

Abstract
Human thymidine kinase 1 (hTK1) and structurally related TKs from other organisms catalyze the initial phosphorylation step in the thymidine salvage pathway. Though ATP is known to be the preferred phosphoryl donor for TK1-like enzymes, its exact binding mode and effect on the oligomeric state has not been analyzed. Here we report the structures of hTK1 and of the Thermotoga maritima thymidine kinase (TmTK) in complex with the bisubstrate inhibitor TP4A. The TmTK-TP4A structure reveals that the adenosine moiety of ATP binds at the subunit interface of the homotetrameric enzyme and that the majority of the ATP-enzyme interactions occur between the phosphate groups and the P-loop. In the hTK1 structure the adenosine group of TP4A exhibited no electron density. This difference between hTK1 and TmTK is rationalized by a difference in the conformation of their quaternary structure. A more open conformation, as seen in the TmTK-TP4A complex structure, is required to provide space for the adenosine moiety. Our analysis supports the formation of an analogous open conformation in hTK1 upon ATP binding.
Keywords: hTK1, TmTK, thymidine kinase, phosphoryl transfer, quaternary structure
Introduction
Deoxythymidine triphosphate (dTTP) is an essential metabolite for DNA synthesis. The intracellular dTTP concentration is cell cycle-dependent such that the level is low in the quiescent state (G0 phase) and increases approximately 20-fold during the DNA synthesis phase (S phase).1 Furthermore, dTTP is an allosteric effector of the enzyme ribonucleotide reductase, stimulating the conversion of GDP to dGDP and inhibiting the conversion of CDP to dCDP.2,3 Therefore, a fine regulation of the dTTP levels is necessary in order to achieve the desired balance in the intracellular nucleotide pool, which is essential for genetic stability.
The two dTTP biosynthetic routes are the de novo and the salvage pathways. Human thymidine kinase 1 (hTK1) catalyzes the first enzymatic step in the salvage pathway converting thymidine into thymidine monophosphate. Transcriptional regulation of hTK1 limits its expression to the S phase of the cell cycle4,5 and transient expression of hTK1 coincides with the oscillation in the intracellular dTTP concentration. Furthermore, several posttranslational events control hTK1 activity. Phosphorylation of serine 13 during mitosis has been linked to a reduction in the catalytic efficiency of hTK1.6,7 Post mitosis, the KEN sequence located at the C-terminal region targets the enzyme for degradation by the anaphase promoting complex/cyclosome (APC/C-Cdh1).8 The importance of the hTK1 degradation mechanisms has been recently highlighted. Cells transfected with a TK1 mutant that cannot be recognized by the APC/C-Cdh1 complex for proteosomal degradation exhibited an abnormal increase in the dTTP concentration, which led to a high frequency of mutations and slow cell growth.9 The existence of several regulatory mechanisms of hTK1 and of TK1-like enzymes from mammals underlines the importance of the salvage pathway for ensuring an appropriate intracellular level of dTTP. The physiological importance of TK1 is also emphasized in studies of TK1 double knockout mice. Such TK1−/− animals have a shorter life span, very low fertility, a compromised immune system and develop fatal kidney disease.10
In addition to the physiological importance of hTK1, there is also a clinical significance for this enzyme. It is required for the activation of nucleoside analogs that are used as antiviral drugs such as AZT and d4T. Furthermore, certain pathogens rely on the salvage pathway to fulfill their needs for dTTP, making this enzyme an attractive drug target for some infectious diseases.11,12 Finally, since hTK1 levels are significantly higher in cancer cells,13,14 inhibitors of this enzyme may be promising anticancer compounds.
The recently reported crystal structures of the human TK1 and Ureaplasma urealyticum TK12,15,16 show that they differ from other deoxyribonucleoside kinases. Due to this structural difference, the deoxyribonucleoside kinases have been divided in two families: one that includes deoxycytidine kinase, deoxyguanosine kinase, mitochondrial thymidine kinase 2 and the herpes thymidine kinases, and another that includes the cytosolic thymidine kinase 1 from mammals and their homologous TKs from other organisms. In earlier manuscripts detailing TK1-like enzymes, the ATP binding site was invariably unoccupied, and previous attempts at visualizing ATP binding were unsuccessful.16 We decided to exploit the tight binding of the bisubstrate inhibitor P1-(5′-adenosyl)P4-(5′-(2′-deoxy-thymidyl)) tetraphosphate (TP4A) for the purpose of elucidating the ATP binding mode in TK1-like enzymes. Here we report the three-dimensional TK structures of the hyperthermophilic bacterium Thermotoga maritima (TmTK) and of Homo sapiens in complex with TP4A. Our results expose the ATP binding site in TK1-like enzymes, and reveal the change in quaternary structure conformation required for phosphoryl donor binding. Furthermore, the TmTK structure represents the first extensively characterized deoxynucleoside kinase from an extremophile. A comparison of the TK structures of these two distant organisms provides valuable insight into the mode of substrate binding, the oligomerization interfaces of these homotetrameric enzymes, and the residues involved in catalysis.
Results
Overall structure
All previously analyzed structures of TK1-like proteins contain the nucleoside thymidine/nucleotide dTTP at the phosphoryl acceptor binding-site (dTTP acts a feedback inhibitor of TK-like enzyme), whereas the phosphoryl donor site is unoccupied12,15,16 (note that the structure of the Clostridium acetobutylicum TK1-like enzyme (CaTK) in complex with ADP, solved by the Northeast Structural Genomics consortia, was deposited (PDB ID 1XX6) prior to this work, without an accompanying analysis). Yet, elucidation and analysis of the ATP binding mode is essential for a full understanding of the structural and functional characteristics of this enzyme family. In order to gain information on the mode of ATP binding we first crystallized human thymidine kinase 1 (hTK1) in the presence of ADP plus thymidine, or with the ATP analog AppNHp plus thymidine. However, despite the excellent electron density observed for the thymidine molecule in both of these hTK1-complexes, there was no detectable electron density for the ADP/AppNHp molecules (data not shown). This suggested that under the employed crystallization conditions there was insufficient occupancy for the nucleotides in the ATP binding site. To increase the likelihood of full occupancy at the ATP binding site we exploited the high binding affinity of the bisubstrate analog TP4A to hTK1.17 We were able to obtain crystals of hTK1 and of its T. maritima homolog (TmTK) in complex with TP4A that diffracted to 2.3 Å and 1.5 Å, respectively. The structure of the human enzyme was solved by the single-wavelength anomalous diffraction method using a platinum derivative. The TmTK structure was solved by the molecular replacement method using the CaTK (40% sequence identity to TmTK) as the search model (PDB ID 1XX6) (see Table 1 for data collection and refinement statistics).
