

Abstract
Palmitoylation enhances membrane association and plays a role in the subcellular trafficking and signaling function of proteins. Unlike other forms of protein lipidation, such as prenylation and myristoylation, palmitoylation is reversible and can therefore play a regulatory role. Enzyme activities have recently been described in mammals and yeast that carry out the palmitoylation of protein substrates. Protein acyltransferases (PATs) transfer a palmitoyl moiety derived from palmitoyl-CoA to a free thiol of a substrate protein to create a labile thioester linkage. Biochemical characterization and kinetic analysis of this new family of enzymes requires methods to purify PATs and their substrates, as well as methods to assay PAT activity. We describe a series of methods using yeast and bacterial expression systems to study protein acyltransferases.
Keywords: DHHC domain, palmitoylation, Coenzyme A, Lipidation, Ras, Vac8, Pfa3
1. Introduction
Protein palmitoylation refers to the transfer of a palmitoyl moiety from palmitoyl-CoA to a cysteine of a recipient protein to form a thioester linkage (Fig 1). The reaction can occur spontaneously under basic pH conditions or enzymatically via a family of protein acyl transferases (PATs). Hydrolysis of the palmitoyl thioester can likewise occur spontaneously at basic pH or enzymatically via protein palmitoylthioesterases (PPTs) (Fig. 1). The identification of PATs in yeast established the first causal link between the DHHC domain (zfDHHC, pfam PF01529) and protein palmitoylation. Yeast encode a total of seven DHHC proteins, four of which have been shown have PAT activity toward specific protein substrates. Akr1 palmitoylates the yeast type I casein kinase Yck2 and the sphingoid long-chain base kinase, Lcb4 (1, 2). Pfa3 palmitoylates Vac8, a myristoylated, vacuole associated protein required for vacuole fusion (3). The protein complex, Erf2-Erf4/Shr5, which together constitute a PAT, catalyzes the addition of palmitate to yeast Ras proteins (4). Swf1 appears to be responsible for palmitoylating the yeast SNARES Snc1, Syn8 and Tlg1 at cysteine residues near the cytoplasmic side of their single transmembrane span (5).
Fig. 1. Diagram illustrating protein palmitoylation and hydrolysis.
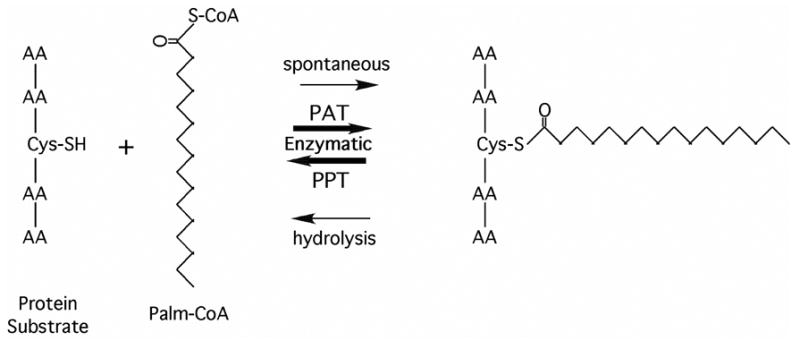
A free thiol group of a substrate protein can undergo palmitoylation from a palmitoyl-CoA donor by either non-ezymatic (spontaneous) or enzymatic (PAT) mechanisms. Palmitoylprotein thioesterase (PPT) enzymatically cleaves the thioester linked palmitate from proteins.
Although the significance of the DHHC residues is presently unclear, the DHHC domain appears to be the signature of one class of PATs. Using the DHHC domain as the query, a large family PATs have been uncovered in all eukaryotes (6). In mammals there are approximately 23 DHHC genes, some of which have been shown to participate in sub-cellular trafficking and modulation of functionally diverse membrane associated proteins. In the cases where it has been investigated, several mammalian DHHC proteins, like their yeast counterparts, have been shown to have PAT activity. Examples include DHHC9, which together with GCP16, a Golgi associated protein, palmitoylates H- and N-Ras protein (7). In neurons, palmitoylation of the huntingtin protein results in its proper trafficking and localization. The huntingtin interacting protein, HIP14, has been linked to the palmitoylation of the huntingtin protein (8) and the postsynaptic density protein PSD95, as well as other proteins including H-Ras (9). Another mammalian DHHC protein, GODZ (DHHC3), has been implicated in the palmitoylation of the GABAA λ2 receptor subunit (10). The discovery of PATs has for the first time provided the opportunity to characterize the substrate specificity and mechanism of enzyme-catalyzed palmitoylation. Here we describe methods for the expression, solubilization and measurement of PAT activity of yeast DHHC proteins, using the Ras PAT, Erf2/Erf4, and Vac8 PAT, Pfa3, as examples. The methods should be applicable to all members of the DHHC PAT family and their substrates.
2. Isolation of PAT substrates
In many cases, protein substrates for palmitoylation require prior lipid modification. For example, Ras and other CaaX box proteins must be prenylated to serve as an effective substrate for palmitoylation. Some Gα subunits of heterotrimeric G proteins are myristoylated prior to undergoing palmitoylation. This presents a challenge to purify sufficient quantities of protein substrates to study palmitoylation. Standard bacterial expression systems are not well suited for the purification of eukaryotic membrane proteins and lack the enzymes required for prenylation and myristoylation. Methods are described here for the purification of Ras substrates from yeast and Vac8 substrate from N-myristoyltransfersase (NMT)-expressing bacterial cells.
