared from wild type/pTE3, amplified with rpsF-specific primers; lane 4, cDNA prepared from wild type/pTE3::syrA, amplified with rpsF-specific primers.
The nod3 and syrM genes are not required for SyrA-mediated transcriptional regulation of lpsS.
The finding that overexpression of syrA can influence transcription was surprising, in that SyrA shows no sequence identity to known transcriptional regulators (1). Furthermore, analysis of its sequence predicted the presence of a cleavable signal sequence and a transmembrane region, which would be expected to direct the protein to a location in the cytoplasmic membrane. While membrane-associated transcriptional regulators are not unprecedented (44), it seemed likely that SyrA affected transcription in an indirect manner. One possibility was that SyrA might somehow affect the expression or activity of either NodD3 or SyrM, which would subsequently upregulate transcription. Therefore, I examined the effect of SyrA on expression of the lpsS::uidA transcriptional fusion in backgrounds inactivated for either nodD3 or syrM. Inactivation of either nodD3 or syrM did not affect the ability of overexpressed syrA to mediate an increase in transcription of lpsS::uidA (Fig. 1A). Therefore, SyrA upregulates transcription of lpsS and exo genes through a mechanism independent of SyrM and NodD3 (although SyrM and NodD3 are clearly required for the symbiotic expression of syrA [1, 46]).
Expression of lpsS is upregulated in exoR::Tn5 and exoS::Tn5 mutants.
The finding that SyrA could influence the expression of the lpsS, exoY, exoH, and exoF fusion suggested that SyrA upregulates transcription of lpsS and exo genes through a common mechanism. The exo genes are known to be transcriptionally upregulated in two mutants: exoR::Tn5, which encodes a poorly understood negative regulator of transcription (17, 52), and exoS::Tn5, which affects the sensor kinase of the ExoS/ChvI two-component regulator system (10, 17). As reported previously (28), expression of the lpsS::uidA fusion was upregulated in both the exoS::Tn5 and exoR::Tn5 mutants (Fig. 1A) compared to wild type. The exoS::Tn5 and exoR::Tn5 mutants also exhibited a 3-fold and 1.5-fold increase in LPS sulfotransferase activity, respectively (Fig. 2A). The modest increase in LPS sulfotransferase activity was surprising, given the large increase in expression of the lpsS::uidA transcriptional fusion. The reason for this is unknown but may result from the massive amounts of succinoglycan (which has an overall inhibitory effect on the in vitro sulfotransferase assay) that accumulate in the exoR::Tn5 and exoS::Tn5 strains. No increase in LPS sulfation was observed in vivo in either the exoR::Tn5 or exoS::Tn5 mutants (Fig. 2B). In order to test whether the exoS::Tn5 mutant also failed to upregulate LPS sulfation due to a limitation for PAPS, I introduced plasmid pGTO101 harboring nodPQ into the exoS::Tn5 mutant. Introduction of plasmid pGTO101 into the exoS::Tn5 mutant resulted in a 4.6-fold increase in LPS sulfation compared to strains harboring vector alone (Fig. 2D). Thus, limiting PAPS appeared to prevent an increase in LPS sulfation from being observed in the exoS::Tn5 mutant background. Interestingly, I was unable to introduce the plasmid pGTO101 into the exoR::Tn5 strain for reasons that are unknown.
Transcriptional upregulation of lpsS by SyrA requires wild-type chvI.
The exoS::Tn5 mutant shows upregulation of lpsS and exo genes, suggesting the possibility that ExoS/ChvI and SyrA cooperate to regulate transcription of lpsS and exo genes. I thus asked whether the ExoS/ChvI two-component system is required for SyrA-mediated upregulation of lpsS and exo gene transcription. Null mutations in exoS or chvI have not been reported (10), likely because both genes are essential for the growth of S. meliloti in laboratory medium (the exoS::Tn5 mutation discussed above is an insertion at the N terminus of the protein which results in an N-terminal truncation of the protein and what is believed to result in a constitutively active form of the ExoS/ChvI two-component system [10]). However, a recent study led to the identification of a mutation (D. H. Wells, E. J. Chen, and S. R. Long, submitted for publication), which encodes a K214T change in the ChvI protein. The K214T mutation affects the proposed DNA binding domain of ChvI and has been hypothesized to alter its ability to function as a transcriptional regulator (Wells et al., submitted). Utilizing a derivative of the plasmid pDW33 (Wells et al., submitted) I introduced the chvI(K214T) mutation into the genome of S. meliloti and asked whether it affected the ability of SyrA to upregulate transcription of lpsS::uidA and exoY::uidA fusions. Expression of lpsS::uidA in the chvI(K214T) mutant harboring the vector control pTE3 showed a slight increase with respect to the lpsS::uidA in a wild-type background (Fig. 4A). The reason for this increase is not known. However, expression of the lpsS::uidA fusion in the chvI(K214T) mutant did not increase and, in fact, decreased 55% in the presence of multicopy syrA (Fig. 4A). RT-PCR analysis of mRNA from strains carrying the chvI(K214T) mutation showed no upregulation of lpsS or exoY in the presence of multicopy syrA (Fig. 4B). I also measured LPS sulfotransferase activity in the presence of the chvI(K214T) mutation and observed no increase in LPS sulfotransferase activity in the presence of multicopy syrA (Fig. 2A).