Table 1.
Data collection and refinement statistics
| Human TK1-TP4A | Thermotoga TK-TP4A | |
|---|---|---|
| APS (SERCAT) | λ = 1.072Å | λ = 1.00Å |
| Space group | P3121 | C2 |
| Unit cell (Å, °) |
a = b= 81.0, c = 156.3
α = β=90, γ = 120 |
a=102.6, b=59.6, c = 61.4
α=90, β = 103.1, γ=90 |
| Content of asymmetric unit | Dimer | Dimer |
| Resolution (Å) | 2.3 | 1.5 |
| Rsyma,b (%) | 7.3 (26.4) | 6.2 (39.5) |
| I/σIa | 12.3 (4.2) | 10.44 (2.9) |
| No. of observed reflections | 141,810 | 170,775 |
| No. of unique reflections | 35,363 | 55,419 |
| Redundancy | 4.0 | 3.1 |
| Completenessa (%) | 97.8 (97.2) | 94.4 (79.0) |
| Figure of meritc | 0.26/0.56 | - |
| No. of heavy atom sites | 2 | - |
| Rcrysd (%) | 19.7 | 16.8 |
| Rfreed (%) | 24.9 | 20.8 |
| r.m.s.d. bond lengthe (Å) | 0.016 | 0.015 |
| r.m.s.d. bond anglee (°) | 1.78 | 1.80 |
| Number of atoms | ||
| Protein | 2479 | 2694 |
| Nucleotide | 66 | 102 |
| Water | 160 | 370 |
| Zinc | 2 | 2 |
| Mean | ||
| B-factors (Å2) | ||
| Protein atoms | 57 | 19 |
| Nucleo tides | 59 | 20 |
| Water molecules | 63 | 35 |
| Zinc atoms | 64 | 20 |
| Ramachandran plot (%) | ||
| Most allowed | 89 | 95 |
| Additional allowed | 11 | 4. |
| Generously allowed | 0 | 0.3 |
| Disallowed | 0 | 0 |
Data for the highest resolution shell are given in parenthesis.
Rsym = Σ | Ii − <Ii> |/Σ <Ii>
Rcryst = Σ || Fobs | − | Fcalc ||/Σ | Fobs |. Rfree is for 10% of the reflections excluded from the refinement.
Root mean square deviations (r.m.s.d.) from ideal values calculated with Refmac.30
Human TK1 and TmTK exhibit 36% identity and 55% similarity at the sequence level (Figure 1(a)). However, the human TK1 contains N-terminal and C-terminal extensions of 17 and 40 amino acid residues, respectively. These regions of hTK1 have regulatory roles and are not required for the catalytic function of the enzyme.18 In fact, the previously reported structures of hTK1 made use of truncation mutants that lacked either this C terminus12 or both the N and C-terminal extensions.15 For the hTK1 structure reported here we used the full-length protein. Despite this fact, our structure lacks electron density for the first 17 residues and the final 43 residues. The part of hTK1 that we observe corresponds almost completely to the size of the full- length TmTK enzyme. For both enzymes several residues between strands 2 and 3 could not be modeled (Figure 1(a)). T. maritima is a eubacterium that belongs to the Thermotogales family, which has been proposed as one of the more primitive branches of prokaryotes.19,20 Therefore, our results allow for a structural comparison of the TK1 homologs from two evolutionary distant organisms, providing insight into the degree of structural and functional conservation in this enzyme family.
Figure 1.