2.1 Purification of prenylated Ras substrates to study Ras PATs
In yeast, Ras proteins are prenylated by Ram1/Ram2, -aaX box proteolyzed by Rce1, carboxymethylated by Ste14, and palmitoylated by the Erf2/Erf4 Ras PAT (11). The substrate for Ras PAT is the prenylated, -aaX cleaved, and carboxymethylated Ras protein. Purification of this substrate is accomplished by expressing amino-terminal epitope-tagged Ras from a galactose-inducible promoter in a yeast strain in which one or both of the endogenous Ras PAT genes (ERF2 ERF4) are deleted to reduce the level of palmitoylation of the Ras substrate. The hydrophobic nature of the processing steps prior to palmitoylation cause GST-Ras2(HV) to associate with the membrane fraction, which provides a convenient way to enrich for the proper substrate prior to affinity purification.
Methods to optimize prenylated, but not palmitoylated Ras substrate have been developed using the yeast strain RJY1627 (MAT a leu2?1, ura3-52, lys2-80, ade2-101, trp1?63, his3?200 erf2::KANr). RJY1627 was constructed from YPH499 (MAT a leu2?1, ura3-52, lys2-80, ade2-101, trp1?63, his3?200) (Stratagene) (12), by replacing the coding region of ERF2 with the KANr using single step gene disruption (13). A Ras expression plasmid was constructed by fusing the C-terminal hypervariable (HV) domain and CaaX box (CCaaX) of Ras2 (residues 287–322) to GST coding region of pEG(KG) (14) to create pEG(KG)Ras2(HV)CCaaX. To further increase the expression of the GST chimera, the Gal4 transcription activator was simultaneously overexpressed from pMA210, a multi-copy plasmid that overexpresses GAL4 from an ADH1 promoter (15). Yeast harboring pEG(KG)-Ras2(HV)CCaaX and pMA210 plasmids were grown on synthetic media (SC) containing a non-fermentable carbon source such as ethanol/glycerol to prevent expression of GAL4 prior to induction. The plasmids are transformed into yeast strains using the EZ Yeast Transformation II method (Zymo Research). Overexpression of pEG(KG)-Ras2(HV)CCaaX results in the production of a prenylated GST-Ras2 substrate.
Transformants of YPH1627 harboring the plasmids pEG(KG)-Ras2(HV)CCaaX and pMA210 were grown at 30°C in 10 ml of selection media (SC -ura, -his/2% ethanol/2% glycerol) to an OD600 of 0.5–0.8. This culture was used to inoculate 600 ml of selection media and the culture was grown to 0.5–0.8 OD600. Galactose (40% sterile stock) solution was added to a final concentration of 4% and the cultures allowed to grow for an additional 15–18 hrs at 30°C. Following induction, cells were collected by centrifugation and washed once with TE buffer (20 mM Tris HCl, pH 8, 1 mM EDTA) and the cell pellets were stored at −80°C prior to lysis.
GST-Ras2 was purified from a crude membrane fraction. Unless otherwise indicated, all steps are carried out at 4°C. The cell pellet was suspended in TBS (50 mM Tris HCl, pH 7.4 with 150 mM NaCl) containing 5 mM EDTA, 5 mM DTT, protease inhibitors (Roche) and 8 μl/ml PMSF at a ratio of 7 ml per 1 gram cells. The cells were lysed by passage (4 times) through an Emulsiflex homogenizer (27,000 psig). For smaller cultures, 0.5 gram cells can be suspended in 1 ml buffer and broken by vigorous vortexing with 400–600 μm glass beads. Approximately 70% of the cells were lysed with these methods. Unbroken cells, nuclei and large debris were removed by centrifugation at 3,000 x g for 15 min. A crude membrane preparation was then obtained by high-speed centrifugation (Type 55 Ti rotor, 45,000 rpm for 2 h). The membrane-associated GST-Ras2 protein was solubilized by suspending the pellet in TBS containing 0.6% Triton X-100 and Roche protease inhibitors (5 ml/g of original cell pellet) using a Dounce homogenizer. Detergent solubilization was carried out at 4°C for 1.5 h with gentle agitation, and the solution is clarified by centrifugation at 10,000 x g for 20 min. Affinity purification of GST-Ras2 was performed by incubating the supernatant with TBS-washed GSH-agarose (Pierce) for 4 h at 4°C. The beads were washed three times with TBS and then washed briefly with 50 mM Tris, 50 mM NaCl, 0.05% Triton X-100, 50 mM glutathione, pH 3.5. The protein was eluted overnight at 4°C in elution buffer (50 mM Tris, 50 mM NaCl, 0.05% Triton X-100, 50 mM glutathione, pH 8.4). The beads were rinsed with elution buffer and the pooled elutions were concentrated using an iCon concentrator (9,000 MW cut-off, Pierce). The purification was monitored by SDS-PAGE and immunoblotting. The yield of GST-Ras2 is generally ~0.5 mg Ras/1 liter of starting culture.