FIG. 4.
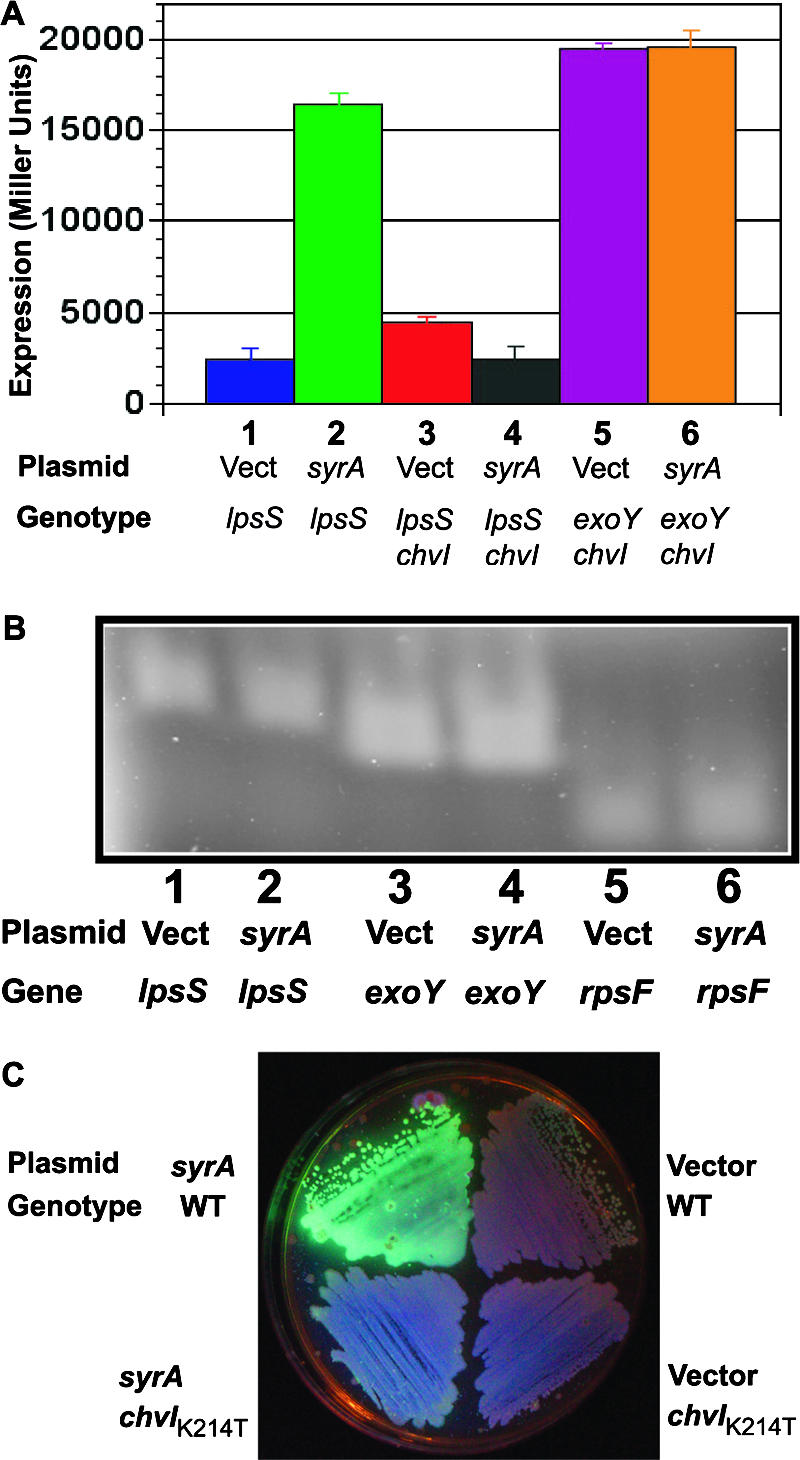
Upregulation of lpsS and exo genes by SyrA requires wild type chvI. A K214T mutation in chvI was introduced by transduction into lpsS::pVO155 strains (which contain an lpsS::uidA fusion) harboring vector alone or overexpressing SyrA. (A) Expression of lpsS::uidA fusion. Strains were grown to saturation (OD600 of 2.5) and assayed for β-glucuronidase activity as described in Materials and Methods. Error bars represent standard deviations of experiments carried out in triplicate. Activity is in Miller units. Lane 1, lpsS::pVO155/pTE3 (vector); lane 2, lpsS::pVO155/pTE3::syrA; lane 3, lpsS::pVO155 chvI(K214T)/pTE3; lane 4, lpsS::pVO155 chvI(K214T)/pTE3::syrA; lane 5, exoY::pVO155 chvI(K214T)/pTE3; lane 6, exoY::pVO155 chvI(K214T)/pTE3::syrA. (B) Measurement of expression by RT-PCR. Lane 1, cDNA prepared from chvI(K214T)/pTE3, amplified with lpsS-specific primers; lane 2, cDNA prepared from chvI(K214T)/pTE3::syrA, amplified with lpsS-specific primers; lane 3, cDNA prepared from chvI(K214T)/pTE3, amplified with exoY-specific primers; lane 4, cDNA prepared from chvI(K214T)/pTE3::syrA, amplified with exoY-specific primers; lane 5, cDNA prepared from chvI(K214T)/pTE3, amplified with rpsF-specific primers; lane 6, cDNA prepared from chvI(K214T)/pTE3::syrA, amplified with rpsF-specific primers. (C) Upregulation of succinoglycan production by SyrA requires wild-type chvI. Strains were streaked out on plates containing calcofluor to determine succinoglycan production and photographed under UV light.
