

Abstract
The extracellular signal regulated protein kinases (ERK1/2) are essential for normal development and functional plasticity of the central nervous system. However, a growing number of recent studies in models of cerebral ischemia, brain trauma and neurodegenerative diseases implicate a detrimental role for ERK1/2 signaling during oxidative neuronal injury. Neurons undergoing oxidative stress-related injuries typically display a biphasic or sustained pattern of ERK1/2 activation. A variety of potential targets of reactive oxygen species and reactive nitrogen species could contribute to ERK1/2 activation. These include cell surface receptors, G proteins, upstream kinases, protein phosphatases and proteasome components, each of which could be direct or indirect targets of reactive oxygen or nitrogen species, thereby modulating the duration and magnitude of ERK1/2 activation. Neuronal oxidative stress also appears to influence the subcellular trafficking and/or localization of activated ERK1/2. Differences in compartmentalization of phosphorylated ERK1/2 have been observed in diseased or injured human neurons and in their respective animal and cell culture model systems. We propose that differential accessibility of ERK1/2 to downstream targets, which is dictated by the persistent activation of ERK1/2 within distinct subcellular compartments, underlies the neurotoxic responses that are driven by this kinase.
Keywords: Alzheimer’s disease, cerebral ischemia, mitogen activated protein kinases, neurodegeneration, neuronal cell death, oxidative stress, Parkinson’s disease, phosphatases, reactive oxygen species, traumatic brain injury
ERK1/2 activation in central nervous system diseases: Promoters of cell death?
The mitogen activated protein kinases (MAPK) comprise a ubiquitous group of signaling proteins that play a prominent role in regulating cell proliferation, differentiation and adaptation. Members of each major MAPK subfamily, the extracellular signal regulated protein kinases (ERK), c-Jun N-terminal kinases and p38 MAPK, have been implicated in neuronal injury and disease (reviewed in [1,2]). The MAPK signaling module is defined by a three-tiered kinase cascade, resulting in phosphorylation of a conserved Thr-X-Tyr activation motif by an upstream dual specificity MAPK kinase (Fig. 1). In particular, ERK1 and ERK2, which are activated by the MAPK/ERK kinase-1/2 (MEK1/2), are emerging as important regulators of neuronal responses to both functional (learning and memory) and pathologic (regulated cell death) stimuli. While ERK signaling plays a beneficial, neuroprotective role in many systems (see companion reviews [2a,2b]), there is growing evidence implicating these kinases in the promotion of cell death in both neurons and other cell types.
Fig. 1. Schematic diagram illustrating potential redox-sensitive ERK regulatory components.
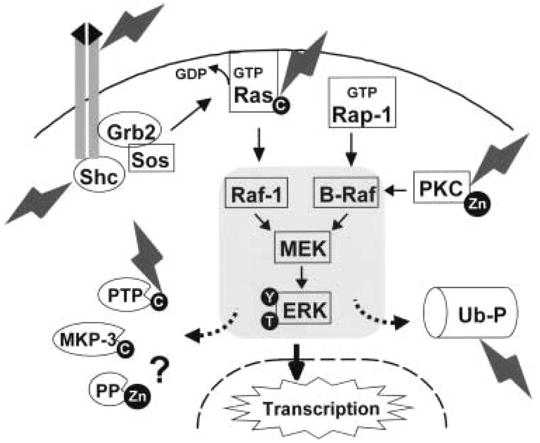
The three-tiered ERK signaling module, involving sequential activation of Raf (MAPK kinase kinase), MEK (MAPK kinase) and ERK (MAPK) is shown in the shaded rectangle. ERK activation during oxidative neuronal injury could result through redox cysteine switch mechanisms at several different levels including growth factor receptors, adapter proteins (Shc), G proteins (Ras) or upstream kinases (e.g. protein kinase C; PKC). Protein tyrosine phosphatases (PTP) and dual specificity MAPK phosphatases (MKP) contain an active site cysteine that is susceptible to oxidative inactivation. While PKC is regulated by redox mediated Zn release in hippocampal preparations, it is unknown whether Zn release may contribute to inactivation of serine/threonine protein phosphatases (PP) as well. Likewise, ubiquitin-proteasome (Ub-P) mediated degradation of phospho-ERKs may become impaired. Several of these mechanisms may coexist, mediating the sustained ERK signaling observed during oxidative neuronal injury.
Initial indications that ERK1/2 activation may contribute to central nervous system (CNS) disease pathogenesis were noted in studies of diseased human brain tissues using antibodies that recognize the active, phosphorylated form of both ERK1 and ERK2. For example, Smith and colleagues noted aberrant neuronal expression of phosphorylated ERK1/2 and other MAPKs in Alzheimer’s disease patients’ brains in association with markers of oxidative stress (reviewed in [2]). MAPK phosphorylation was also noted in a variety of sporadic and familial neurodegenerative diseases characterized by tau deposits [3]. Phospho-ERK1/2 is increased in substantia nigra neurons of patients with Parkinson’s disease and other Lewy body diseases, and the midbrains of these patients show elevated ERK activity [4]. In addition to chronic neurodegenerative diseases, increased ERK1/2 phosphorylation has been noted in the vulnerable penumbra following acute ischemic stroke in humans [5]. While the association between ERK1/2 phosphorylation and vulnerable neurons in human CNS diseases seems compelling, it is difficult to ascribe functionality from expression analysis alone, as kinase activation may simply reflect a cellular response to stress and not necessarily contribute to ensuing mobilization of cell death or survival pathways.