2.2 Purification of myristoylated Vac8, the substrate of the yeast Pfa3
Vac8 is another example of a protein that requires prior lipid modification to be a substrate for palmitoylation (16). In this case, Vac8 must undergo myristoylation via an N-terminal glycine amide. This can be accomplished by expressing Vac8 in bacteria expressing eukaryotic N-myristoyltransferase (NMT) (17). To this end, a bacterial expression vector was constructed with a C-terminally Myc-tagged Vac8 just upstream of a hexahistidine motif (VAC8-MYC-6xHIS). The E. coli strain JM109, was transformed with this construct (pML658) and a vector carrying the yeast NMT1 (pBB131) (3, 17). It was confirmed that Vac8 is myristoylated by Nmt1 in E. coli by examining incorporation of [3H]-myristate into Vac8 (3).
For large-scale purification of myristoylated Vac8 (myr-Vac8-myc-6xHis), the bacterial culture was grown at 37°C to an OD600 of 0.400, and induced with 0.3 mM IPTG in the presence of 1 μg/ml chloramphenicol. The culture was incubated for 20 h at 30°C. Cells were harvested, washed, and suspended in lysis buffer (50 mM Tris, pH 8.0, 5 mM EDTA, 1 mM DTT) 3 ml per 1 gram of cell pellet. Protease inhibitors (Roche) were included during the lysis step. Cells were lysed using a French press at 1000 psi for 3 passages and cleared by centrifugation at 25,400 x g for 30 min.
Unfortunately, myr-Vac8-myc-6xHis was unable to bind to a nickel resin despite the presence of a hexahistidine tag. Therefore, conventional chromatography was used to purify the protein. The cleared lysate was applied to an 8 ml Q-sepharose column (1.5 cm × 10 cm) pre-equilibrated with 80 ml of lysis buffer. The column was washed with 80 ml of buffer A (50 mM Tris, pH 8.0, 1 mM EDTA, 1 mM DTT) and protein was eluted with 160 ml of an ascending NaCl gradient (0–350 mM) in buffer A. Fractions containing myr-Vac8-myc-6xHis were identified by Western blot, pooled and applied to a 20 ml hydroxylapatite column (2.5 cm × 20 cm) that was pre-equilibrated with 200 ml of buffer B (20 mM Tris, pH 8.0, 1 mM DTT, 100 mM NaCl). The column was washed with 200 ml of buffer B and protein was eluted with 300 ml of an ascending potassium phosphate gradient (0–150 mM) in buffer B. Fractions containing myr-Vac8-myc-6xHis were pooled and subjected to buffer exchange into buffer C (20 mM Hepes, pH 7.2, 1 mM EDTA, 1mM DTT) in an Amicon ultrafiltration device (YM30 membrane). The sample was applied to a 4 ml Q-sepharose column (1 cm × 5 cm) that was pre-equilibrated with 40 ml of buffer C. The column was washed with 60 ml of buffer C and protein was eluted with 80 ml of an ascending NaCl gradient (0–300 mM) in buffer C. Fractions containing myr-Vac8-myc-6xHis were pooled and concentrated using an Amicon ultrafiltration device. The sample was next subjected to gel filtration using a HiLoad 16/60 Superdex 200 size-exclusion column using a fast protein liquid chromatography system (Amersham). The sample was eluted using Buffer D (50 mM Tris, pH 8.0, 1 mM EDTA, 1 mM DTT, 100 mM NaCl) and fractions containing myr-Vac8-myc-6xHis were pooled and concentrated in a Centricon-30 centrifilter (Amicon). Typically, one liter of E. coli yielded 750 μg of full-length, myristoylated Vac8 as determined by Coomassie blue staining. Using mobility shift analysis we determined that the majority of the purified Vac8-myc-6xHis was myristoylated, making it a suitable substrate for in vitro assays (18).
2.3 Preparation of 3H Palmitoyl-CoA
Tritiated palmitoyl-CoA can be purchased (American Radiolabeled Chemicals, Inc,) or prepared from 3H-palmitate and CoenyzmeA using Acyl CoA synthetase (Sigma) essentially as described (19). Briefly, the reaction involves adding acyl CoA synthetase (2.1 U/ml) to a solution of 15 μM 3H-palmitate (Perkin-Elmer Life Sciences), 1.8 mM Coenzyme A, 4.5 mM ATP, 4.5 mM MgCl2, in 17.5 mM Tris HCl, pH 7.8 containing 0.04% Triton X-100. The reaction is carried out at room temperature for 3 h. The product is purified from the remaining reactants by solid-phase extraction (Alltech C8 Maxi-Clean Cartridge) using a step gradient of acetonitrile in 50 mM ammonium acetate pH 5.3. 3H Palmitoyl-CoA elutes with 40% acetonitrile and is concentrated by lyophilization and dissolved in ammonium acetate (50 mM), pH 5.3 containing 15% acetonitrile and 0.05% Triton X-100 to give a final concentration of 30 μM.
3. Expression and characterization of recombinant yeast PATs in yeast membranes
This assay can be used to measure palmitoylation of various substrates in crude membrane fractions isolated from yeast expressing palmitoyltransferases from high copy number plasmids. This provides a rapid method to screen a putative PAT for autoacylation and thereby establish whether it is likely to have PAT activity. This method will be illustrated using 5 yeast DHHC proteins, Erf2, Akr1, Pfa3, Pfa4, and Pfa5.