I also tested the effect of the chvI(K214T) mutation on the transcriptional upregulation of exo genes. The chvI(K214T) mutation prevented an increase in exo expression (as judged by exoY::uidA fusions) (Fig. 4A) in the presence of multicopy syrA, which was confirmed by RT-PCR (Fig. 4B). It should be noted that the overall level of expression of the exoY::uidA fusions was higher in strains carrying the chvI(K214T) compared to the exoY::uidA fusion in a wild-type background. This apparent increase in expression likely results from the reduced amount of tetracycline in the medium used for the chvI(K214T) mutant strains (see below). The lack of transcriptional upregulation by SyrA in the presence of the chvI(K214T) mutation was also reflected in reduced upregulation of succinoglycan biosynthesis, as judged by a lack of mucoidy (my unpublished data) and decreased fluorescence when cultured on medium containing calcofluor (which fluoresces in the presence of succinoglycan [21, 33]) (Fig. 4C). Therefore, upregulation of lpsS and exo gene transcription by overexpressed SyrA is greatly reduced in strains harboring a mutant version of chvI.
As discussed above, the chvI gene is believed to be essential for growth of S. meliloti, and, not surprisingly, the chvI(K214T) mutant shows a number of growth phenotypes. For example, the chvI(K214T) mutant harboring the vector control pTE3 (which encodes tetracycline resistance) is unable to grow on LB plates containing 10 μg/ml tetracycline (although it will grow at 2 μg/ml tetracycline). Introduction of multicopy syrA restored the ability of the chvI(K214T) mutant to grow on medium containing 10 μg/ml tetracycline (Fig. 5A). Thus, overexpression of syrA can suppress the growth phenotype of the chvI(K214T) mutant.
FIG. 5.

Phenotypes of chvI exoS::Tn5 strains overexpressing syrA. (A) Overexpression of syrA restores resistance of chvI(K214T) mutants to high concentrations of tetracycline. Wild type, strains carrying the chvI(K214T) mutation, and either vector control or vector carrying syrA were cultured on LB medium in the presence of 10 μg/ml tetracycline, and the plates were then photographed. (B) The exoS::Tn5 mutation prevents upregulation of the lpsS::uidA transcriptional fusion in the presence of overexpressed syrA. Vector control and vector containing syrA were introduced into wild-type S. meliloti and the exoS::Tn5 mutation containing the lpsS::DW33 insertion (which results in an lpsS::uidA transcriptional fusion). The extracts were then assayed for β-glucuronidase activity as described in Materials and Methods. Lane 1, lpsS::pDW33 harboring pTE3; lane2, lpsS::pDW33 harboring pTE3::syrA; lane 3, lpsS::pDW33 exoS::Tn5 harboring pTE3; lane 4, lpsS::pDW33 exoS::Tn5 harboring pTE3::syrA.
The exoS::Tn5 mutation prevents upregulation of lpsS by SyrA.
The chvI(K214T) mutant prevents transcriptional regulation by SyrA, and overexpression of syrA can suppress the growth phenotype of the chvI(K214T) mutation. Therefore, SyrA could function through manipulation of the functionality of the ExoS/ChvI two-component system. If SyrA affects the activity of the ExoS/ChvI two-component system, then constitutively active forms of the ExoS/ChvI two-component system would be expected to be unable to carry out SyrA-mediated upregulation of lpsS and exo gene expression. I introduced a vector alone and a multicopy form of syrA into the exoS::Tn5 mutant and measured their ability to upregulate expression of the lpsS::uidA transcriptional fusion. Although expression of the lpsS::uidA fusion was elevated in the exoS::Tn5 mutant background with respect to the wild type, the overall level of expression was lower than was observed in the absence of plasmids. The reason for this is unknown but may result from the effect of tetracycline (required for retention of the plasmids) in the medium. More importantly, the expression of lpsS::uidA did not increase in the presence of multicopy syrA (Fig. 5B). Therefore, the exoS::Tn5 mutant appears to prevent upregulation of transcription by SyrA.
The N terminus of SyrA contains a signal sequence.
