

Abstract
Cidofovir (HPMPC) is a broad-spectrum anti-viral agent whose potential, particularly in biodefense scenarios, is limited by its low oral bioavailability. Two prodrugs (3, 4) created by conjugating ethylene glycol-linked amino acids (L-Val, L-Phe) with the cyclic form of cidofovir (cHPMPC) via a P-O ester bond were synthesized and their pH dependent stability (3, 4) potential for in vivo reconversion to drug (3), and oral bioavailability (3) were evaluated. The prodrugs were stable in buffer between pH 3–5, but underwent rapid hydrolysis in liver (t½ = 3.7 min), intestinal (t½ = 12.5 min) and Caco-2 cell homogenates (t½ = 20.2 min). In vivo (rat), prodrug 3 was >90% reconverted to cHPMPC. The prodrug was 4x more active than ganciclovir (IC50 value, 0.68 μM vs 3.0 μM) in a HCMV plaque reduction assay. However, its oral bioavailability in a rat model was similar to the parent drug. The contrast between the promising activation properties and unenhanced transport of the prodrug is briefly discussed.
Keywords: Prodrug, cHPMPC, HPMPC, biotransformation, HCMV, anti-viral, biodefense
Cidofovir (Vistide®, HPMPC, 1, Fig. 1) is a broad-spectrum anti-viral agent that is used in the treatment of cytomegalovirus (CMV) retinitis in AIDS patients. Cidofovir also has therapeutic potential in the treatment of other herpes and DNA viruses including polyoma-, papilloma-, adeno-, and poxvirus infections.1, 2 Recently, cidofovir has become of particular interest as a potential emergency therapy for orthopox virus infections.3 An important limitation of cidofovir and analogous nucleotide drugs in a therapeutic role is their low oral bioavailability and poor transport into cells, necessitating intravenous administration. In an emergency situation such as a smallpox outbreak, an orally available form of cidofovir would thus be highly advantageous.
Figure 1.
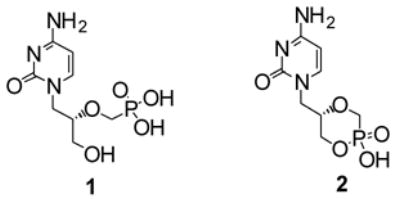
Chemical structures of cidofovir (HPMPC) 1 and cyclic cidofovir (cHPMPC) 2.
Our current research is focused on improving cidofovir’s oral bioavailability. The phosphonic diacid group of cidofovir is ionized under physiological conditions, which substantially accounts for its low bioavailability (<5%).4, 5 Cyclic cidofovir (cHPMPC, 2, Fig. 1), undergoes biotransformation when exposed to cCMP phosphodiesterase to generate the parent drug, HPMPC6, 7. Severe nephrotoxic effects have been associated with cidofovir treatment8 while cHPMPC has been reported to be significantly less nephrotoxic.6 cHPMPC, also exhibits low oral bioavailability, indicating a need to mask its remaining anionic charge at physiological pH.
Prodrugs9 have proven valuable for increasing drug bioavailability as well as for drug targeting after internal absorption.10 The prodrug strategy is based on chemical modification of the drug, often achieved by conjugating a pro-moiety to the pharmacophore. The modification should not only confer enhanced transport, but must also be reversible via biotransformation in vivo to release the parent drug.
We have previously reported the synthesis of dipeptide conjugates of cidofovir as prodrugs targeting a peptide transporter (PepT1) present in the small intestine. 11–13 Several of these conjugates showed significantly enhanced intestinal uptake in a rat model. 11–13 Gly-sar, a known PepT1 substrate, inhibited uptake in the same model. Attaching the pro-moiety of the prodrug through the phosphonic acid group on cyclic cidofovir achieves complete masking of cidofovir’s original phosphonic diacid group. On exposure of these cyclic cidofovir dipeptide conjugates to biological fluids, the parent drug was rapidly released from the biologically benign dipeptide pro-moiety. 11–13 Other research groups have recently published different types of cidofovir prodrugs designed for enhanced absorption in the intestine,14, 15 such as ether-lipid-conjugates of cidofovir and cyclic cidofovir, in which the pro-moiety resembles a natural lipid.16 Alternative prodrug approaches have been documented in the literature.17–21
The successful valyl ester prodrug of acyclovir (valacyclovir)22 stimulated our interest in exploring the chemical and biological properties of a single amino acid conjugate of cyclic cidofovir. Several structural requirements must be satisfied in masking the phosphonic diacid group of cidofovir and targeting the active transporter PepT1 present in the small intestine. One crucial structural requirement for optimal recognition by PepT1 is a free -NH2 group.23, 24 Therefore, the synthetic strategy presented herein involves P-O conjugation between cyclic cidofovir and an ethylene glycol ester-linked amino acid, which masks the negative charge on the cyclic cidofovir (and the amino acid carboxyl group), leaving the N-terminal of the amino acid free for possible involvement as a substrate recognition site for PepT1 (Fig. 2).