3.1 Expression and membrane isolation
YPH499 was independently transformed with FLAG epitope tagged versions of Akr1, Pfa3, Pfa4, Pfa5, or Erf2 (with GST-Erf4) pESC-TRP expression (Stratagene) constructs. Transformants were grown in selective media with 2% ethanol and 2% glycerol as the carbon sources. Upon reaching an OD600 of 0.4–0.6, the culture was induced by addition of galactose (3% final concentration) and grown for 16 hours at 23°C. Cells were collected at 1000 x g for 5 min at 4°C. The pellet was washed with cold water and stored at –80°C. To lyse the cells, the pellet was suspended in 400 μl of Lysis Buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, 1 mM EGTA, 1 mM EDTA, 15 μM pepstatin A, 10 μM leupeptin, 0.3 μM aprotinin, 1 mM PMSF), 400 μl of glass beads were added, and the suspension vortexed vigorously at 4°C for 2 h. The lysate was collected and the beads washed with an additional 500 μl of lysis buffer. The lysate and wash were pooled and centrifuged at 1000 x g for 5 min at 4°C to remove unbroken cells. The membrane fraction was prepared by centrifugation (200,000 x g for 20 min). The crude membrane fraction was suspended in 400 μl lysis buffer by gently passing through a series of syringe needles (18, 23 and 25 gauge). Protein concentration was determined by a Bradford assay.
3.2 A membrane-based PAT assays
In preparation for the assay, PAT-containing membranes were diluted to a total protein concentration of 2 mg/ml in PAT membrane dilution buffer (50 mM Tris HCl, pH 8.0, 1 mM EDTA, 1 mM DTT, 0.05% Triton X-100), for a final concentration of 0.4 mg/ml in the assay (assay volume, 50 μl). All steps were performed on ice unless otherwise indicated. The protein substrate (GST-Ras2 or myr-Vac8-myc-6xHis) was diluted to 1.6 μM in the PAT membrane dilution buffer, for a final concentration of 0.32 μM in the assay. Immediately prior to initiating the reaction, a stock solution of 3H palmitoyl-CoA was diluted to a final concentration of 1.7 μM in PAT reaction buffer (167 mM MES, pH 6.4, 1 mM DTT). For each enzyme preparation, it is important to include a control without substrate in order to distinguish palmitoylation of the substrate from palmitoylation of proteins in the membrane preparation (Fig. 2). The reactions were initiated by adding 30 μl of the diluted 3H palmitoyl-CoA and incubated at 30°C for the indicated time. The reactions were terminated by addition of 12.5 μl 5x SDS sample buffer (250 mM Tris pH 6.8, 50% glycerol, 5% SDS, 10 mg/ml Bromophenol Blue, 10 mM DTT) and heating (100°C, 60 seconds). Proteins were resolved by SDS-PAGE. The dye front, which contains unincorporated radioactive palmitoyl-CoA, was cut from the gel prior to staining. The gel was placed in Coomassie blue stain for 20 min, destained (10% acetic acid, 30% methanol) for 90 min, and treated with 1M sodium salicylate in 15% methanol for 30 min. The gel was dried onto filter paper and exposed to film (Kodak BioMax XAR) at −80°C for 2–7 days.
Fig. 2.
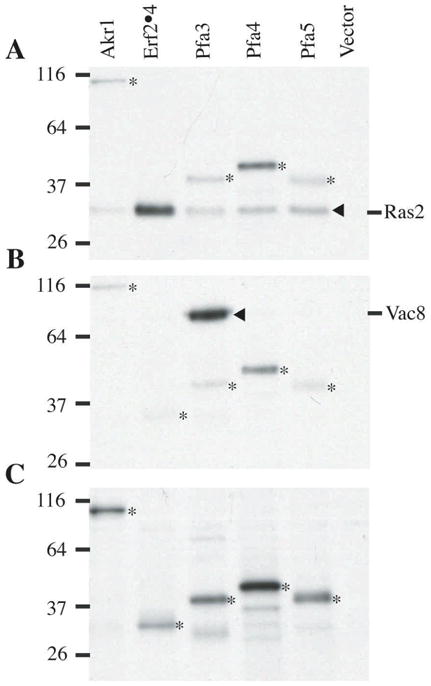
Evaluation of the substrate specificity of yeast DHHC PATs. Yeast membranes (0.4 mg/ml) were prepared as described in the text from strains expressing either Akr1-Flag, Flag-Erf2/GST-Erf4, Pfa3-Flag, Pfa4-Flag, or Pfa5-Flag. The crude membrane fractions were incubate with either 320 nM GST-Ras2(HV)CaaX (A), 320 nM Vac8-myc (B), or buffer alone (C) and 0.8 mM 3H-palmitoyl-CoA. Films were exposed for 2 days (A and B) or 7 days (C). Asterisk denotes autoacylation of the DHHC PATs. The position of migration of Ras2 and Vac8 are indicated in panels A and B, respectively.
As an illustration of this method, the specificity of five yeast DHHC PATs (Akr1, Erf2/Erf4, Pfa3, Pfa4, and Pfa5) toward two different palmitoylated proteins (Ras2 and Vac8) was examined (Fig. 2). As seen in Panel A, Erf2/Erf4 preferentially palmitoylates Ras2, with the other PATs showing very little activity. In contrast, Pfa3 is the only PAT with measurable activity toward Vac8 (Panel B). These results correspond to the in vivo specificities of Ras PAT and Vac8 PAT, respectively. A longer exposure of the gel reveals that all five PATs exhibit autopalmitoylation, presumably representing an intermediate in the transferase reaction.