As mentioned above, inspection of the SyrA amino acid sequence strongly suggested the presence of a N-terminal signal sequence (residues 1 to 31) and a transmembrane sequence. Thus, the protein would be expected to be located in the cytoplasmic membrane. I constructed a translational fusion of a fragment of syrA encoding residues 1 to 31 to a deleted form of phoA that encodes a mutant alkaline phosphatase lacking the N terminus (residues 1 to 24) (25, 26, 39, 40, 42, 43). The truncated N-terminal region of PhoA removes the leader peptide that directs delivery to the periplasm. Accordingly, the leader peptide mutant of PhoA would be expected to remain cytoplasmic and would be inactive (15). If the N terminus of SyrA contains a signal sequence, it would be expected to facilitate delivery of the leaderless PhoA out of the cytoplasm, resulting in active alkaline phosphatase. The translational fusion containing residues 1 to 31 (SyrA1-31) with the leaderless PhoA resulted in alkaline phosphatase activity (as judged by blue color on medium containing 5-bromo-4-chloro-3-indolyl phosphate, which was not seen in the strains overexpressing syrA or the vector alone) (Fig. 6). Thus, the N terminus of SyrA encodes a functional signal sequence. I also constructed a translational fusion of the leaderless phoA to full-length syrA. In contrast to the fusion to residues 1 to 31 of SyrA, this fusion did not result in alkaline phosphatase activity, as evidenced by a lack of blue color on medium containing 5-bromo-4-chloro-3-indolyl phosphate (Fig. 6). Because it was possible that the fusion resulted in an unstable form of the protein, I assayed the ability of the full-length syrA::phoA translational fusion to upregulate transcription of lpsS. The fusion was active as a transcriptional activator (my unpublished results). Therefore, these data suggest that the N terminus is capable of mediating the translocation of a heterologous protein from the cytoplasm, but the C terminus of SyrA remains cytoplasmic.
FIG. 6.
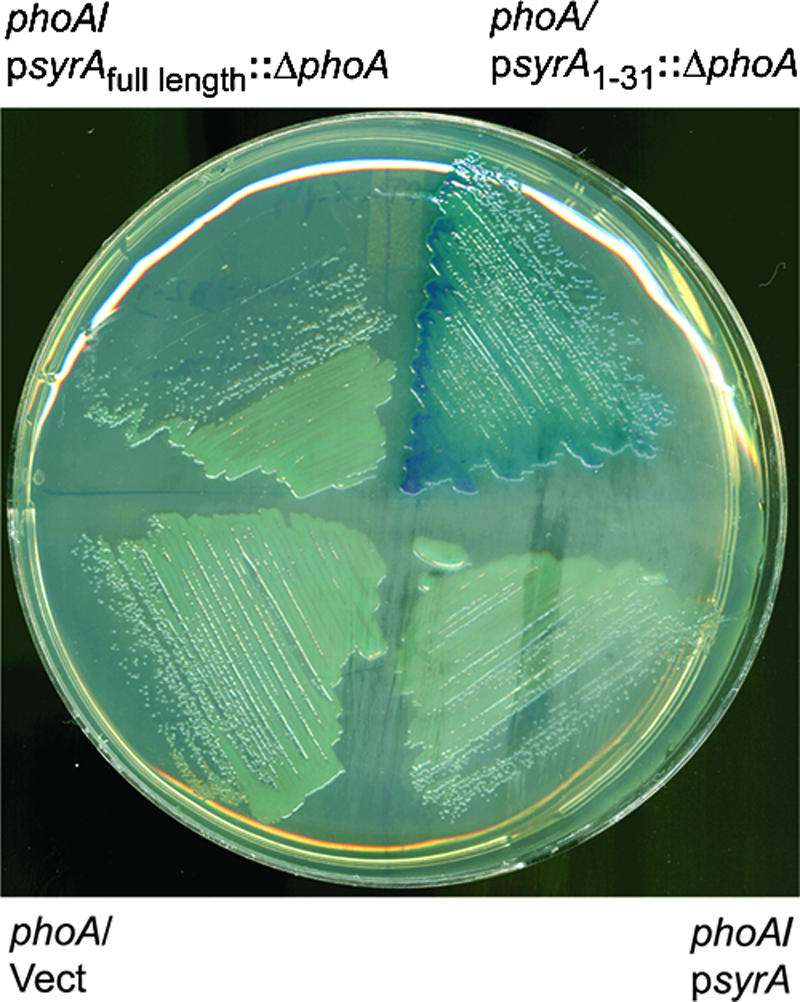
The N terminus of SyrA contains a signal sequence. Either wild-type syrA or a fragment of syrA encoding residues 1 to 31 was translationally fused to a mutant form of phoA lacking residues 1 to 23 of the open reading frame. The plasmids were then introduced into S. meliloti strain Rm8002 (which shows greatly reduced alkaline phosphatase activity), streaked onto plates containing 5-bromo-4-chloro-3-indolyl phosphate), and photographed.
DISCUSSION
S. meliloti lpsS mutants exhibit an altered symbiosis, suggesting that lpsS is necessary under symbiotic conditions and that its expression might be symbiotically regulated. Using a combination of transcriptional fusions, RT-PCR, and assays of enzyme activity, I have shown that transcription of lpsS is upregulated by the symbiotic regulator SyrA. SyrA also upregulates the transcription of multiple exo genes, suggesting that it is a global regulator of gene transcription in S. meliloti. SyrA-mediated upregulation of lpsS and exo genes was prevented in strains harboring the chvI(K214T) mutation or in strains harboring the exoS::Tn5 mutation, implying an involvement of the global ExoS/ChvI two-component system in transcriptional upregulation by SyrA.