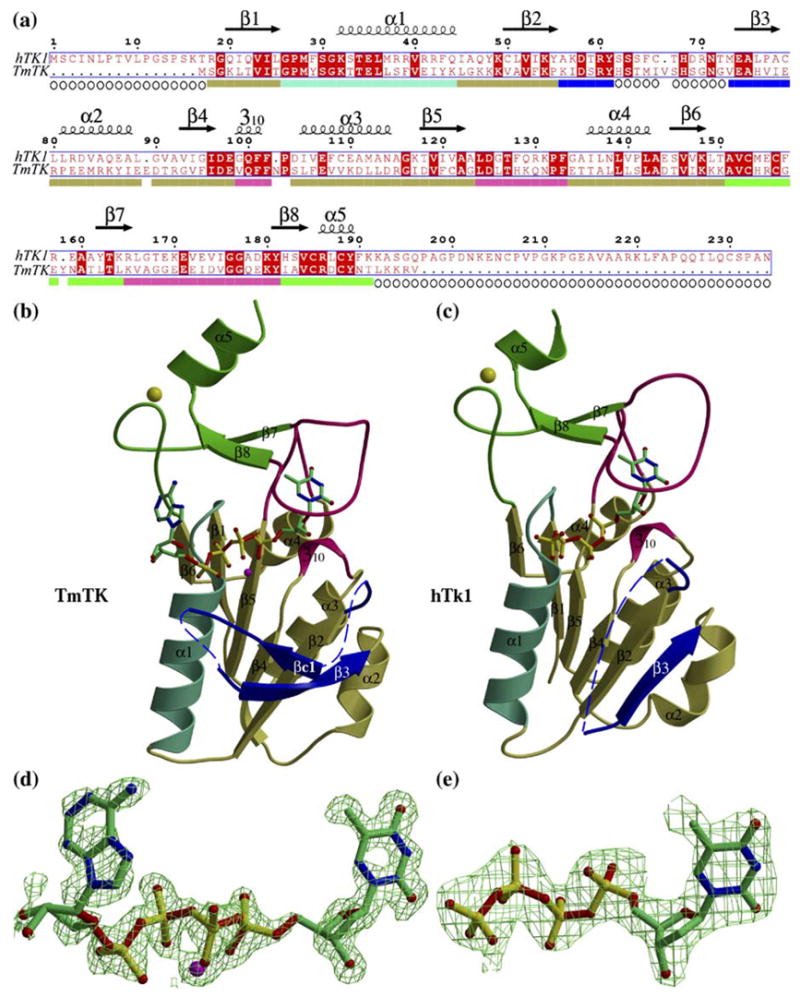
The overall fold of hTK1 and TmTK is similar, (a) Sequence alignment of the two kinases. The displayed secondary structure is that for hTK1, which is basically identical to that of TmTK apart from lacking strand (βc1 and its following loop (residues 48 to 58 of TmTK). The coloring scheme below the sequence alignment corresponds to the regions shown in (b). Open circles indicate residues that could not be modeled due to lacking electron density, (b) Ribbon diagram of a TmTK monomer with the TP4A molecule shown in a ball-and-stick representation. The observed magnesium ion is depicted as a purple sphere. The N-terminal domain (in olive-green) is composed of a β-sheet and four helices. Of those, helix αl (cyan) contributes to the ATP-binding site upon oligomerization. Strand (β3 (blue) is observed in a different conformation to that in hTK1, and is preceded by a region that lacked electron density (broken line). The C-terminal domain (green) is also composed of the lasso loop (magenta). It is this loop that via its main chain atoms interacts with the thymidine. The zinc atom is displayed as a yellow sphere, (c) Ribbon diagram of the hTK1 monomer with the part of the TP4A molecule that displayed good density shown as ball-and-stick. Color scheme is as in (b). (d) The simulated-annealing omit map (F0 − Fc) electron density surrounding the TP4A molecule with its magnesium ligand (purple) contoured at 2.0 sigma. (e) The analogous map for the part of the TP4A molecule that was possible to model in the hTK1-TP4A complex structure. Note the excellent density for the fourth phosphate, but the lack of density for the adenosine moiety of TP4A.
The overall fold of hTK1 and TmTK is similar, with a root mean square deviation (rmsd) of 0.87 Å over 127 Cα atoms. Of the two domains present in these enzymes, the larger N-terminal α/β domain forms the central core and is built by an open β sheet that is enclosed by one long α helix on one side and by three helices on the other side (Figure 1(b) and (c)). The smaller C-terminal domain begins with a 15-residue loop that contains two cysteine residues that participate in zinc binding. The zinc-binding loop is followed by strand (β7, which is connected to strand (β8 by the so-called “lasso” loop12 that is involved in thymidine binding. A short α helix, at the end of the C-terminal domain contributes the remaining two zinc-binding cysteine residues. The zinc atom was proposed to confer structural rigidity to the C-terminal domain.15
In the case of the complex with TmTK we observed excellent electron density for the entire TP4A molecule (Figure 1(d)). In contrast, for the hTK1 complex the electron density was clear only for the thymidine moiety and the four phosphate groups of the TP4A molecule (Figure 1(e)). Thus, even the use of the bisubstrate analog TP4A, which assures complete occupancy of the binding sites, could not solve the problem of disorder for the adenosine moiety. The possible reasons for this apparently high disorder of the adenosine part of the TP4A molecule in the case of the human enzyme are discussed later.
In addition to prominent differences in conformation of residues located between (β2 and α2, the main structural differences between hTK1 and TmTK are localized to the zinc-binding loop and the lasso loop (Figure 2). Despite containing the same number of residues, the lasso loop in TmTK adopts a slightly different conformation from that seen in the human enzyme. The function of the lasso loop, to bind the thymidine nucleoside, is accomplished by several main chain interactions, which is consistent with the low sequence conservation of this region (Figure 1(a), magenta). The difference in the conformation of the zinc-binding loop between hTK1 and TmTK can be directly related to the difference in conformation of their quaternary structure. The hTK1 tetramer is more closed than the TmTK tetramer. The zinc-binding loop is at the monomer-monomer interface that is sensitive to the presence of ATP. In the TmTK structure, where the adenosine moiety of TP4A is specifically bound, the protein adopts the more open conformation. Thus, the conformational difference observed in the zinc-binding loop seems to be involved in the quaternary structure rearrangement from the closed to the open state.
Figure 2.
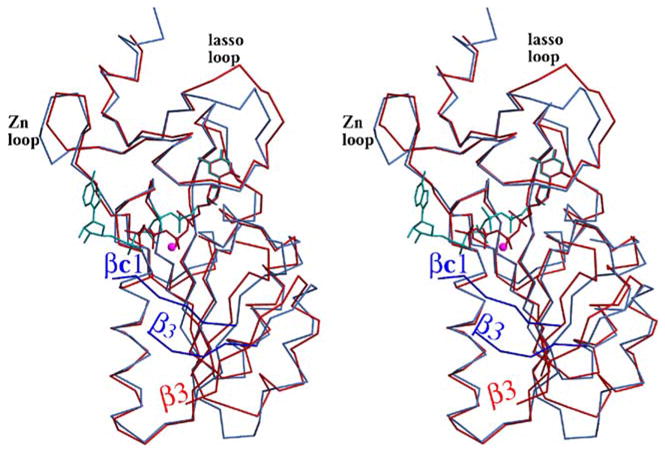
Overlay of hTK1 (red) and TmTK (blue) demonstrates the similarities between the two enzymes. Significant differences are in the position of strand (β3, the presence of strand (βc1 only in TmTK, and in the conformation of the zinc-binding and lasso loops.