Neuroprotective effects of ERK1/2 inhibition in vivo
A series of reports by Alessandrini and colleagues in models of cerebral ischemia-reperfusion injury provided the first in vivo evidence that activation of the MEK-ERK1/2 signaling pathway may contribute to acute brain injuries (for example [6]). In these studies, ERK1/2 activation was blocked using pharmacologic inhibitors of MEK1/2 and led to reduced neuronal injury and loss of function in mice and gerbils. These findings have been confirmed by similar studies from other groups [7,8]. Prominent ERK1/2 activation is also observed after neonatal hypoxic-ischemic injury [9]. In addition, ERK1/2 activation may contribute to traumatic brain injury, possibly through activation of matrix metalloproteinases [10]. It is interesting to note that different regions of the hippocampus show preferential susceptibility to ischemic vs. traumatic injuries, and that neuronal ERK1/2 phosphorylation occurs in regions that subsequently undergo neuronal cell death [11]. Although the MEK1/2 inhibitor studies offer compelling evidence supporting a detrimental role for ERK signaling in acute brain injuries, other studies indicate that ERK may promote functional recovery following mild trauma [12]. The accompanying review by Hetman discusses studies using MEK1/2 inhibitors to implicate a neuroprotective effect for ERK1/2 [2a].
What accounts for the seemingly contradictory effects of MEK1/2 inhibition on neuronal cell survival following acute injury? Differences in outcome resulting fromMEK1/2 inhibition may depend not only upon the nature and severity of injury, but also upon drug dosing regimens or the cell type expressing activated ERK1/2. Although most acute neuronal injury studies focus upon neuronal expression of phospho-ERK1/2, activation of this kinase in surrounding glial or endothelial cells could also impact on neuronal survival. For example, persistent astroglial expression of phosphorylated ERK1/2 is observed after stab injuries to the mouse brain [13]. Moreover, ERK1/2 activation in microglia results in release of inflammatory mediators detrimental to substantia nigra neurons [14]. Until cell type-specific inhibition of ERK1/2 activation can be attained, themechanism responsible for the neuroprotective in vivo effects of MEK1/2 inhibition will remain unresolved.
Neuroprotective effects of ERK1/2 inhibition in vitro
The activation of MAPKs including ERK1/2 has been extensively studied with regard to cellular proliferation and responses to growth factors or prosurvival hormones. Seminal experiments by the Greenberg group in the PC12 pheochromocytoma cell line established protective effects of activated ERK1/2 against apoptosis induced by neurotrophic factor withdrawal (reviewed in [15] and accompanying reviews by Hetman and Cavanaugh [2a,2b]). Many studies ensued that further substantiated the neuroprotective effect of ERK1/2 in neuronal cell lines and primary neuron cultures [2a]. However, even in the PC12 neurotrophic factor withdrawal model, a MEK1/2 inhibitor could exert a partial protective effect [16]. More compelling results indicating neuroprotective effects from inhibiting ERK1/2 activation were subsequently obtained in hippocampal slice cultures where protein phosphatase inhibition was used to induce cell death [17,18].
In subsequent years, a number of groups have used similar approaches to reveal protective effects of blocking ERK1/2 activation in both established cell lines and primary neurons subjected to a variety of insults. These include toxicity induced by peroxynitrite [19], mechanical trauma [20], glutathione depletion [21–23], zinc [24,25], amyloid beta peptide plus iron [26], the parkinsonian neurotoxins MPP+ [27] and 6-hydroxydopamine [28,29] and other miscellaneous insults [30,31]. The potential positive contribution of ERK1/2 activation to cell death is not limited to neurons as MEK1/2 inhibitors have been found to block cell death in astrocytes [32], oligodendroglia-like cells [33], vascular smooth muscle [34], fibroblasts [35] and renal epithelial cells [36]. It is interesting to note that oxidative stress often plays a role in both neuronal and non-neuronal model systems in which ERK1/2 contributes to injury. Indeed, redox mechanisms have also been implicated in cell death models elicited by nonoxidative stimuli, such as those based on altered growth factor levels [31,37].
It is important to recognize that in cultured cell lines and enriched primary neuron cultures, direct effects of pharmacological inhibitors of ERK1/2 activation can be established, as contributions of glial cell derived cytokines or vascular effects of the inhibitors are not a factor. However, the possibility of other kinases also being affected by the inhibitors cannot be excluded. Molecular manipulation of select components of the ERK1/2 signaling pathway will be required to definitively establish specific effects of ERK1/2 on neurotoxicity. Indeed, a recent study used si RNA to demonstrate a specific role for ERK2, but not ERK1, in a non-apoptotic form of regulated cell death [37a].