Figure 2.
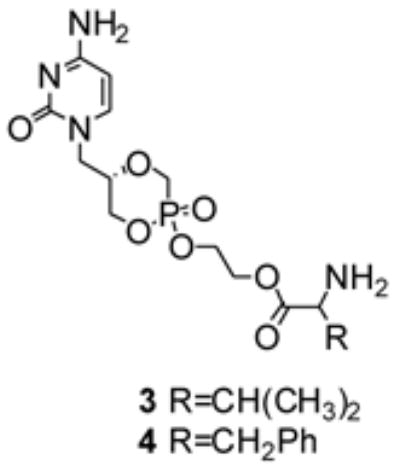
Structure of the synthesized ethylene glycol-linked amino acid conjugates of cyclic cidofovir 3 and 4.
(S)-HPMPC was synthesized by modification11 of a previously described25 method. The ethylene glycol-linked amino acids were synthesized by conjugating t-BOC protected amino acids (L-Val, 3, or L-Phe, 4, 1 eq) to ethylene glycol (4 eq) using DCC in the presence of a catalytic amount of DMAP (Scheme 1).
Scheme 1.
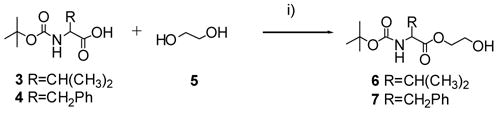
Synthesis of ethylene glycol-linked amino acids 6 and 7. Reagents and conditions: (i) DMAP (0.15 eq), DCC (1.3 eq), CH2Cl2, rt, 18 h, 48–84%.
After purification of 6 (48%) and 7 (84%), by silica gel column chromatography, the ethylene glycol-linked amino acids were conjugated to cidofovir using 2.5 eq PyBOP as the condensing agent (Scheme 2). Concurrent conversion of HPMPC to cHPMPC occurs in a 1-pot reaction.11 The reaction is easily monitored by 31P NMR: cyclic cidofovir has a chemical shift of ~5 ppm while the ethylene glycol-linked amino acid conjugates show resonance between 13–15 ppm. The t-BOC protected conjugates (8 and 9) were purified by silica gel column chromatography (CH2Cl2: acetone: methanol, 6:3:1) and deprotected by exposure to trifluoroacetic acid (TFA). The final compounds, 3 and 4, were obtained as a diastereoisomeric mixture (diastereomeric ratios of 1:0.8), after purification by preparative TLC on silica in overall yields of 8% and 25%, respectively. 26, 27
Scheme 2.
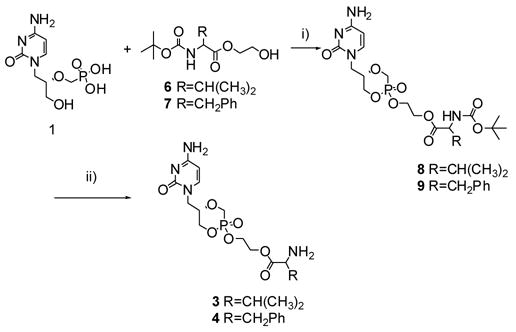
Synthesis of 3 and 4: ethylene glycol-linked amino acid conjugates of cyclic cidofovir. Reagents and conditions: (i) PyBOP (2.5eq), DIEA, DMF, 40°C, 2.5–4h, 30–56%. (ii) TFA, CH2Cl2, rt 4h.
The stability of cHPMPC, 3 and 4 in buffered aqueous solution at 37°C was evaluated by incubating each compound (500 μM) in 50 mM phosphate buffer in which the pH was adjusted by HCl to 3.0, 4.0, 5.0, 6.0, or 7.0. Aliquots (0.25 mL) were removed at 0, 1, 2, and 3 hours and immediately combined with an equal volume of ice-cold 10% TFA to minimize degradation prior to HPLC analysis. The rate of disappearance was estimated by HPLC and the half-life (t½) was determined from apparent first order rate constant derived from linear regression analysis (r2 ≤ 0.95) of pseudo-first order plots of prodrug concentration versus time (Table 1).