As mentioned before, a significant difference between hTK1 and TmTK is manifested in the flexible region located between strand (β2 and helix α2 (shown in blue, Figure 1(b) and (c)): 11 of 24 residues of this flexible region had clear electron density and could be modeled in hTK1, which contains strand (β3 and two short loops, while 16 of 24 residues were modeled in TmTK. The (β3-strand in hTK1 is completely parallel to the (β-strands of the central core, thereby incorporating into the core (β-sheet (Figure 1(c)). In contrast to the human enzyme, the analogous (β3 strand in TmTK only partially aligns to the (β-sheet of the core. Instead the (β3 strand of TmTK also interacts with a short strand that we designated (βc1 (Figure 1(b)), which is positioned at an approximately 90° angle to the core (β-sheet. Strand (βc1 is missing in the human structure, and may reflect the fact that in the human TK1 the ATP-binding site is not occupied by the ribose and the adenine of the TP4A molecule, while in TmTK the ATP-binding site is fully occupied. In other words, it seems likely that strand (βc1 adopts a more rigid structure only when ATP is correctly bound. Consistent with this interpretation is the fact that this segment was only partially modeled in previously reported hTK1 structures that lack ATP.12,15 Interestingly, in the structure by Birringer et al.15 one of the eight monomers in the asymmetric unit did allow for complete tracing of this segment. In fact, the conformation of the 11 residues that we observe in our structure of hTK1 is very similar to that modeled by Birringer et al. Thus, it appears that dTTP whose base and deoxyribose moieties were located in the nucleoside binding site12 cannot mimic the effects of ATP on the conformation of this region.
The finding that the conformation of this flexible region could be dependent on the presence of ATP, combined with the presence of a conserved arginine residue (Arg60 in hTK1), suggests that this region may be important for catalysis. Thus, in an analogous fashion to the so-called “Lid” region in other nucleoside kinases, which functions to supply positively charged residues upon ATP binding, it is likely that the above flexible region in TK1-like kinases fulfills a similar function. The conclusion from these results is that most of the differences between the TmTK and hTK1 structures (i.e. in strands (β3 and βc1, and in the zinc-binding loop) are due to the presence of the phosphoryl donor, mimicked by the TP4A molecule in TmTK and the ensuing difference in the conformation of their quaternary structure. This view is supported by the CaTK structure in complex with ADP (PDB ID 1XX6): the protein adopts an open conformation where the zinc-binding and the putative catalytic loop (i.e. βc1) are in a very similar conformation to that in the TmTK structure.
Quaternary structure
hTK1 elutes as a tetramer from a size-exclusion chromatography column.18 The X-ray structures of both enzymes show the homotetrameric organization of their subunits. In our structures, the asymmetric unit of both the hTK1 and TmTK crystals consists of a single dimmer, in which the two monomers are virtually identical (r.m.s.ds between molecule A and molecule B are 0.28 Å over 160 Cα atoms and 0.23 Å over 164 Cα atoms for hTK1 and TmTK, respectively). Crystallographic 2-fold symmetry forms the tetrameric structure in both hTK1 and TmTK. The overall organization of the subunits of the homotetramer is the same in hTK1 and TmTK. This homotetrameric organization of TK1-like proteins has been reported in other TK1-like structures.12,15,16 An overlay of the two tetramers is shown in Figure 3(a) and in Supplementary Data Figure S1.
Figure 3.
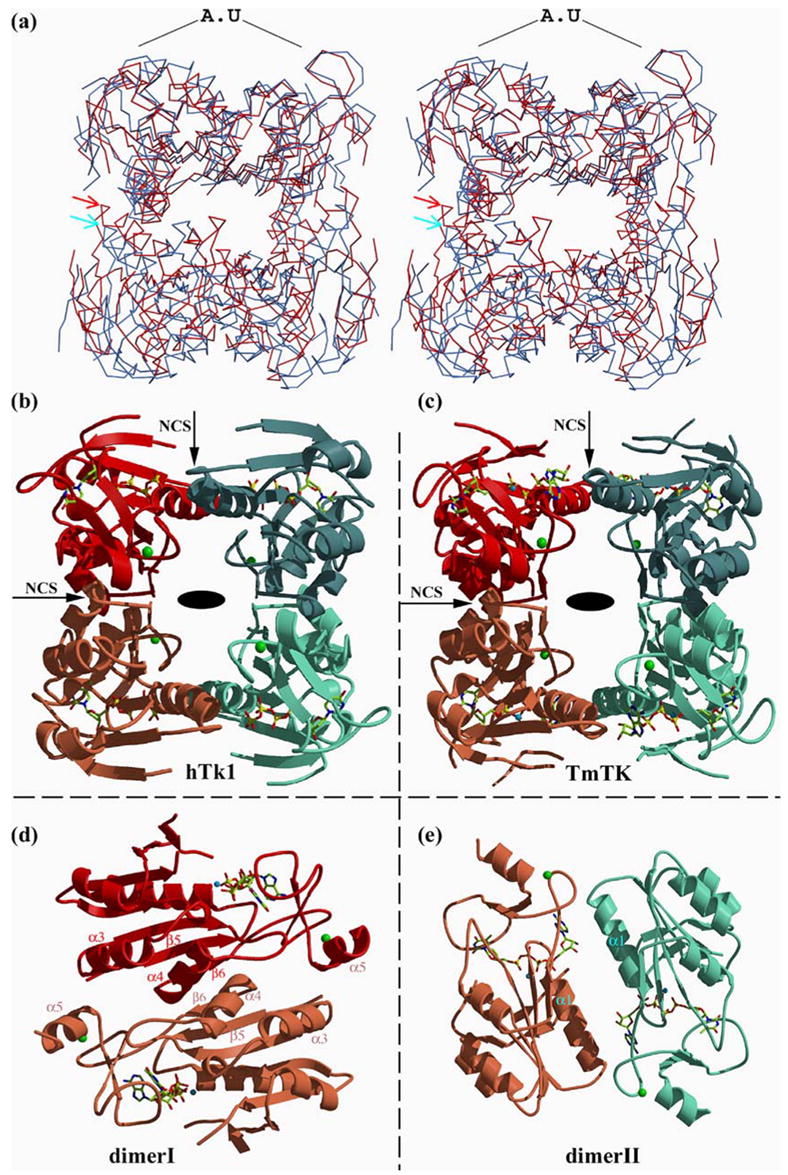
The homotetrameric TK proteins are composed of a dimer-of-dimers. (a) Overlay of the hTK1 (red) and TmTK (blue) tetramers based on the superposition of the corresponding asymmetric unit (AU) dimers. The positions of the helices at the dimer-dimer interface sensitive to ATP are indicated with arrows, and demonstrate the more open conformation of the TmTK enzyme, (b) Ribbon representation of an hTK1 tetramer, where each monomer is colored differently. The part of the TP4A molecules observed is shown, as are the four zinc atoms (green spheres). The crystallographic 2-fold that builds the tetramer by relating the dimer painted in red to the dimer painted in blue is