The observation that the expression of lpsS was increased by multicopy SyrA was surprising, in that SyrA had not previously been implicated as a transcriptional regulator. The syrA gene was originally identified through its ability to upregulate the biosynthesis of succinoglycan (1, 46). Analysis of its open reading frame predicted the presence of a leader peptide, a transmembrane region, and the lack of a DNA binding domain. Furthermore, the authors of the previous study did not observe a change in expression of exoP and exoF transcriptional fusions and therefore concluded that SyrA upregulated succinoglycan biosynthesis via a posttranscriptional mechanism (1). I demonstrated an increase in expression of exoY::uidA, exoH::uidA, and exoF::uidA transcriptional fusions in the presence of multicopy syrA. Although these data differ from previous results regarding the expression of the exo genes (1), the clear effect of multicopy syrA on expression of exo fusions, steady-state levels of mRNA, and succinoglycan production leads to the interpretation that syrA affects transcription of exo genes. Furthermore, the finding that regulation via SyrA requires the ExoS/ChvI two-component system (a known transcriptional regulator of exo genes [10, 17]) further strengthens this conclusion. A recent study of global transcription in S. meliloti reported that overexpression of nodD3 resulted in increased transcription of syrM and syrA, as well as exo genes and lpsS (2). In fact, overexpression of nodD3 was shown in this study to upregulate >70 genes, and repress ca. 100 genes through what the authors suggest is an indirect mechanism. The elevated levels of SyrA observed under conditions of nodD3 overexpression would be expected to mimic the situation observed in strains that overexpress syrA, leading to increased transcription of exo genes and lpsS. Thus, it seems possible that SyrA was responsible for the indirect global regulation observed in strains that overexpress nodD3.
Although I observed an increase in lpsS transcription and LPS sulfotransferase activity in strains harboring multicopy syrA, this increased activity was not reflected in a detectable increase in sulfated LPS. However, increased LPS sulfation was detected in strains that overexpressed syrA as well as nodPQ, which is responsible for the biosynthesis of the activated sulfate donor (PAPS) (57-59). Measurements in wild-type S. meliloti showed that the intracellular PAPS concentration was below the limit of detection, suggesting that limiting PAPS prevents an increase in LPS sulfation under conditions of SyrA overexpression. During symbiosis, syrA transcription is predicted to be upregulated as part of a regulatory loop that also leads to an increase in nodD3 transcription (60). NodD3 can activate transcription of the nodPQ (one of two copies of the nodPQ genes) genes, which encode the enzymes necessary to synthesize PAPS (2, 46). Thus, syrA transcription in planta would be expected to result in an increase in LpsS activity, an elevated intracellular pool of PAPS, and increased LPS sulfation.
Examination of the primary sequence of SyrA predicts that the N terminus contains a cleavable signal peptide (residues 1 to 21) and a transmembrane sequence (residues 40 to 62). Thus, cleavage of the signal peptide upon translocation would result in a periplasmic N terminus (residues 22 to 39), followed by a transmembrane region (residues 40 to 62), and a cytoplasmic C terminus (residues 63 to 81). The alkaline phosphatase data presented here are consistent with the predicted topology. While membrane-associated transcriptional regulators have been described previously (44), SyrA is a very small protein (ca. 9 kDa) that shows no similarity to known DNA binding proteins. Furthermore, these studies show that SyrA functionality requires the ExoS/ChvI two-component system. The exoS::Tn5 mutant (which is believed to result in a constitutively active form of ExoS/ChvI [10]) has recently been reported to affect the expression of ca. 250 genes, both positively and negatively (65). Thus, it was not surprising that lpsS transcription was upregulated in the exoS::Tn5 mutant. It was surprising however to find that the chvI(K214T) mutant prevented transcriptional upregulation via SyrA. Although the mechanism of upregulation remains unclear, several lines of evidence suggest that SyrA could function via manipulation of the activity of the ExoS/ChvI two-component system. First, although only a subset of genes has been tested, all genes that are regulated by SyrA are also controlled by ExoS/ChvI. Second, the exoS::Tn5 mutation is unable to undergo SyrA-mediated upregulation of lpsS. Third, SyrA-mediated upregulation of lpsS is blocked in the chvI(K214T) mutant. Fourth, overexpression of syrA allows growth of the chvI(K214T) mutant on LB medium containing 10 μg/ml tetracycline. However, overexpression of SyrA will not allow the construction of null mutations in chvI, suggesting that overexpression of syrA cannot functionally replace ExoS/ChvI (my unpublished results). Collectively, the data presented here are consistent with a model where SyrA alters the functionality of the ExoS/ChvI two-component system.
Finally, although it does not resemble known transcriptional regulators, SyrA bears significant sequence identity to ExoX from S. meliloti (36% identity over the N-terminal half of the protein) (51, 66) and strain NGR234 (22), as well as Psi (42% identity over the central part of the protein) from Rhizobium etli (5, 6, 32). Interestingly, ExoX was also identified as a regulator of succinoglycan biosynthesis. A previous study reported that ExoX did not affect the expression of a translational fusion to the exoP gene, implying a posttranslational mechanism of regulation (66). However, considering the sequence similarities between ExoX and SyrA and the complex regulation of exo genes, it seems possible that ExoX could operate through a similar mechanism. Experiments to address this possibility are under way.
Acknowledgments
I thank Guy Townsend for helpful comments on the manuscript, Melanie Barnett for plasmids pS73 and pMB89, Esther Chen for strain EC69, Derek Wells for plasmid pDW33 and strains DW223 and DW226, and Erika Piedras-Rentería for advice regarding the RT-PCR assays. I also thank Derek Wells, Esther Chen, and Sharon Long for communicating results prior to publication.