4. Purification and characterization of DHHC PATs from yeast expression systems
The purification of the Erf2/Erf4 Ras PAT will be used as an example of using high copy number, galactose inducible yeast expression systems to purify PATs. The divergent GAL1,10 galactose inducible promoters on the shuttle plasmid pESC-LEU were used (Stratagene) (Fig. 3). The coding region of the ERF4 gene was amplified by PCR from yeast genomic DNA and ligated into the BglII-PacI site of pESC-LEU. The ERF2 coding region was amplified from yeast genomic DNA, incorporating a N-terminal 6x His tag into the oligonucleotide primer used for PCR. The ERF2 fragment was ligated into the BamHI-XhoI site of the vector pESC-LEU-Flag-Erf4. This resulted in the construction of the vector pESC-LEU-6XHis-Erf2/Flag-Erf4 in which Flag-Erf4 is expressed off the GAL10 promoter and 6XHis-Erf2 is expressed from the GAL1 promoter. The yeast strain YPH499 is co-transformed with pMA210 and pESC-LEU-6XHis-Erf2/Flag-Erf4 to further increase the expression of the GAL promoters by increasing the level of Gal4 activator protein as described earlier ref?.
Fig. 3.
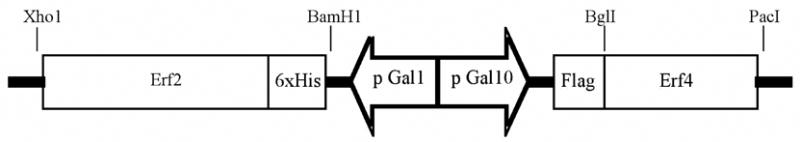
Schematic of the divergent promoter sytems used to co-express Erf2 and Erf4 in yeast. DNA fragments expressing a 6xHis tagged version of Erf2 and Flag tagged Erf4 were ligated into the promoter control region of the yeast expression vector, pESC-LEU, as diagramed. Plasmid construction details can be found in the text.
The growth, induction, lysis, and membrane fractionation of YPH499 expressing Erf2 and Erf4 was done as described for the production of GST-Ras2 in Section 2.1. The membrane fraction was suspended TBS buffer using a Dounce homogenizer to obtain a total protein concentration of approximately 15 mg/ml. The membrane fraction can be stored at −80°C for several months without significant loss of PAT activity.
4.1.2 Detergent solubilization
Solubilization of Erf2/Erf4 from the membrane fraction was done by incubating with detergent (0.75% (w/v)) in TBS (50 mM Tris HCl, pH 7.4 with 150 mM NaCl) at 4°C for 2 h followed by centrifugation (Type 55 Ti rotor, 45,000 rpm for 2 h) to remove insoluble material. The effectiveness of solubilization was assessed by SDS-PAGE followed by immunobloting for 6XHis-Erf2 and Flag-Erf4 with anti-His and anti-Flag antibodies, respectively. Several detergents were examined: dodecylmaltoside (DDM), deoxycholic acid (DCA), Triton X-100, octylglucoside, and CHAPS. Using 0.75% (w/v) detergent, DDM was found to be the most effective at solubilizing Erf2/Erf4 as a complex from the membrane, followed by Triton X-100, and DCA(Table 1). To determine the effect of detergent extraction on enzyme activity, a PAT assay was performed using Ras2 and 3H-palmitate (preparation described in Sections 2.1 and 2.3). The specific activity of membranes prior to extraction was 114 pmol Ras2 palmitoylated/min/mg. Extraction with DDM led to an increase in specific activity, whereas extraction with Triton X-100 did not increase the specific activity and extraction with DCA led to a decrease in specific activity. DDM was therefore chosen for future experiments
4.1.3 Ras PAT Purification
Ras PAT was purified to homogeneity by nickel affinity and anion exchange chromatography. Membranes (P100) isolated from cells expressing 6XHis-Erf2/Flag-Erf4 were resuspended in TBS and fresh protease inhibitors (see above) were added. The membrane was solubilized by adding DDM (0.7%), NaCl (400 mM final), imidazole (1 mM), Urea (2 M final), and β-mercaptoethanol (10 mM). The pH was adjusted 8.5 with Tris base. Following incuation at 5°C (1 hr), the solution was spun (10,000 x g, 20 min) to remove insoluble material.. The Ni-NTA resin was pre-equilibrated in 50 mM Tris HCl, pH 8.5 with 300 mM NaCl and 0.08% DDM prior to addition to the solubilized membranes. The mixture was incubated 1 h at 4°C and then the slurry was loaded into a column and washed using 3 sequential wash solutions of 30 column volumes each: (1) 50 mM Tris HCl pH 8.5, 300 mM NaCl, 0.08% DDM, 5 mM β-mercaptoethanol, (2) 50 mM Tris HCl pH 8.5, 150 mM NaCl, 0.08% DDM, 5 mM β-mercaptoethanol, 5% glycerol, and (3) 50 mM Tris HCl pH 8.5, 150 mM NaCl, 0.08% DDM, 5 mM β-mercaptoethanol, 5% glycerol, 10 mM imidazole. Erf2/Erf 4 was eluted using 8–10 column volumes of 50 mM Tris HCl pH 8.5, 150 mM NaCl, 0.08% DDM, 5 mM β-mercaptoethanol, 5% glycerol, and 250 mM imidazole.