Redox regulation of ERK1/2 activation kinetics
Divergent effects of the ERK1/2 signaling pathway on neuronal cell survival are not surprising, as previous studies have demonstrated diverse biological effects of ERK1/2 even within a single cell type (reviewed in [38]). In fact, understanding the underlying mechanisms responsible for generating unique cellular responses by a limited set of signaling molecules (i.e. the MAPKs) has been the goal of many studies in neuronal and non-neuronal cells alike [38]. One important component of the ERK1/2 signaling network that appears to play a critical role in dictating cellular response is the precise kinetics of ERK1/2 activation. It is interesting to note that many model systems that implicate ERK1/2 in a detrimental role are associated with a delayed, sustained phase of ERK1/2 activation [4,21,29, 34,37]. As the impact of ERK1/2 activation kinetics on neurodegeneration has been discussed in a recent review [15]), we will focus our discussion on the role of redoxsensitive mechanisms in ERK1/2 activation.
It is well accepted that neurodegenerative diseases and ischemia-reperfusion injury to most organs share a common dependence upon generation of reactive oxygen species (ROS)/reactive nitrogen species (RNS). Therefore, it is probable that in vitro studies that examine ERK1/2 activation in response to oxidative stress will reveal important details relevant to neuronal cell injury in vivo. Indeed, ERK1/2 activation appears to be mediated by redox mechanisms in both acute neuronal injuries [21] and in models of neurodegeneration [4,26], including transmissible spongi form encephalopathies [39]. Both transgenic animal studies [40] and cell culture studies [4,29] suggest that inhibition of ERK1/2 signaling comprises an important mechanism by which antioxidants confer protection.
Redox sensitivity of upstream activators of ERK1/2
Classic receptor-regulated activation of ERK1/2 signaling occurs through recruitment of cytoplasmic adaptor proteins and the small G protein Ras to the membrane (Fig. 1). GTP-loaded Ras promotes activation of the MAPK kinase kinase Raf-1. Raf-1 then activates the MAPK kinase MEK1/2, leading to ERK1/2 phosphorylation. In addition, MEK1/2 can be activated by B-Raf, a neuron-enriched isoform of Raf, which is in turn activated by a cAMP responsive Ras homologue called Rap-1 [41]. Heterotrimeric G proteins are also involved in regulating ERK1/2 signaling through effects on scaffolding functions of β-arrestins, or by modulating the activity of protein kinase C, which can activate Raf isoforms at the apex of the ERK1/2 module (reviewed in [42]).
Redox regulation at each of these steps has been demonstrated, predominantly in non-neuronal systems (reviewed in [43]). Receptor tyrosine kinases can be activated through hydrogen peroxide mediated oxidation of cysteine residues or through covalent oxidative stabilization of receptor dimers [44]. Adaptor proteins such as Shc can be activated in a monoamine oxidase dependent manner [45]. The small GTP-binding protein Ras contains a surface redox-sensitive cysteine residue whose oxidation results in activation of the ERK1/2 pathway [43]. Heterotrimeric GTP-binding proteins contain redox-sensitive cysteine residues that when modified result in ERK1/2 activation [46]. Members of the protein kinase C family, which are capable of activating Raf, can be activated or inactivated by redox modification of thiol residues in different domains of the enzyme [43,47]. While reversible cysteine switches appear to form a general facet of normal trophic factor induced ERK1/2 activation, mechanisms underlying patterns of activation observed in pathologic situations remain to be elucidated.
Redox sensitivity of downstream inactivators of ERK1/2
The pathologically sustained ERK1/2 activity observed in injured neuronal cells probably reflects impairment of negative feedback regulators that normally function to terminate signaling responses (Fig. 1). Regulation of kinase signaling involves coordinated input from both upstream activators and inactivating phosphatases [48,49]. In addition, alterations in cellular degradation pathways may contribute to injury. Alterations in both the ubiquitinproteasome and autophagolysosome systems have been implicated in neurodegenerative diseases [50,51]. In degenerating neurons, phosphorylated ERK1/2 is observed in autophagocytosed mitochondria [52]. Moreover, ERK1/2 can be targeted for proteasomal degradation [53], and delayed sustained patterns of ERK1/2 phosphorylation can be elicited by proteasome inhibitors [54].
Phosphatases capable of inactivating ERK1/2 include serine/threonine directed protein phosphatases (PPs), protein tyrosine phosphatases (PTPs) and dual-specificity phosphatases (DSPs; which include the MKPs–MAP kinase phosphatases) [55]. PTPs and DSPs share a HC(X)5R motif that is critical for enzymatic activity, and this catalytic cysteine residue is particularly susceptible to oxidation (Fig. 1). Indeed, transient oxidative inactivation of PTPs represents an important normal mechanism of signal transduction, involving conversion of the active-site cysteine into a metastable sulfenic acid (Cys-SOH) that is reversed by reaction with glutathione (reviewed in [56]). Progression to irreversible sulfonic acid (Cys-SO2H) or sulfonic acid (Cys-SO3H) forms may underlie pathologically sustained ERK1/2 responses. Although redox regulation of metallophosphatases have not been as intensively studied, oxidative modification of the metal binding residues could hypothetically affect serine/threonine PP activity as well.