This study was funded by grant 2005-35319-15304 from the U.S. Department of Agriculture.
Footnotes
Published ahead of print on 5 January 2007.
REFERENCES
- 1.Barnett, M. J., J. A. Swanson, and S. R. Long. 1998. Multiple genetic controls on Rhizobium meliloti syrA, a regulator of exopolysaccharide abundance. Genetics 148:19-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnett, M. J., C. J. Toman, R. F. Fisher, and S. R. Long. 2004. A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc. Natl. Acad. Sci. USA 101:16636-16641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beringer, J. E. 1974. R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84:188-198. [DOI] [PubMed] [Google Scholar]
- 4.Bochner, B. R., and B. N. Ames. 1982. Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J. Biol. Chem. 257:9759-9769. [PubMed] [Google Scholar]
- 5.Borthakur, D., R. F. Barker, J. W. Latchford, L. Rossen, and A. W. Johnston. 1988. Analysis of pss genes of Rhizobium leguminosarum required for exopolysaccharide synthesis and nodulation of peas: their primary structure and their interaction with psi and other nodulation genes. Mol. Gen. Genet. 213:155-162. [DOI] [PubMed] [Google Scholar]
- 6.Borthakur, D., and A. W. Johnston. 1987. Sequence of psi, a gene on the symbiotic plasmid of Rhizobium phaseoli which inhibits exopolysaccharide synthesis and nodulation and demonstration that its transcription is inhibited by psr, another gene on the symbiotic plasmid. Mol. Gen. Genet. 207:149-154. [DOI] [PubMed] [Google Scholar]
- 7.Brewin, N. J. 1991. Development of the legume root nodule. Annu. Rev. Cell Biol. 7:191-226. [DOI] [PubMed] [Google Scholar]
- 8.Brewin, N. J. 1992. Nodule formation in legumes, p. 229-248. In In J. Lederberg (ed.), Encyclopedia of microbiology, vol. 3. Academic Press, San Diego, CA. [Google Scholar]
- 9.Cedergren, R. A., J. Lee, K. L. Ross, and R. I. Hollingsworth. 1995. Common links in the structure and cellular localization of Rhizobium chitolipooligosaccharides and general Rhizobium membrane phospholipid and glycolipid components. Biochemistry 34:4467-4477. [DOI] [PubMed] [Google Scholar]
- 10.Cheng, H. P., and G. C. Walker. 1998. Succinoglycan production by Rhizobium meliloti is regulated through the ExoS-ChvI two-component regulatory system. J. Bacteriol. 180:20-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cronan, G. E., and D. H. Keating. 2004. Sinorhizobium meliloti sulfotransferase that modifies lipopolysaccharide. J. Bacteriol. 186:4168-4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis, R. W., D. Botstein, and J. R. Roth. 1980. Advanced bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 13.de Bruijn, F. 1991. Biochemical and molecular studies: symbiotic nitrogen fixation. Curr. Opin. Biotechnol. 2:184-192. [DOI] [PubMed] [Google Scholar]
- 14.Denarie, J., and J. Cullimore. 1993. Lipo-oligosaccharide nodulation factors: a new class of signaling molecules mediating recognition and morphogenesis. Cell 74:951-954. [DOI] [PubMed] [Google Scholar]
- 15.Derman, A. I., W. A. Prinz, D. Belin, and J. Beckwith. 1993. Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science 262:1744-1747. [DOI] [PubMed] [Google Scholar]
- 16.Ditta, G., S. Stanfield, D. Corbin, and D. R. Helinski. 1980. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc. Natl. Acad. Sci. USA 77:7347-7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doherty, D., J. A. Leigh, J. Glazebrook, and G. C. Walker. 1988. Rhizobium meliloti mutants that overproduce the R. meliloti acidic calcofluor-binding exopolysaccharide. J. Bacteriol. 170:4249-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Downie, J. A. 1994. Signalling strategies for nodulation of legumes by rhizobia. Trends Microbiol. 2:318-324. [DOI] [PubMed] [Google Scholar]
- 19.Egelhoff, T. T., and S. R. Long. 1985. Rhizobium meliloti nodulation genes: identification of nodDABC gene products, purification of nodA protein, and expression of nodA in Rhizobium meliloti. J. Bacteriol. 164:591-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrhardt, D. W., E. M. Atkinson, K. F. Faull, D. I. Freedberg, D. P. Sutherlin, R. Armstrong, and S. R. Long. 1995. In vitro sulfotransferase activity of NodH, a nodulation protein of Rhizobium meliloti required for host-specific nodulation. J. Bacteriol. 177:6237-6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finan, T. M., A. M. Hirsch, J. A. Leigh, E. Johansen, G. A. Kuldau, S. Deegan, G. C. Walker, and E. R. Signer. 1985. Symbiotic mutants of Rhizobium meliloti that uncouple plant from bacterial differentiation. Cell 40:869-877. [DOI] [PubMed] [Google Scholar]