Ras PAT was further purified by anion exchange purification using a HiTrap Q FF (Amersham) FPLC column. The Ni-NTA eluate was diluted 1:2 (vol/vol) with FPLC Buffer A (20 mM Tris HCl, pH 8.8, 0.1% DDM, and 5% glycerol) and loaded onto a 1 ml HiTrap Q FF column, which had been pre-equilibrated in FPLC Buffer A. The column was eluted using a linear gradient of FPLC Buffer B (FPLC Buffer A containing 1 M NaCl). The column was initially washed with 2 column volumes (cv) 0% Buffer B (0.8 ml/min), then with a linear gradient to 18% Buffer B over 5 cv, followed by 12 cv at 18% Buffer B at a reduced flow rate (0.3 ml/min). The Erf2/Erf4 complex was eluted by increasing the Buffer B concentration from 18% to 40% over 15 cv (0.3 ml/min). The Erf2/Erf4 complex eluted at approximately 250 mM NaCl. The addition of 5% glycerol (final) to the ion exchange buffers helped to sharpen the peaks. Fractions (1 ml) were monitored by UV detection (280 nm), SDS-PAGE, and PAT activity (Fig. 4). The Erf2/Erf4 Ras PAT is stable frozen (−80°C) and tolerates freeze-thawing well, however it loses activity rapidly at room temperature.
Fig. 4.
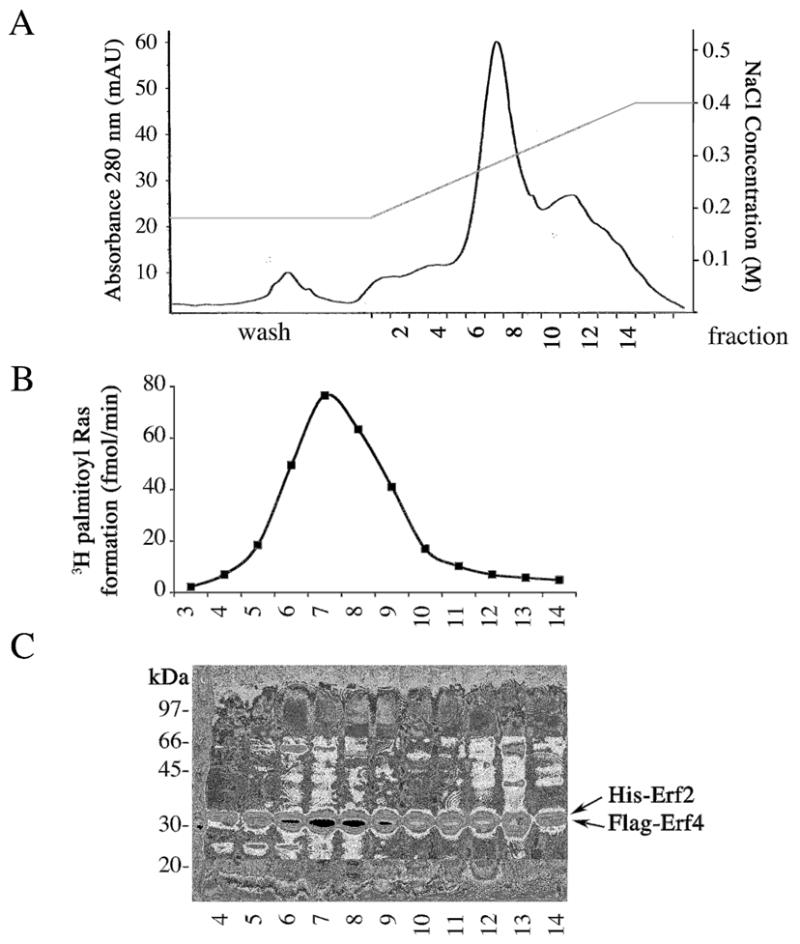
Purification of the Erf2/Erf4 PAT complex by FPLC using anion exchange chromatography. (Panel A) UV 280 nm from anion exchange chromatography showing fractions containing His-Erf2/Flag-Erf4. (Panel B) Ras PAT activity of anion exchange fractions. (Panel C) Anion exchange fractions on 10% SDS PAGE with Coomassie staining. 6xHis-Erf2 and Flag-Erf4 comigrate at 32 kDa under these conditions, as verified by immunoblotting.
4.2 Characterization of Erf2/Erf4 PAT
The availability of purified Ras PAT provides an opportunity to characterize the basic features of the palmitoylation reactions. In this section, pH dependence, detergent dependence, and sensitivityof Ras PAT to inhibition by 2-bromopalmitate is presented.
Ras PAT activity is quantitated by resolving the reaction mixture by SDS-PAGE, excising the 3H palmitoyl-Ras band, and measuring the counts by liquid scintillation counting. A typical reaction consists of 15 μl PAT Buffer (240 mM Tris, pH 7.9, 100 mM NaCl, 1 mM DTT), €5.0 μl 0.5 mg/ml yeast GST Ras2HV (3.7 μM) and 3.0 μl 1.7 μg/ml yeast Erf2/Erf4. The reaction was initiated by adding 2.0 μl 30 μM 3H palmitoyl-CoA (2.4 μM) followed by incubation at 30°C (typically 10–15 min). The reaction was stopped by the addition of 6.5 μl 5x SDS loading buffer lacking reducing agent and the sample was loaded directly onto the gel without applying heat. The loading buffer should be near neutral pH and should not contain high concentrations (>10 mM) of reducing agents (DTT, βME, etc.), as the palmitoyl Ras thioester is unstable in the presence of reducing agents, extreme pH (<5 or >9), or extended heating. The addition of SDS-PAGE loading buffer is sufficient to completely inhibit any further reaction without destabilizing the thioester product. Following SDS-PAGE, the gel was stained with Coomassie stain to visualize the GST-Ras2 product and the bands were excised from the gel. The gel pieces were dissolved in 0.5 ml Soluene 350 (Perkin Elmer Life Science) for 3 h at 50°C or overnight at room temperature, after which scintillation cocktail was added for liquid scintillation counting.