While oxidative stress in neurons during reperfusion injury results in induction of several ERK-directed phophatases [57], ERK1/2 phosphorylation is increased, not decreased [9,40]. This apparent dissociation suggests that either the phosphatases are inactivated or they are unable to access their target, perhaps due to altered trafficking or sequestration of the phosphorylated ERK1/2. Many ERK-directed phosphatases, especially those in the MKP family, share a docking domain with high affinity for binding ERK1/2 [58]. Several of these phosphatases are restricted either to the cytoplasm (e.g. MKP3) or to the nucleus (e.g. MKP1). Transfection studies with mutated MKPs suggest that inactivated phosphatases can serve as passive anchors for ERK1/2 (reviewed in [49]). Such a mechanism may explain divergent patterns of sustained cytoplasmic vs. nuclear localization of phospho-ERK1/2 under neuropathological conditions that involve oxidative stress.
Location, location, location
Regulation of ERK1/2 subcellular localization
Subcellular localization of activated MAPKs influences resultant cellular responses in a variety of cell types (reviewed in [49]). For example, rapid nuclear translocation of activated ERK1/2 following growth factor stimulation is essential for stimulating progression through the cell cycle [49]. Because ERK1/2 substrates are found in various subcellular compartments (for review see [59]), the biological outcome of ERK1/2 activation will depend in part upon the localization of ERK1/2 and its accessibility to potential substrates within that compartment. It is also probable that molecular scaffolds, which direct the action of MAPK to specific substrates [60], will have unique compositions within distinct compartments and cell types, adding to the flexibility of downstream signaling that results from ERK1/2 activation. Model studies in yeast and mammalian cells have identified regulators of ERK1/2 compartmentalization including ‘anchoring proteins’ that restrict active ERK1/2 to the cytoplasmic or nuclear compartment [49]. As mentioned above, specific MKPs may serve a dual role in regulating ERK1/2 activity within a specific compartment by either terminating kinase action through dephosphorylation or restricting the subcellular trafficking of ERK1/2 through formation of relatively stable complexes. The observation that phosphorylated ERK1/2 displays distinct patterns of subcellular localization in ischemic or degenerating human neurons provides insight into potential mechanisms mediating beneficial vs. detrimental effects of these ubiquitous kinases (Fig. 2).
Fig. 2. Distinct subcellular localization patterns for phosphorylated ERK1/2: Mechanistic implications.
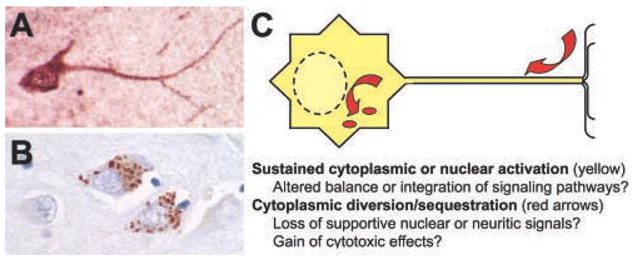
Neurons exhibiting ERK1/2 activation typically display several general immunohistochemical patterns including diffuse labeling restricted to processes or soma and diffuse nuclear and cytoplasmic staining, as observed in this ischemic human neuron (A). Degenerating populations of neurons may also display discrete, cytoplasmic labeling for phospho-ERK1/2, as illustrated by these hippocampal neurons from a patient with dementia (B). Hypothetical effects related to differences in subcellular localization of activated ERK1/2 are schematically illustrated (C). Although compartmentalization of activated ERK1/2 does not necessarily imply that downstream signaling effects are limited to these compartments, it is probable that different subsets of scaflolding anchors, target proteins and regulatory phosphatases are involved. Cellular integration of divergent downstream effects will determine the favorable vs. detrimental outcomes to neuronal injury involving ERK1/2 signaling pathways. In addition, the potential contribution of ERK1/2 activation in non-neuronal CNS cell types must be considered at the organism or tissue level.
Sustained nuclear localization of ERK1/2 in acute neuronal injury: Beneficial or detrimental?
Neuronal cell function is also dramatically influenced by the subcellular localization of activated ERK1/2. Marshall and colleagues performed classic experiments showing that differential responses of PC12 cells to epidermal growth factor (i.e. proliferative) vs. nerve growth factor (differentiation) were triggered by alternative kinetics of ERK1/2 activation and compartmentalization, with sustained nuclear localization of active ERK1/2 associated with differentiation (reviewed in [15,59]). Subsequently, sustained nuclear localization of ERK1/2 has been found to be critical for long-term potentiation [61]. While these studies reveal conditions where nuclear localization of active ERK1/2 promotes physiological function, this property of ERK1/2 may not strictly apply to injured neurons.