- 22.Gray, J. X., M. A. Djordjevic, and B. G. Rolfe. 1990. Two genes that regulate exopolysaccharide production in Rhizobium sp. strain NGR234: DNA sequences and resultant phenotypes. J. Bacteriol. 172:193-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higashi, S. 1993. (Brady)Rhizobium-plant communications involved in infection and nodulation. J. Plant Res. 106:206-211. [Google Scholar]
- 24.Hirsch, A. M. 1992. Developmental biology of legume nodulation. New Phytol. 122:211-237. [DOI] [PubMed] [Google Scholar]
- 25.Inouye, H., W. Barnes, and J. Beckwith. 1982. Signal sequence of alkaline phosphatase of Escherichia coli. J. Bacteriol. 149:434-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito, K., P. J. Bassford, Jr., and J. Beckwith. 1981. Protein localization in E. coli: is there a common step in the secretion of periplasmic and outer-membrane proteins? Cell 24:707-717. [DOI] [PubMed] [Google Scholar]
- 27.Jefferson, R. A., S. M. Burgress, and D. Hirsch. 1986. β-Glucuronidase from Escherichia coli as a gene-fusion marker. Proc. Natl. Acad. Sci. USA 83:8447-8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keating, D. H. 2007. The Sinorhizobium meliloti ExoR protein is required for the downregulation of lpsS transcription and succinoglycan biosynthesis in response to divalent cations. FEMS Microbiol. Lett. 267:23-29. [DOI] [PubMed] [Google Scholar]
- 29.Keating, D. H., M. G. Willits, and S. R. Long. 2002. A Sinorhizobium meliloti lipopolysaccharide mutant altered in cell surface sulfation. J. Bacteriol. 184:6681-6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kijne, J. W., A. A. N. Bakhuizen, H. C. J. van Brussel, C. L. CanterCremers, B. S. Diaz, de Pater, G. Smit, H. P. Spaink, S. Swart, C. A. Wiffelman, and B. J. J. Lugtenberg. 1992. The Rhizobium trap: root hair curling in root-nodule symbiosis. Perspectives in plant cell recognition. Soc. Exp. Biol. Semin. Ser. 48:267-284. [Google Scholar]
- 31.Kondorosi, E., M. Buire, M. Cren, N. Iyer, B. Hoffmann, and A. Kondorosi. 1991. Involvement of the syrM and nodD3 genes of Rhizobium meliloti in nod gene activation and in optimal nodulation of the plant host. Mol. Microbiol. 5:3035-3048. [DOI] [PubMed] [Google Scholar]
- 32.Latchford, J. W., D. Borthakur, and A. W. Johnston. 1991. The products of Rhizobium genes, psi and pss, which affect exopolysaccharide production, are associated with the bacterial cell surface. Mol. Microbiol. 5:2107-2114. [DOI] [PubMed] [Google Scholar]
- 33.Leigh, J. A., E. R. Signer, and G. C. Walker. 1985. Exopolysaccharide-deficient mutants of Rhizobium meliloti that form ineffective nodules. Proc. Natl. Acad. Sci. USA 82:6231-6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lerouge, P., P. Roche, C. Faucher, F. Maillet, G. Truchet, J. C. Prome, and J. Denarie. 1990. Symbiotic host-specificity of Rhizobium meliloti is determined by a sulphated and acylated glucosamine oligosaccharide signal. Nature 344:781-784. [DOI] [PubMed] [Google Scholar]
- 35.Leyh, T. S., J. C. Taylor, and G. D. Markham. 1988. The sulfate activation locus of Escherichia coli K12: cloning, genetic, and enzymatic characterization. J. Biol. Chem. 263:2409-2416. [PubMed] [Google Scholar]
- 36.Long, S., S. McCune, and G. C. Walker. 1988. Symbiotic loci of Rhizobium meliloti identified by random TnphoA mutagenesis. J. Bacteriol. 170:4257-4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maillet, F., F. Debelle, and J. Denarie. 1990. Role of the nodD and syrM genes in the activation of the regulatory gene nodD3, and of the common and host-specific nod genes of Rhizobium meliloti. Mol. Microbiol. 4:1975-1984. [DOI] [PubMed] [Google Scholar]
- 38.Maniatis, T. E., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 39.Manoil, C., and J. Beckwith. 1986. A genetic approach to analyzing membrane protein topology. Science 233:1403-1408. [DOI] [PubMed] [Google Scholar]
- 40.Manoil, C., and J. Beckwith. 1985. TnphoA: a transposon probe for protein export signals. Proc. Natl. Acad. Sci. USA 82:8129-8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meade, H. M., S. R. Long, G. B. Ruvkun, S. E. Brown, and F. M. Ausubel. 1982. Physical and genetic characterization of symbiotic and auxotrophic mutants of Rhizobium meliloti induced by transposon Tn5 mutagenesis. J. Bacteriol. 149:114-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michaelis, S., L. Guarente, and J. Beckwith. 1983. In vitro construction and characterization of phoA-lacZ gene fusions in Escherichia coli. J. Bacteriol. 154:356-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michaelis, S., H. Inouye, D. Oliver, and J. Beckwith. 1983. Mutations that alter the signal sequence of alkaline phosphatase in Escherichia coli. J. Bacteriol. 154:366-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller, V. L., R. K. Taylor, and J. J. Mekalanos. 1987. Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell 48:271-279. [DOI] [PubMed] [Google Scholar]