perpendicular to the page (depicted by an oval). The two non-crystallographic symmetry 2-folds are indicated by arrows. Note, there is no 4-fold axis, (c) The analogous representation for the TmTK enzyme, (d) Shown is the dimer with the significantly greater monomer-monomer interactions (see also Table 2), designated as dimer I. Coloring is as in (c), but the view is after a 90° rotation. In this type of dimer, the adenosine moiety is solvent-exposed. The structural elements labeled are those involved in monomer-monomer interactions, (e) The dimer with the smaller interface and fewer inter-subunit polar interactions, designated as dimer II, is shown after a 90° relative rotation to that shown in (c). Residues from both monomers in this dimer flank the adenosine moiety. See the text for details.
The homotetramers possess D2 symmetry (Figure 3(b) and (c)), which indicates that the tetramer is formed by a dimer-of-dimers. Such an arrangement implies the presence of two different types of monomer-monomer interfaces. This is true for both the more open TmTK and the more closed hTK1 enzymes. The monomers in the first type of dimer, dimer I, are related by 2-fold non-crystallographic symmetry (NCS), indicated by the horizontal arrow in Figure 3(b) and (c). The monomers in the second type of dimer, dimer II, are related by another 2-fold NCS indicated by the vertical arrows. In the more open quaternary conformation, as seen in the TmTK-TP4A structure, the monomer-monomer interface in dimer I is significantly more extensive than that present in dimer II, as evaluated by both the surface area buried and the number of polar interactions (Table 2). However, we observe in this structure that the adenosine moiety serves to bridge the two monomers that form dimer II. In fact, the majority of interactions stabilizing dimer II are not direct, but rather go through the adenosine moiety of TP4A and a water network present at the interface of the two monomers.
Table 2.
Interface area and polar interactions of type I and II dimers
| hTK1
|
TmTK
|
|||
|---|---|---|---|---|
| Interface area (Å2) | No. of polar/salt-bridge interactions | Interface area (Å2) | No. of polar/salt-bridge interactions | |
| Dimer I | 1229 | 21 | 1228 | 18 |
| Dimer II | 844 | 5 | 350 (760)a | 2 |
Dimer interface area taking into consideration the contributions of ATP.
Notably, the two adenosine moieties present at the dimer II interface contribute 410 Å2 of the total 760 Å2 interface area. In contrast, in the hTK1-TP4A structure, where the adenosine moiety of TP4A is disordered, the two analogous monomers that form dimer II are closer to each other (Figure 3 (a)) and make several interactions that are absent in the TmTK-TP4A structure. The dimer interface of dimer I still remains larger than that of dimer II, despite the adoption of a more closed quaternary conformation in the hTK1-TP4A structure (Table 2). We conclude that the monomer-monomer interaction is appreciably stronger in the type I dimer (in both structures), and that it is the weaker type II dimer interface that is sensitive to the presence of the phosphoryl donor. This provides a structural explanation to the previous biochemical observation that at low hTK1 concentration (130 nM) ATP induces tetramerization.21,22
The thymidine-binding site
The thymidine-binding sites of hTK1 and TmTK are very similar, as is also reflected by similar Km values for thymidine of 0.5 μM for both enzymes (S. L. et al., unpublished results and21). The thymidine-binding pocket for the two enzymes is presented in Figure 4(a) and (b). As mentioned before, both enzymes have a lasso loop of 17 residues in their C-terminal domain that forms interactions through its main chain with the thymidine molecule. Since the thymidine binding site for hTK1 has been recently described,12,15 we focus on the thymidine-binding site of the thermophilic TmTK.
Figure 4.
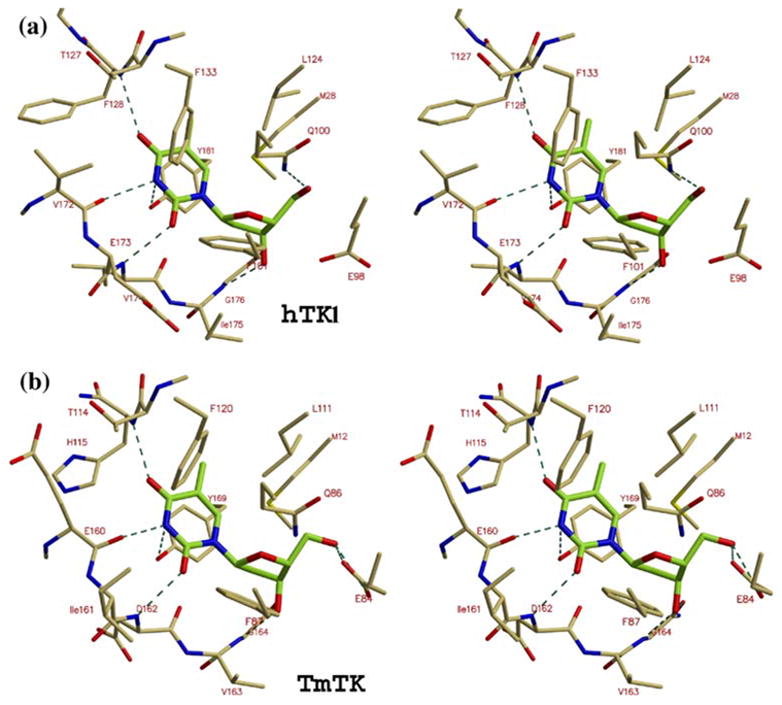
The thymidine-binding site. Stereo views of the thymidine moiety of TP4A (green) bound to (a) hTK1 and (b) TmTK. A special feature of TK1-like kinases is the involvement of main chain atoms in binding the thymine base (hydrogen bonds between the nucleoside and the enzyme are depicted as broken lines). The putative role of the conserved carboxylic acid, Glu98 in hTK1 and Glu84 in TmTK, is to activate the thymidine 5′-hydroxyl for nucleophilic attack on the ATP γ-phosphate.