Table 1.
Half-lives (t½ in hours) for cHPMPC, 3 and 4 presented in hours at various pHs.a
| t½(h) | |||||
|---|---|---|---|---|---|
| pH 3.0 | pH 4.0 | pH 5.0 | pH 6.0 | PH 7.0 | |
| cHPMPC | st | st | st | st | St |
| 3 | st | st | st | 16.0 | 3.5 |
| 4 | st | st | st | nd | 3.7b |
t½ of 500μM cHPMPC, 3 and 4 in an aqueous solution at 37°C as a function of pH. (st=stable, nd = not done)
pH = 6.5
The ethylene glycol-linked valine and phenylalanine conjugates of cyclic cidofovir (3 and 4) showed no evidence of hydrolysis over 3 hours at pH values 5.0. Although 3 and 4 were stable in moderately acidic solutions (3.0–5.0) noticeable but slow hydrolysis was observed at pH 6.0–7.0. These observations were in agreement with the chemical stability observed for the cyclic cidofovir dipeptide derivatives previously reported.11 Further investigations focused on the L-Val conjugate 3. The activation of 3 in the presence of endogenous hydrolases was investigated by exposing the compound (control cHPMPC) to cellular (Caco-2 and HFF) and tissue homogenates (rat liver and intestine) as well as to an intestinal perfusate obtained from Sprague Dawley rats. cHPMPC and 3 were exposed to the various homogenates at 37°C and samples taken at several time intervals between 0 and 3 hours were analyzed by HPLC. The half-lives of cHPMPC and 3 when exposed to homogenates were determined as described for the buffer pH stability data. The results are summarized in Table 2.
Table 2.
Half-lives (t½ in minutes) for HPMPC and 3 in cellular and tissue homogenates.a
Estimated t½ of cidofovir and 3 in homogenate solutions at 37°C. (st=stable).
Perfusate represents intestinal luminal contents and is prepared by passing buffered solution (pH=6.5) through an isolated intestinal segment as described elsewhere. 28
When the t½ values for 3 obtained from the buffer pH and homogenate stability studies are compared, it is apparent that although 3 is stable to moderately acidic conditions and only slowly hydrolyzed in neutral buffer, it is rapidly metabolized by endogenous liver, intestinal and cellular enzymes. The activation pathway for 3 and the relevant enzymes involved are not yet known.
To obtain a marker for its anti-viral activity, 3 was evaluated in a HCMV plaque reduction assay using the Towne strain, plaque purified isolate Po of HCMV (kindly provided by Dr. Mark Stinski, University of Iowa). HFF cells seeded in 24-well cluster dishes were infected with approximately 100 p.f.u. of HCMV per cm2 cell sheet. Following virus adsorption, the compounds, prepared as 10 mg/mL stock solutions in DMSO, were diluted with growth medium and added to duplicate wells at 4 to 8 concentrations. After incubation at 37°C for 7 to 10 days, cell sheets were fixed, stained with crystal violet and the microscopic plaques enumerated.29 Drug effects were calculated as a percentage of reduction in the number of plaques present at each drug concentration compared to the number observed in the absence of the drug. Fifty percent inhibitory concentrations (IC50) were calculated from the linear portions of the regression lines and are presented in Table 3.30 Compared to the control drug (ganciclovir), 3 was found to be 4-fold more potent.
Table 3.
IC50 data for HPMPC and 3 obtained in a HCMV plaque reduction assay conducted in HFF cells.
| Drug | IC50 (μM) |
|---|---|
| 3 | 0.68 |
| Controla | 3.0 |
ganciclovir served as the control.
The cytotoxicity of 3 was evaluated in KB cells. Cells were grown logarithmically for two days in the presence of each compound (in triplicate) and evaluated by spectrophotometric quantitation of crystal violet dye eluted from stained KB cells.29 It was found that cHPMPC as well as 3 showed little to no cytotoxicity in KB cells up to a concentration of 100 μM.