- 45.Mougous, J. D., R. E. Green, S. J. Williams, S. E. Brenner, and C. R. Bertozzi. 2002. Sulfotransferases and sulfatases in mycobacteria. Chem. Biol. 9:767-776. [DOI] [PubMed] [Google Scholar]
- 46.Mulligan, J. T., and S. R. Long. 1989. A family of activator genes regulates expression of Rhizobium meliloti nodulation genes. Genetics 122:7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niehaus, K., A. Largares, and A. Puhler. 1998. A Sinorhizobium meliloti lipopolysaccyharide mutant induces effective nodules on the host plant Medicago sativa (alfalfa) but fails to establish a symbiosis with Medicago truncatula. Mol. Plant Microbe Interact. 11:906-914. [Google Scholar]
- 48.Oke, V., and S. R. Long. 1999. Bacterial genes induced within the nodule during the Rhizobium-legume symbiosis. Mol. Microbiol. 32:837-849. [DOI] [PubMed] [Google Scholar]
- 49.Peters, N. K., J. W. Frost, and S. R. Long. 1986. A plant flavone, luteolin, induces expression of Rhizobium meliloti nodulation genes. Science 233:977-980. [DOI] [PubMed] [Google Scholar]
- 50.Phillips, D. A., C. M. Joseph, and C. A. Maxwell. 1992. Trigonelline and stachydrine released from alfalfa seeds activate NodD2 protein in Rhizobium meliloti. Plant Physiol. 99:1526-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reed, J. W., M. Capage, and G. C. Walker. 1991. Rhizobium meliloti exoG and exoJ mutations affect the exoX-exoY system for modulation of exopolysaccharide production. J. Bacteriol. 173:3776-3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reed, J. W., J. Glazebrook, and G. C. Walker. 1991. The exoR gene of Rhizobium meliloti affects RNA levels of other exo genes but lacks homology to known transcriptional regulators. J. Bacteriol. 173:3789-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reuhs, B. L., R. W. Carlson, and J. S. Kim. 1993. Rhizobium fredii and Rhizobium meliloti produce 3-deoxy-d-manno-2-octulosonic acid-containing polysaccharides that are structurally analogous to group II K antigens (capsular polysaccharides) found in Escherichia coli. J. Bacteriol. 175:3570-3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ridge, R. 1992. A model for legume root hair growth and Rhizobium infection. Symbiosis 14:359-373. [Google Scholar]
- 55.Rivera-Marrero, C. A., J. D. Ritzenthaler, S. A. Newburn, J. Roman, and R. D. Cummings. 2002. Molecular cloning and expression of a novel glycolipid sulfotransferase in Mycobacterium tuberculosis. Microbiology 148:783-792. [DOI] [PubMed] [Google Scholar]
- 56.Rougeaux, H., J. Guezennec, R. W. Carlson, N. Kervarec, R. Pichon, and P. Talaga. 1999. Structural determination of the exopolysaccharide of Pseudoalteromonas strain HYD 721 isolated from a deep-sea hydrothermal vent. Carbohydr. Res. 315:273-285. [DOI] [PubMed] [Google Scholar]
- 57.Schwedock, J., and S. R. Long. 1990. ATP sulphurylase activity of the nodP and nodQ gene products of Rhizobium meliloti. Nature 348:644-647. [DOI] [PubMed] [Google Scholar]
- 58.Schwedock, J. S., C. Liu, T. S. Leyh, and S. R. Long. 1994. Rhizobium meliloti NodP and NodQ form a multifunctional sulfate-activating complex requiring GTP for activity. J. Bacteriol. 176:7055-7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwedock, J. S., and S. R. Long. 1992. Rhizobium meliloti genes involved in sulfate activation: the two copies of nodPQ and a new locus, saa. Genetics 132:899-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swanson, J. A., J. T. Mulligan, and S. R. Long. 1993. Regulation of syrM and nodD3 in Rhizobium meliloti. Genetics 134:435-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swanson, J. A., J. Tu, J. Ogawa, R. Sanga, R. F. Fisher, and S. R. Long. 1987. Extended region of nodulation genes in Rhizobium meliloti 1021. I. Phenotypes of Tn5 insertion mutants. Genetics 117:181-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Townsend, G. E., II, L. S. Forsberg, and D. H. Keating. 2006. Mesorhizobium loti produces nodPQ-dependent sulfated cell-surface polysaccharides. J. Bacteriol. 188:8560-8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vijn, I., L. das Nevas, A. van Kammen, H. Franssen, and T. Bisseling. 1993. Nod factors and nodulation in plants. Science 260:1764-1765. [DOI] [PubMed] [Google Scholar]
- 64.Wells, D. H., and S. R. Long. 2002. The Sinorhizobium meliloti stringent response affects multiple aspects of symbiosis. Mol. Microbiol. 43:1115-1127. [DOI] [PubMed] [Google Scholar]
- 65.Yao, S. Y., L. Luo, K. J. Har, A. Becker, S. Ruberg, G. Q. Yu, J. B. Zhu, and H. P. Cheng. 2004. Sinorhizobium meliloti ExoR and ExoS proteins regulate both succinoglycan and flagellum production. J. Bacteriol. 186:6042-6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhan, H. J., and J. A. Leigh. 1990. Two genes that regulate exopolysaccharide production in Rhizobium meliloti. J. Bacteriol. 172:5254-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]