The activation of ERK1/2 and other MAPKs in cerebral ischemia was first reported 10 years ago by Hu & Wieloch [62]. ERK1/2 activation has since been observed in various models of focal and global ischemia although the precise kinetics, duration and regional distribution of active ERK1/2 differs in the various models (reviewed in [63]). In some cases, the subcellular localization of activated ERK1/2 in ischemic tissue has also been examined. Thus, while active ERK1/2 persists for up to 24 h within neurons in ‘penumbral- like’ regions following middle cerebral artery occlusion in adult rat, it is mainly localized to the cytoplasm, perikarya or neuropil [63]. In contrast, chronically activated ERK1/2 is retained in the nuclei within neurons from various brain regions following hypoxia/ischemia in neonatal rats [9], and in cell culture models involving oxidative glutamate toxicity [22]. Based upon these apparently conflicting reports, it seems reasonable to conclude that ERK1/2 has the capacity to promote cell death in neurons through its effects on either cytoplasmic or nuclear targets, depending upon the nature of the acute insult and the affected cell types.
Cytoplasmic diversion in neurodegeneration?
In some neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease and Lewy body dementias, ERK1/2 appears to be localized within discrete, cytoplasmic granules [4,64], a pattern that is also observed in 6-hydroxydopamine-treated cells in culture [4]. Although the precise structural or functional interactions underlying these punctate cytoplasmic staining patterns are not well understood, immunofluorescence and ultrastructural studies of degenerating human substantia nigra neurons indicate the presence of phosphorylated ERK1/2 in association with fibrillar bundles, distended mitochondria and autophagosomes [52]. These discrete accumulations of ERK1/2 in the cytoplasm raise the possibility that even though it is phosphorylated, ERK1/2 may not be available to act on potential downstream pro-survival targets (Fig. 2).
Onepotential loss of function pathway that is implicated in Parkinson’s disease by both human tissue studies [4] and a cell culture model (E. M. Chalovich & C. T. Chu, unpublished data) is the ERK-RSK-CREB pathway, which regulates transcription of potentially neuroprotective genes such as Bcl-2 and brain derived neurotrophic factor. In addition, given the normal role of ERK1/2 signaling in regulating synaptic plasticity, it is possible that reduced signaling in this capacity contributes to neurodegeneration, as synaptic dysfunction undoubtedly precedes overt cell death. Indeed, it has recently been shown that alpha-synuclein affects caveolar signaling, and that the resultant dysregulation of ERK1/2 signaling adversely affects neuritic outgrowth [65].
Alternatively, accumulation of phosphorylated ERK1/2 within discrete cytoplasmic bodies may be associated with a toxic gain of cytoplasmic function that somehow contributes to neurodegeneration, perhaps through the activation of cytoplasmic or mitochondrial cell death mediators (Fig. 2). One potentially interesting candidate is calpain, a cysteine protease implicated in both apoptotic and necrotic conditions. Co-localization of phosphorylated ERK1/2 with markers of calpain activation have been observed following neonatal hypoxic ischemic injury in rats [9]. Moreover, calpain, which is increased in Parkinson’s disease neurons [66], appears to be a direct cytoplasmic target of ERK1/2 [67]. Ultimately, the persistence of activated ERK1/2 within any individual compartment (i.e. nucleus or cytoplasm) may disrupt the intricate balance between pro-survival and pro-death signals that are being integrated to elicit a final cellular response.
Conclusions and caveats
As ERK1/2 is a shuttling protein that traffics between the nuclear and cytoplasmic compartments, it may be misleading to associate its predominant localization within a single compartment revealed in fixed cells or tissues with action towards a restricted set of substrates. We also must keep in mind that compartment-specific scaffolding proteins could impact not only the activity of ERK1/2, but also its target protein selection. Furthermore, if ERK1/2-mediated phosphorylation of specific target proteins is not readily reversed by appropriate phosphatases, ERK1/2 effects within a given compartment may persist well beyond the time that active ERK1/2 is resident within that given compartment. Sequestration of ERK1/2 within discrete subcellular bodies could also affect the accessibility of ERK1/2 to its targets. Clearly, a detailed analysis is needed of the targets of ERK1/2 that directly function to promote neuronal cell death in response to various forms of both chronic and acute neuronal cell injury. It therefore seems likely that as various signaling pathways are mobilized in response to neuronal cell injury, the temporal and spatial coincidence between effector kinases (e.g. ERK1/2) and their substrates will play an essential role in directing cells towards a survival pathway or one that leads to their demise.
Acknowledgments
ERK-related research in the authors’ laboratories is supported by grants from the National Institutes of Health (R01 NS40817 to C. T. C., F30 NS43824 to D. J. L. and R01 NS38319 to D. B. D.).