In measuring palmitoylation by this method it is important to distinguish between spontaneous (ie., non-enzymatic) and PAT mediated (enzymatic) transesterification between palmitoyl-CoA and the cysteine thiol of Ras. This is most conveniently done by comparing palmitoylation of Ras before and after heat inactivation of the Ras PAT (95°C, 15 min). The pH of the reaction plays an important role in dictating the ratio of enzymatic to non-enzymatic palmitoylation. By substituting buffers of varying pH for the Tris pH 7.9 in the PAT buffer, the pH effects on both the enzymatic palmitoylation and non-enzymatic palmitoylation of Ras can be determined. For example, alkaline conditions (pH >8) favors spontaneous palmitoylation (Fig. 5A). It is therefore recommended that a non-enzymatic control always be included when measuring palmitoylation, and the non-enzymatic reaction subtracted from the total reaction to obtain a true value for the enzymatic palmitoylation. Alternately, the non-enzymatic reaction can be reduced by conducting the assay at pH <7, even though, in the case of the yeast Ras PAT, the enzymatic reaction is also less efficient at lower pH.
Fig. 5.
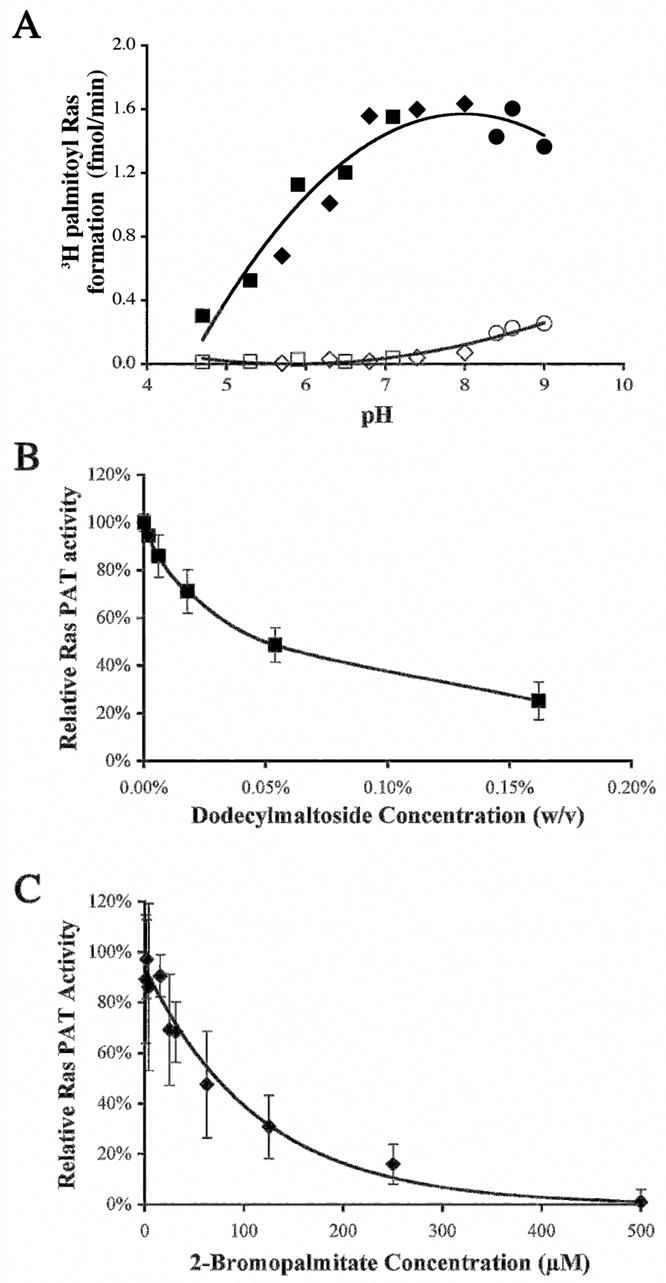
(Panel A) pH profile of enzymatic and non-enzymatic palmitoylation of Ras by purified yeast Erf2/Erf4 Closed symbols indicate enzymatic palmitoylation. Open symbols indicate non-enzymatic palmitoylation. The buffers used were ● Tris HCl, ◆ sodium phosphate and ■ sodium citrate/sodium phosphate. (Panel B). Effect of excess dodecylmaltoside on in vitro Ras palmitoylation by purified Erf2/Erf4. Enzyme activity is expressed relative to that in the absence of added detergent. (Panel C). In vitro inhibition of Ras palmitoylation by 2-bromopalmitate (added from a stock solution in isopropanol). The final isopropanol concentration in reaction was 6%
Detergent solubilization of Erf2/Erf4 Ras PAT from yeast membranes was described in Section 4.1.2 above and dodecylmaltoside was found to be the most effective detergent tested for solubilization of Ras PAT. Therefore, purified Ras PAT is typically stored in 0.08% DDM (2.4 mM) to prevent aggregation. However, the presence of DDM above the critical micelle concentration (0.15 mM) was found to reversibly inhibit the palmitoylation reaction when present in the PAT reaction, possibly by affecting the availability of lipid substrates. To investigate more closely the effect of DDM on Ras PAT activity, the relative PAT activity was measured as a function of DDM concentration (Fig. 5B). Ras PAT activity is quite sensitive to DDM concentration in the reaction buffer, with 50% inhibition of activity occurring at approximately 0.04%. Based on this, Ras PAT is diluted in a typical reaction such that the final DDM concentration is 0.01%.