The lasso loop of the zinc-binding domain, composed by residues 153 to 169, interacts with the 3′-hydroxyl of the deoxyribose and with the polar atoms of the thymidine base. Main chain carbonyl oxygen and nitrogen atoms of the lasso loop make polar interactions with the nucleoside. In addition to these polar interactions, the thymidine moiety of the TP4A molecule packs in a hydrophobic pocket. Notably, the methionine residue, which is located in the P-loop sequence, is conserved in all known TK1-like proteins. The structures of TmTK and hTK1 suggest that this methionine (Met12 in TmTK, Met28 in hTK1) functions to constrain the position of the 5′-hydroxyl of the thymidine, forcing this moiety to approach the conserved Glu84 (equivalent to Glu98 in hTK1). This carboxylic acid is the putative base that acts to increase the nucleophilicity of the thymidine by abstracting a proton from the 5′-hydroxyl group.
The ATP-binding site
The four ATP-binding sites per tetramer are located at subunit interfaces as described above. Only in the TmTK structure could we model the complete TP4A molecule, including the essential magnesium ion, whereas only the four phosphate groups and the thymidine were observed in the hTK1 structure. A possible reason for the lack of electron density for the adenosine moiety of the TP4A molecule is presented in Discussion. The phosphates are bound by the P-loop of one subunit while the ribose and the adenine base are bound between the two helices that build the interface of the second type of dimer (dimer II) in the tetrameric enzyme (Figure 5). Main chain amide groups of the P-loop, which spans residues 10 to 16 in the TmTK sequence, bind the TP4A phosphate groups. The invariant P-loop lysine interacts with one of the oxygen atoms of the β-phosphate.
Figure 5.
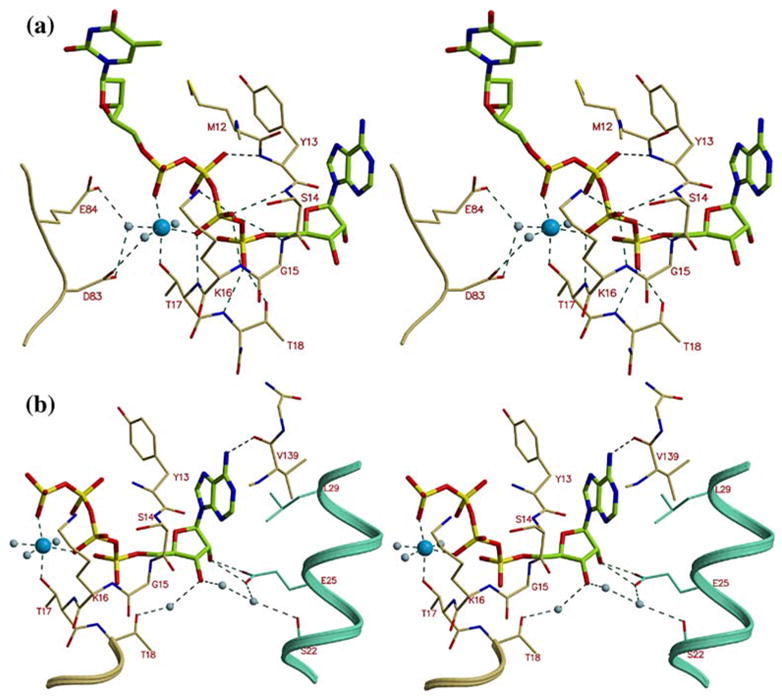
The ATP binding site in TmTK. (a) Stereo view of TP4A bound to TmTK. The phosphate groups bind to the enzyme P-loop (residues 10–16). We observe an octahedrally liganded magnesium atom. The conserved carboxylic acid residues at positions 83 and 84 interact via water molecules with the magnesium, (b) The adenosine moiety is sandwiched between two monomers that make up dimers of type II. The base is flanked by Tyr13 of one monomer and Leu29 of another. Glu25 of the neighboring subunit also directly interacts with the adenosine ribose moiety.
In the hTK1 structure, the phosphate groups of the TP4A molecule are in a different conformation to those of the TP4A bound to TmTK. Overlay of the two structures (Figure 2) shows that the β-phosphate of the TP4A bound to the human enzyme superimposes with the magnesium bound to TmTK. This indicates that the conformation of the phosphate groups adopted in the hTK1-TP4A structure is not compatible with the binding of the divalent cation. Therefore, it is the TmTK-TP4A structure that allows us to visualize all the six ligands in the TK1-like family of proteins. The octahedrally coordinated magnesium ion is liganded by two phosphate groups of the TP4A molecule, the hydroxyl group of Thr17, and three water molecules (Figure 5(a)). The conserved Asp83 and Glu84 position two of the three water molecules that coordinate the divalent cation.