Abbreviations
- CNS
central nervous system
- DSP
dual-specificity phosphatase
- ERK
extracellular signal regulated protein kinase
- MAPK
mitogen activated protein kinase
- MEK1/2
MAPK/ERK kinase-1/2
- MKP
MAP kinase phosphatase
- PP
protein phosphatase
- PTP
protein tyrosine phosphatases
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
References
- 1.Harper SJ, Wilkie N. MAPKs: new targets for neurodegeneration. Expert Opin Ther Targets. 2003;7:187–200. doi: 10.1517/14728222.7.2.187. [DOI] [PubMed] [Google Scholar]
- 2.Zhu X, Lee HG, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals. 2002;11:270–281. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- 2a.Hetman M, Gozdz A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem. 2004;271:2050–2055. doi: 10.1111/j.1432-1033.2004.04133.x. [DOI] [PubMed] [Google Scholar]
- 2b.Cavanaugh JE. Role of extracellular signal regulated kinase 5 in neuronal survival. Eur J Biochem. 2004;271:2056–2059. doi: 10.1111/j.1432-1033.2004.04131.x. [DOI] [PubMed] [Google Scholar]
- 3.Ferrer I, Barrachina M, Tolnay M, Rey MJ, Vidal N, Carmona M, Blanco R, Puig B. Phosphorylated protein kinases associated with neuronal and glial tau deposits in argyrophilic grain disease. Brain Pathol. 2003;13:62–78. doi: 10.1111/j.1750-3639.2003.tb00007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu JH, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated kinase in Lewy body diseases. Am J Pathol. 2002;161:2087–2098. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slevin M, Krupinski J, Slowik A, Rubio F, Szczudlik A, Gaffney J. Activation of MAP kinase (ERK-1/ERK-2), tyrosine kinase and VEGF in the human brain following acute ischaemic stroke. Neuroreport. 2000;11:2759–2764. doi: 10.1097/00001756-200008210-00030. [DOI] [PubMed] [Google Scholar]
- 6.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, Trzaskos JM, Feuerstein GZ. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther. 2003;304:172–178. doi: 10.1124/jpet.102.040246. [DOI] [PubMed] [Google Scholar]
- 8.Wang ZQ, du Wu C, Huang FP, Yang GY. Inhibition of MEK/ERK1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. 2004;996:55–66. doi: 10.1016/j.brainres.2003.09.074. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Zhu C, Qiu L, Hagberg H, Sandberg M, Blomgren K. Activation of ERK1/2 after neonatal rat cerebral hypoxia-ischaemia. J Neurochem. 2003;86:351–362. doi: 10.1046/j.1471-4159.2003.01838.x. [DOI] [PubMed] [Google Scholar]
- 10.Mori T, Wang X, Aoki T, Lo EH. Down-regulation of matrix metalloproteinase-9 and attenuation of edema via inhibition of ERK mitogen activated protein kinase in traumatic brain injury. J Neurotrauma. 2002;19:1411–1419. doi: 10.1089/089771502320914642. [DOI] [PubMed] [Google Scholar]
- 11.Otani N, Nawashiro H, Fukui S, Nomura N, Yano A, Miyazawa T, Shima K. Differential activation of mitogen-activated protein kinase pathways after traumatic brain injury in the rat hippocampus. J Cereb Blood Flow Metab. 2002;22:327–334. doi: 10.1097/00004647-200203000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Dash PK, Mach SA, Moore AN. The role of extracellular signal-regulated kinase in cognitive and motor deficits following experimental traumatic brain injury. Neuroscience. 2002;114:755–767. doi: 10.1016/s0306-4522(02)00277-4. [DOI] [PubMed] [Google Scholar]
- 13.Carbonell WS, Mandell JW. Transient neuronal but persistent astroglial activation of ERK/MAP kinase after focal brain injury in mice. J Neurotrauma. 2003;20:327–336. doi: 10.1089/089771503765172282. [DOI] [PubMed] [Google Scholar]
- 14.Choi SH, Joe EH, Kim SU, Jin BK. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. J Neurosci. 2003;23:5877–5886. doi: 10.1523/JNEUROSCI.23-13-05877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colucci-D’Amato L, Perrone-Capano C, di Porzio U. Chronic activation of ERK and neurodegenerative diseases. Bioessays. 2003;25:1085–1095. doi: 10.1002/bies.10355. [DOI] [PubMed] [Google Scholar]
- 16.Kummer JL, Rao PK, Heidenreich KA. Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J Biol Chem. 1997;272:20490–20494. doi: 10.1074/jbc.272.33.20490. [DOI] [PubMed] [Google Scholar]
- 17.Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc Natl Acad Sci USA. 1998;95:11975–11980. doi: 10.1073/pnas.95.20.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Runden E, Seglen PO, Haug FM, Ottersen OP, Wieloch T, Shamloo M, Laake JH. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci. 1998;18:7296–7305. doi: 10.1523/JNEUROSCI.18-18-07296.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oh-hashi K, Maruyama W, Yi H, Takahashi T, Naoi M, Isobe K. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun. 1999;263:504–509. doi: 10.1006/bbrc.1999.1237. [DOI] [PubMed] [Google Scholar]