The HPMPC compounds were next tested directly for oral availability by direct injection of the drugs into the gastrointestinal tract of rat (Table 4). 3 is efficiently converted to cHPMPC in this in vivo system: >90% of the sample detected after dosing of 3 was cHPMPC. After direct injection of 3 into the GI tract of rat, the oral availability of 3 was estimated to be 1.8%, which was not significantly different from cHPMPC (2.2%). Since improved oral bioavailability was not achieved, separation and evaluation of the 3 individual diastereoisomers were not pursued.
Table 4.
Bioavailability of HPMPC and 3 conducted in rat.
| Dose normalized | ||
|---|---|---|
| mean AUC0-∞ (ng/mL*h) | % Bioavailabilitya | |
| cHPMPC-IV | 472.2 | 100 |
| 3 | 258.5 | 1.8 |
| cHPMPC | 304.6 | 2.2 |
The AUC for 3 was determined by combining the contributions from both the generated cHPMPC and the prodrug. The bioavailability was calculated from the ratio of the dose normalized oral AUC divided by the intravenously (IV) administered cHPMPC AUC data, and a factor to account for the difference in the IV dose vs oral dose (0.1 mg IV vs 3 mg oral).
In conclusion, we have synthesized two novel ethylene glycol-linked amino acid conjugates of cyclic cidofovir. One, the valine conjugate (3) was investigated in detail. 3 was actively metabolized releasing the parent drug in vivo. 3 was also highly active against HCMV in a HFF cell assay showing an IC50 value in the submicromolar range, 4-fold lower than ganciclovir, the positive control. 3 was not cytotoxic at a concentration of 100 μM when evaluated in KB cells. However, the estimated oral bioavailability in rat of 3 was similar to that of the parent drug. This indicated that this ethylene glycol-linked mono-L-Val derivative of cHPMPC, while susceptible to biotransformation in vivo, lacks a structural requirement for enhanced oral bioavailability that is present in the Val-Ser-side chain-linked dipeptide conjugates of cHPMPC. 11
Supplementary Material
I) Detailed synthetic procedures for compounds 3 and 4 , II) NMR spectra for 3 and 4 and III) Methodology for evaluation of the oral bioavailability of 3.
Acknowledgments
We thank the NIH for financial support (R43-AI056864). This research was presented in part at the 19th International Conference on Antiviral Research (ICAR), San Juan, Puerto Rico, May 7th–11th 2006.31
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Clercq E. Clin Microbiol Rev. 1997;10:674. doi: 10.1128/cmr.10.4.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Clercq E, Holy A. Nat Rev Drug Disc. 2005;4:928. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq E. Antivir Res. 2002;55:1. doi: 10.1016/S0166-3542(02)00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wachsman M, Petty BG, Cundy KC, Jaffe HS, Fisher PE, Pastelak A, Lietman PS. Antivir Res. 1996;29:153. doi: 10.1016/0166-3542(95)00829-2. [DOI] [PubMed] [Google Scholar]
- 5.Cundy KC, Li ZH, Hitchcock MJ, Lee WA. Drug Metab Dispos. 1996;24:738. [PubMed] [Google Scholar]
- 6.Bischofberger N, Hitchcock MJM, Chen MS, Barkhimer DB, Cundy KC, Kent KM, Lacy SA, Lee WA, Li ZH, et al. Antimicrob Agents Chemother. 1994;38:2387. doi: 10.1128/aac.38.10.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendel DB, Cihlar T, Moon K, Chen MS. Antimicrob Agents Chemother. 1997;41:641. doi: 10.1128/aac.41.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lalezari JP, Stagg RJ, Kuppermann BD, Holland GN, Kramer F, Ives DV, Youle M, Robinson MR, Drew WL, Jaffe HS. Ann Internal Med. 1997;126:257. doi: 10.7326/0003-4819-126-4-199702150-00001. [DOI] [PubMed] [Google Scholar]