Several monomer–monomer interactions are mediated by the adenosine moiety of TP4A. Specifically, the ribose 2′-hydroxyl group interacts with Glu25 of the neighboring subunit (2.6 Å). In addition, a water network is established between Thr18, the 3′-hydroxyl group of the ribose, and two amino acids of the neighboring monomer, Ser22 and Glu25 (Figure 5(b)). In contrast to the other family of nucleoside kinases, where the adenine base is observed in trans (e.g. in dCK23), the adenine is observed in the syn conformation. In TmTK, the adenine is flanked by Tyr13, located in the P-loop, and by Leu29 of the neighboring subunit. Additionally, the main chain carbonyl oxygen atom of Val139 interacts with N6 (2.9 Å). This interaction clarifies the preference of hTK1-like enzymes for ATP over GTP (Table 3 and S.L. et al, unpublished data). In the case of GTP, the oxygen atom at position six may cause repulsion with the carbonyl oxygen of Val139. An additional potential mechanism that rationalizes the preference of TK1-like kinases for ATP over GTP or pyrimidine nucleoside triphosphates (NTPs) implicates the selection of the base-binding site for the syn conformation of the nucleotide. This conformation is most disfavored by pyrimidine nucleotides, explaining the higher Km for CTP over ATP (Table 3). At the same time, as most of the interactions with the phosphoryl donor take place between the P-loop and the phosphate groups, it is not surprising that these enzymes are able to utilize several NTPs as phosphoryl donors, albeit with lower efficiencies in comparison to ATP.
Table 3.
Steady-state kinetic parameters for hTK1 with various NTPs
| Km(μM) | kcat(S−1) | Hill coef. | |
|---|---|---|---|
| ATP | 45 | 2 | 1.8 |
| GTP | 240 | 1.7 | 1.4 |
| CTP | 150 | 0.3 | 3.4 |
Discussion
This structural study of the T. maritima thymidine kinase and the human thymidine kinase 1 in complex with the bisubstrate inhibitor TP4A analyzes the ATP-binding mode for the thymidine kinase 1-like family. We could fully model the TP4A molecule in the TmTK active site, whereas the adenosine moiety was not visible in the hTK1 complex. The TmTK-TP4A structure reveals that ATP binds at the interface between two monomers of the homotetrameric enzyme (Figure 6(a)). The D2 symmetry present in these enzymes creates two very different interfaces between monomers (Figure 3). ATP binds at the interface with the fewer monomer–monomer interactions (i.e. dimer type II; Figure 3(e)). Each dimer II interface is stabilized by two adenosine moieties, one from each monomer. In fact, most of the interactions between these monomers are mediated via the adenosine moiety. This structural finding is consistent with earlier reports showing that ATP induces tetramerization of human TK1 at very low enzyme concentrations where an equilibrium between dimers and tetramers exists.21
Figure 6.
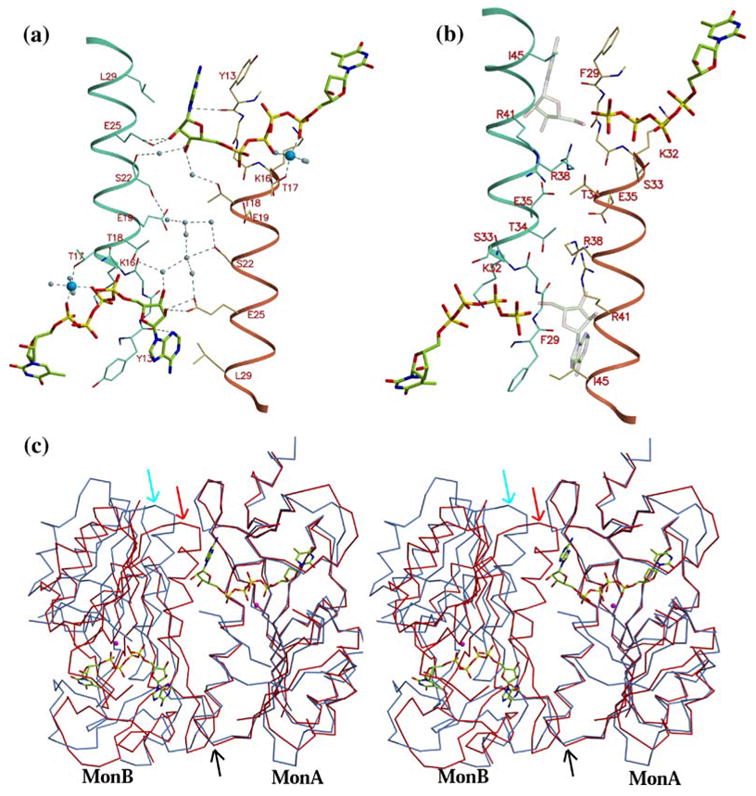
Binding of ATP to TK1-like proteins requires a change in conformation of the quaternary structure to a more open state, (a) TP4A binding to TmTK, looking down a 2-fold axis that relates two binding sites. The adenosine moiety is positioned at the interface of two monomers, mainly composed of helix α1 (shown in cyan and brown). A majority of the monomer–monomer interactions occur via the base and the sugar. The distance between residues that sandwich the adenine ring, originating from different monomers (Tyr13 and Leu29), is ~11 Å. (b) In hTK1, the corresponding distance between Phe29 and Ile45 at the dimer interface is only ~7.5 Å Shown are the thymidine plus four phosphate groups that were observed in the hTK1-TP4A structure. In addition, a model where the adenosine (shown as a transparent object) would lie as based on the TmTK structure demonstrates that the closed quaternary conformation adopted by hTK1 is not compatible with the presence of adenosine at that position, (c) Stereo view showing the superposition of a dimer II of hTK1 (red) and TmTK (blue). The overlay was calculated using Cα atoms only from monomer A, and hence these monomers overlay well (see lower black arrow that points at the coincident interface helix). However, the other monomers that complete the dimer II do not overlay well. This demonstrates that the relative orientation of monA to monB is different in hTK1 versus TmTK (top red and blue arrows point to the corresponding interface helix).