- 20.Mori T, Wang X, Jung JC, Sumii T, Singhal AB, Fini ME, Dixon CE, Alessandrini A, Lo EH. Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J Cereb Blood Flow Metab. 2002;22:444–452. doi: 10.1097/00004647-200204000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri M, Johnson J, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- 22.Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J Biol Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- 23.Satoh T, Nakatsuka D, Watanabe Y, Nagata I, Kikuchi H, Namura S. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci Lett. 2000;288:163–166. doi: 10.1016/s0304-3940(00)01229-5. [DOI] [PubMed] [Google Scholar]
- 24.Park JA, Koh JY. Induction of an immediate early gene egr-1 by zinc through extracellular signal-regulated kinase activation in cortical culture: its role in zinc-induced neuronal death. J Neurochem. 1999;73:450–456. doi: 10.1046/j.1471-4159.1999.0730450.x. [DOI] [PubMed] [Google Scholar]
- 25.Seo SR, Chong SA, Lee SI, Sung JY, Ahn YS, Chung KC, Seo JT. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J Neurochem. 2001;78:600–610. doi: 10.1046/j.1471-4159.2001.00438.x. [DOI] [PubMed] [Google Scholar]
- 26.Kuperstein F, Yavin E. ERK activation and nuclear translocation in amyloid-beta peptide- and iron-stressed neuronal cell cultures. Eur J Neurosci. 2002;16:44–54. doi: 10.1046/j.1460-9568.2002.02056.x. [DOI] [PubMed] [Google Scholar]
- 27.Gomez-Santos C, Ferrer I, Reiriz J, Vinals F, Barrachina M, Ambrosio S. MPP+ increases alpha-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblastoma SH-SY5Y cells. Brain Res. 2002;935:32–39. doi: 10.1016/s0006-8993(02)02422-8. [DOI] [PubMed] [Google Scholar]
- 28.Kulich SM, Chu CT. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: Implications for Parkinson’s disease. J Neurochem. 2001;77:1058–1066. doi: 10.1046/j.1471-4159.2001.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulich SM, Chu CT. Role of reactive oxygen species in ERK phosphorylation and 6-hydroxydopamine cytotoxicity. J Biosci. 2003;28:83–89. doi: 10.1007/BF02970136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du S, McLaughlin B, Pal S, Aizenman E. In vitro neurotoxicity of methylisothiazolinone, a commonly used industrial and household biocide, proceeds via a zinc and extracellular signal-regulated kinase mitogen-activated protein kinase-dependent pathway. J Neurosci. 2002;22:7408–7416. doi: 10.1523/JNEUROSCI.22-17-07408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cha YK, Kim YH, Ahn YH, Koh JY. Epidermal growth factor induces oxidative neuronal injury in cortical culture. J Neurochem. 2000;75:298–303. doi: 10.1046/j.1471-4159.2000.0750298.x. [DOI] [PubMed] [Google Scholar]
- 32.Regan RF, Wang Y, Ma X, Chong A, Guo Y. Activation of extracellular signal-regulated kinases potentiates hemin toxicity in astrocyte cultures. J Neurochem. 2001;79:545–555. doi: 10.1046/j.1471-4159.2001.00590.x. [DOI] [PubMed] [Google Scholar]
- 33.Bhat NR, Zhang P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J Neurochem. 1999;72:112–119. doi: 10.1046/j.1471-4159.1999.0720112.x. [DOI] [PubMed] [Google Scholar]
- 34.Gurjar MV, Deleon J, Sharma RV, Bhalla RC. Role of reactive oxygen species in IL-1 beta-stimulated sustained ERK activation and MMP-9 induction. Am J Physiol Heart Circ Physiol. 2001;281:H2568–H2574. doi: 10.1152/ajpheart.2001.281.6.H2568. [DOI] [PubMed] [Google Scholar]
- 35.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramachandiran S, Huang Q, Dong J, Lau SS, Monks TJ. Mitogen-activated protein kinases contribute to reactive oxygen species-induced cell death in renal proximal tubule epithelial cells. Chem Res Toxicol. 2002;15:1635–1642. doi: 10.1021/tx0200663. [DOI] [PubMed] [Google Scholar]
- 37.Subramaniam S, Strelau J, Unsicker K. Growth differentiation factor-15 prevents low potassium-induced cell death of cerebellar granule neurons by differential regulation of Akt and ERK pathways. J Biol Chem. 2003;278:8904–8912. doi: 10.1074/jbc.M210037200. [DOI] [PubMed] [Google Scholar]
- 37a.Castro-Obregón S, Rao RV, del Rio G, Chen SF, Poksay KS, Rabizadeh S, Vesce S, Zhang X, Swanson RA, Bredesen DE. Alternative, nonapoptotic programmed cell death: Mediation by arrestin 2, ERK2, and Nur77. J Biol Chem. 2004;279:17543–17553. doi: 10.1074/jbc.M312363200. [DOI] [PubMed] [Google Scholar]
- 38.Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider B, Mutel V, Pietri M, Ermonval M, Mouillet-Richard S, Kellermann O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc Natl Acad Sci USA. 2003;100:13326–13331. doi: 10.1073/pnas.2235648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noshita N, Sugawara T, Hayashi T, Lewen A, Omar G, Chan PH. Copper/zinc superoxide dismutase attenuates neuronal cell death by preventing extracellular signal-regulated kinase activation after transient focal cerebral ischemia in mice. J Neurosci. 2002;22:7923–7930. doi: 10.1523/JNEUROSCI.22-18-07923.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 42.Luttrell LM. ‘Location, location, location’: activation and targeting of MAP kinases by G protein-coupled receptors. J Mol Endocrinol. 2003;30:117–126. doi: 10.1677/jme.0.0300117. [DOI] [PubMed] [Google Scholar]