- 9.Albert A. Nature. 1958;182:421. doi: 10.1038/182421a0. [DOI] [PubMed] [Google Scholar]
- 10.Ettmayer P, Amidon GL, Berndt C, Testa B. J Med Chem. 2004;47:2393. doi: 10.1021/jm0303812. [DOI] [PubMed] [Google Scholar]
- 11.McKenna CE, Kashemirov BA, Eriksson U, Amidon GL, Kish PE, Mitchell S, Kim JS, Hilfinger JM. J Organomet Chem. 2005;690:2673. [Google Scholar]
- 12.McKenna CE, Kashemirov BA, Peterson LW, Eriksson U, Saejueng K, Kim JS, Mitchell S, Kijek P, Hilfinger JM, Drach JC. Antivir Res. 2006;70:A37. [Google Scholar]
- 13.Kim JS, Mitchell S, Kijek P, McKenna CE, Kashemirov BA, Eriksson U, Breitenbach J, Borysko K, Gordon A, Drach J, Hilfinger J. Antivir Res. 2006;70:A58. [Google Scholar]
- 14.Bidanset DJ, Beadle JR, Wan WB, Hostetler KY, Kern ER. J Infect Dis. 2004;190:499. doi: 10.1086/421912. [DOI] [PubMed] [Google Scholar]
- 15.Quenelle DC, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Kern ER. Antimicrob Agents Chemother. 2004;48:404. doi: 10.1128/AAC.48.2.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Painter GR, Hostetler KY. Trends Biotechnol. 2004;22:423. doi: 10.1016/j.tibtech.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Ballatore C, McGuigan C, De Clercq E, Balzarini J. Bioorg Med Chem Lett. 2001;11:1053. doi: 10.1016/s0960-894x(01)00128-7. [DOI] [PubMed] [Google Scholar]
- 18.Meier C. Eur J Org Chem. 2006:1081. [Google Scholar]
- 19.Lee WA, Martin JC. Antivir Res. 2006;71:254. doi: 10.1016/j.antiviral.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 20.Oliyai R, Arimilli MN, Jones RJ, Lee WA. Nucleos Nucleot Nucl. 2001;20:1411. doi: 10.1081/NCN-100002566. [DOI] [PubMed] [Google Scholar]
- 21.Holy A. Curr Pharm Des. 2003;9:2567. doi: 10.2174/1381612033453668. [DOI] [PubMed] [Google Scholar]
- 22.Beauchamp LM, Orr GF, De Miranda P, Burnette T, Krenitsky TA. Antiviral Chem Chemother. 1992;3:157. [Google Scholar]
- 23.Bailey PD, Boyd CAR, Bronk JR, Collier ID, Meredith D, Morgan KM, Temple CS. Angew Chem Int Ed. 2000;39:506. doi: 10.1002/(sici)1521-3773(20000204)39:3<505::aid-anie505>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 24.Rubio-Aliaga I, Daniel H. Trends Pharmacol Sci. 2002;23:434. doi: 10.1016/s0165-6147(02)02072-2. [DOI] [PubMed] [Google Scholar]
- 25.Brodfuehrer PR, Howell HG, Sapino C, Jr, Vemishetti P. Tetrahedron Lett. 1994;35:3243. [Google Scholar]
- 26.3: 1H NMR (CD3OD, ppm): 0.98 (6H, m), 2.21 (1H, m), 3.67–4.43 (12H, m), 5.85 (1H, m), 7.54–7.62 (1H, 2d, J=7.1). 31P NMR (CD3OD, ppm):13.91 (1P, s) and 15.16 (1P’, s). 4: 1H NMR (CD3OD, ppm):3.04–4.40 (14H, m), 5.82 (1H, d, J=7.2), 5.86 (1H, d, J=8.28), 7.15–7.30 (5H, m), 7.53 (1H, d, J=7.2), 7.63 (1H, d, J=8.28). 31P NMR (CD3OD, ppm): 13.87 (1P, s) and 15.23 (1P’, s).
- 27.Eriksson U. PhD Thesis. University of Southern California; Dec, 2005. [Google Scholar]
- 28.Kim J-S, Mitchell S, Kijek P, Tsume Y, Hilfinger J, Amidon GL. Mol Pharm. 2006 doi: 10.1021/mp060042f. (accepted for publication) [DOI] [PubMed] [Google Scholar]
- 29.Prichard MN, Turk SR, Coleman LA, Engelhardt SL, Shipman C, Jr, Drach JC. J Virol Methods. 1990;28:101. doi: 10.1016/0166-0934(90)90091-s. [DOI] [PubMed] [Google Scholar]
- 30.Goldstein A. Biostatistics, an introductory text. New York: Macmillan; 1964. [Google Scholar]
- 31.Eriksson U, Peterson LW, Kim J-s, Mitchell S, Kijek P, Hilfinger JM, Drach JC, Kashemirov BA, McKenna CE. Antivir Res. 2006;70:A58. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
I) Detailed synthetic procedures for compounds 3 and 4 , II) NMR spectra for 3 and 4 and III) Methodology for evaluation of the oral bioavailability of 3.