The availability of purified Ras PAT or other DHHC PATs provides an opportunity to identify and characterize palmitoylation inhibitors. 2-bromopalmitate (2BP) is widely believed to be a palmitoylation inhibitor. Addition of 2BP to cells results in decrease in palmitoylation of proteins. However, the specificity of 2BP as an S-PAT inhibitor is somewhat uncertain, given that it is also used in vivo as an inhibitor of fatty acid oxidation. In fact, it has been reported that 2BP inhibits fatty acid-CoA ligase, acyl CoA oxidase and carnitine-O-palmitoyl transferase, as well as other enzyme activities (20). To examine inhibition of purified Erf2/Erf4 PAT by 2BP, we used purified Ras PAT and performed quantitative PAT assays. 2BP was added from a stock solution in isopropanol (2 μl), adjusting the PAT buffer volume to keep the final reaction volume at 25 μl. Controls were done using only the carrier to account for any solvent deactivation of the enzyme. After the addition of the Ras substrate and enzyme, the mixture is pre-incubated at 4°C for 10 min prior to the addition of 3H palmitoyl-CoA. Using purified Erf2/Erf4 palmitoyltransferase in the Erf2/Erf4 Ras PAT assay, 2BP was found to be an inhibitor of Ras S-palmitoylation with an IC50 of 60 μM (Fig. 5C). This result confirms that 2BP can be used as an effective inhibitor of S-palmitoylation, but the relatively low affinity leaves open the possibility of discovering specific, high affinity PAT inhibitors.
Acknowledgments
Work in the authors’ laboratories is supported by NIH grants GM051466 (MEL) and CA050211 (RJD) and the Showhouse Breast Cancer Research Fund (RJD). We would like to thank David Mitchell and other members of our groups for reading this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roth AF, Feng Y, Chen L, Davis NG. J Cell Biol. 2002;159:23–8. doi: 10.1083/jcb.200206120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kihara A, Kurotsu F, Sano T, Iwaki S, Igarashi Y. Mol Cell Biol. 2005;25:9189–97. doi: 10.1128/MCB.25.21.9189-9197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smotrys JE, Schoenfish MJ, Stutz MA, Linder ME. J Cell Biol. 2005;170:1091–9. doi: 10.1083/jcb.200507048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lobo S, Greentree WK, Linder ME, Deschenes RJ. J Biol Chem. 2002;277:41268–73. doi: 10.1074/jbc.M206573200. [DOI] [PubMed] [Google Scholar]
- 5.Valdez-Taubas J, Pelham H. Embo J. 2005;24:2524–32. doi: 10.1038/sj.emboj.7600724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. J Lipid Res 2006 [Google Scholar]
- 7.Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, Linder ME. J Biol Chem. 2005;280:31141–8. doi: 10.1074/jbc.M504113200. [DOI] [PubMed] [Google Scholar]
- 8.Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, Orban PC, Mullard A, Cowan CM, Raymond LA, Drisdel RC, Green WN, Ravikumar B, Rubinsztein DC, El-Husseini A, Hayden MR. Nat Neurosci. 2006;9:824–31. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Neuron. 2004;44:987. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 10.Keller CA, Yuan X, Panzanelli P, Martin ML, Alldred M, Sassoe-Pognetto M, Luscher B. J Neurosci. 2004;24:5881–91. doi: 10.1523/JNEUROSCI.1037-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright LP, Philips MR. J Lipid Res. 2006;47:883–91. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Sikorski RS, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothstein RJ. MethEnzymol. 1983;101:202–11. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell DA, Marshall TK, Deschenes RJ. Yeast. 1993;9:715–22. doi: 10.1002/yea.320090705. [DOI] [PubMed] [Google Scholar]
- 15.Ma J, Ptashne M. Cell. 1987;48:847–53. doi: 10.1016/0092-8674(87)90081-x. [DOI] [PubMed] [Google Scholar]
- 16.Wang YX, Catlett NL, Weisman LS. J Cell Biol. 1998;140:1063–74. doi: 10.1083/jcb.140.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duronio RJ, Jackson-Machelski E, Heuckeroth RO, Olins PO, Devine CS, Yonemoto W, Slice LW, Taylor SS, Gordon JI. ProcNatlAcadSciUSA. 1990;87:1506–10. doi: 10.1073/pnas.87.4.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smotrys JE, Linder ME. Annu Rev Biochem. 2004;73:559–87. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- 19.Taylor DC, Weber N, Hogge LR, Underhill EW. Anal Biochem. 1990;184:311–6. doi: 10.1016/0003-2697(90)90686-4. [DOI] [PubMed] [Google Scholar]
- 20.Coleman RA, Rao P, Fogelsong RJ, Bardes ES. Biochimica et Biophysica Acta. 1992;1125:203–9. doi: 10.1016/0005-2760(92)90046-x. [DOI] [PubMed] [Google Scholar]