- 43.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 44.Zhang P, Wang YZ, Kagan E, Bonner JC. Peroxynitrite targets the epidermal growth factor receptor, Raf-1, and MEK independently to activate MAPK. J Biol Chem. 2000;275:22479–22486. doi: 10.1074/jbc.M910425199. [DOI] [PubMed] [Google Scholar]
- 45.Vindis C, Seguelas MH, Lanier S, Parini A, Cambon C. Dopamine induces ERKactivation in renal epithelial cells through H2O2 produced by monoamine oxidase. Kidney Int. 2001;59:76–86. doi: 10.1046/j.1523-1755.2001.00468.x. [DOI] [PubMed] [Google Scholar]
- 46.Nishida M, Schey KL, Takagahara S, Kontani K, Katada T, Urano Y, Nagano T, Nagao T, Kurose H. Activation mechanism of Gi and Go by reactive oxygen species. J Biol Chem. 2002;277:9036–9042. doi: 10.1074/jbc.M107392200. [DOI] [PubMed] [Google Scholar]
- 47.Knapp LT, Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J Biol Chem. 2000;275:24136–24145. doi: 10.1074/jbc.M002043200. [DOI] [PubMed] [Google Scholar]
- 48.Bhalla US, Ram PT, Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science. 2002;297:1018–1023. doi: 10.1126/science.1068873. [DOI] [PubMed] [Google Scholar]
- 49.Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2002;64:755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- 50.Ding Q, Keller JN. Proteasomes and proteasome inhibition in the central nervous system. Free Radic Biol Med. 2001;31:574–584. doi: 10.1016/s0891-5849(01)00635-9. [DOI] [PubMed] [Google Scholar]
- 51.Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002;17:897–908. doi: 10.14670/HH-17.897. [DOI] [PubMed] [Google Scholar]
- 52.Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–481. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu Z, Xu S, Joazeiro C, Cobb MH, Hunter T. The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol Cel. 2002;9:945–956. doi: 10.1016/s1097-2765(02)00519-1. [DOI] [PubMed] [Google Scholar]
- 54.Hashimoto K, Guro G, Katagiri Y. Delayed and sustained activation of p42/p44mitogen-activated protein kinase induced by proteasome inhibitors through p21 (ras) in PC12 cells. J Neurochem. 2000;74:92–98. doi: 10.1046/j.1471-4159.2000.0740092.x. [DOI] [PubMed] [Google Scholar]
- 55.Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 56.Gabbita SP, Robinson KA, Stewart CA, Floyd RA, Hensley K. Redox regulatory mechanisms of cellular signal transduction. Arch Biochem Biophys. 2000;376:1–13. doi: 10.1006/abbi.1999.1685. [DOI] [PubMed] [Google Scholar]
- 57.Takano S, Fukuyama H, Fukumoto M, Hirashimizu K, Higuchi T, Takenawa J, Nakayama H, Kimura J, Fujita J. Induction of CL100 protein tyrosine phosphatase following transient forebrain ischemia in the rat brain. J Cereb Blood Flow Metab. 1995;15:33–41. doi: 10.1038/jcbfm.1995.4. [DOI] [PubMed] [Google Scholar]
- 58.Tanoue T, Nishida E. Molecular recognitions in the MAP kinase cascades. Cell Signal. 2003;15:455–462. doi: 10.1016/s0898-6568(02)00112-2. [DOI] [PubMed] [Google Scholar]
- 59.Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- 60.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 61.Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- 62.Hu BR, Wieloch T. Tyrosine phosphorylation and activation of mitogen-activated protein kinase in the rat brain following transient cerebral ischemia. J Neurochem. 1994;62:1357–1367. doi: 10.1046/j.1471-4159.1994.62041357.x. [DOI] [PubMed] [Google Scholar]
- 63.Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- 64.Pei JJ, Braak H, An WL, Winblad B, Cowburn RF, Iqbal K, Grundke-Iqbal I. Up-regulation of mitogen activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Brain Res Mol Brain Res. 2002;109:45–55. doi: 10.1016/s0169-328x(02)00488-6. [DOI] [PubMed] [Google Scholar]
- 65.Hashimoto M, Takenouchi T, Rockenstein E, Masliah E. Alpha-synuclein up-regulates expression of caveolin-1 and down-regulates extracellular signal-regulated kinase activity in B103 neuroblastoma cells: role in the pathogenesis of Parkinson’s disease. J Neurochem. 2003;85:1468–1479. doi: 10.1046/j.1471-4159.2003.01791.x. [DOI] [PubMed] [Google Scholar]
- 66.Mouatt-Prigent A, Karlsson JO, Agid Y, Hirsch EC. Increased M-calpain expression in the mesencephalon of patients with Parkinson’s disease but not in other neurodegenerative disorders involving the mesencephalon: a role in nerve cell death? Neuroscience. 1996;73:979–987. doi: 10.1016/0306-4522(96)00100-5. [DOI] [PubMed] [Google Scholar]
- 67.Glading A, Uberall F, Keyse SM, Lauffenburger DA, Wells A. Membrane-proximal ERK signaling is required for M-calpain activation downstream of EGF receptor signaling. J Biol Chem. 2001;276:23341–23348. doi: 10.1074/jbc.M008847200. [DOI] [PubMed] [Google Scholar]