The lack of electron density for the adenosine moiety of the TP4A molecule bound to hTK1 is surprising, taking into account the very high affinity of the bisubstrate analog for hTK1, with a Kd of 29 nM.17 Moreover, the excellent electron density for the thymidine moiety and the four phosphate groups of TP4A confirm the presence of the inhibitor (Figure l(e)). Therefore we are compelled to address the question of why the adenosine moiety of TP4A is not visible in the hTK1 electron density map. The answer lies in differences in the conformation of the quaternary structures of hTK1 and TmTK.
Although the overall tetrameric organization is similar in hTK1 and TmTK, an important distinction is present. The inter-subunit distances between the dimerization helices (α1) are larger in TmTK than in hTK1. The inter-subunit distance between the ct-carbon atoms of the residues Tyr13 and Leu29 that flank the adenine base is 11.3 Å in TmTK (Figure 6(a)). The separation between the equivalent residues in hTK1, Phe29 and Ile45, is 7.5 Å (Figure 6(b)). The consequence of the reduction in separation between the two helices α1 in hTK1 is a lack of space to accommodate the adenosine moiety. Modeling in the hTK1 structure the adenosine moiety of TP4A as observed in the complex with TmTK reveals that Ile45 would clash with the adenine base (Figure 6(b)). In other words, the dimer-of-dimers that forms the tetramer is more open in the case of TmTK in comparison to hTK1, and it is only this open state that is compatible with binding to the adenosine moiety of TP4A.
The inability to observe hTK1 in the open state could be due to the crystallization conditions. These apparently favor the adoption of a closed conformation in the hTK1 tetramer, leaving no space for specific binding of the adenosine group. As a consequence, the adenosine moiety is disordered in our structure, being indicative of its weak binding to hTK1. Based on analogy to TP4A, for ATP the majority of binding energy seems to come from the interactions between the alpha and beta phosphate groups and the enzyme P-loop. Indeed, in TmTK only two hydrogen bonds are formed between the enzyme and the adenosine group, while multiple interactions occur with the phosphate groups.
An additional strong evidence for a conserved mode of ATP binding between hTK1, TmTK, and TK1-like proteins in general, comes from the CaTK structure in complex with ADP (PDB ID 1XX6). While containing the same arginine cluster in helix al, as in the human enzyme, the CaTK-ADP structure reveals that the ADP molecule is bound in a similar fashion as the ADP part of the TP4A molecule bound by TmTK. As predicted by the above discussion, the CaTK adopts the open quaternary structure, which creates the required space for adenosine to bind in the interface of the subunits. Consequently, we propose that for the hTK1 structure, the inability to observe the adenosine group in the ATP-binding site is due to the fact that the enzyme crystallizes in a closed conformation, which is sterically not compatible with ATP/ADP binding. Furthermore, this analysis predicts that hTK1 does indeed adopt the open quaternary state upon ATP binding.
Interestingly, five of the seven available TK1-like structures were obtained in the closed conformation. These include the two previously reported human TK1 structures,12,15 the two TK1-like structures from U. urealyticum12,16 plus the hTK1 structure reported here. Notably, the latest U. urealyticum TK structure reported by Kosinska et al.16 is in the closed conformation, even though the authors attempted to crystallize the enzyme in the presence of thymidine and the ATP analog AppCH2p. Not surprisingly, due to the closed conformation, there was a lack of electron density for the ATP analog while the electron density for the thymidine molecule was clear. Although crystallization was carried out at a pH below 7 for the above structures, this alone cannot explain the predominance of the closed state, since the TmTK-TP4A crystals were obtained at pH 5 but yet revealed the open state.
The closed conformation presumably is inactive due to steric hindrance for binding of the adenosine moiety of ATP; its significance for the enzymatic mechanism of TK1-like proteins is unclear. However, it is possible to foresee a continuous oscillation in the quaternary structure of the enzyme between the closed and open conformation of the tetrameric structure along the catalytic cycle. This oscillation would be possible due to the rather weak interactions between the dimers that build the ATP-binding site (dimer II). Indeed, the limited number of contacts between the subunits that form the ATP-binding site could allow for subunit rearrangement to take place upon ATP binding. Thus, this could provide the structural explanation for the reported modulation of hTK1 activity by ATP at low protein concentrations (by stabilizing the active tetrameric state) and the observed positive cooperativity for ATP (Table 3), by inducing the open state of the tetramer.
Materials and Methods
Protein expression and purification
The human thymidine kinase coding region was cloned in the bacterial expression vector pET14b and expressed as an N-terminal His6-tag fusion protein in Escherichia coli strain BL21(DE3). The cloned hTK1 region encoded the full-length 234 amino acid residue protein. Surface residues Lys203 and Glu204 were substituted by alanine during a screening process for a suitable construct for crystallization.24 This hTK1 mutant has similar kinetic and oligomeric properties to wild-type hTK1 (data not shown).
The bacterial cells were grown at 37 °C until an absorbance of 0.6 at 600 nm was reached. Before induction the temperature was decreased to 25 °C, and 0.3 mM IPTG were then added to the culture in order to induce expression of hTK1. The cells were pelleted by centrifugation, resuspended in lysis buffer (300 mM NaCl, 10% (v/v) glycerol, 25 mM Tris-HCl (pH 7.5), 1% Triton X-100,2 mM MgCl2, 1 mM PMSF) and lysed by sonication. The lysate was centrifuged at 40,000g for 1 h at 4 °C. The supernatant was loaded onto a metal chelating Sepharose column charged with nickel ions (GE Healthcare), and pre-equilibrated with 0.5 M NaCl, 25 mM Tris-HCl (pH 7.5), 2 mM MgCl2. The column was washed with this buffer, and the bound protein was eluted with a step gradient of imidazole. The eluted protein was dialyzed against 0.3 M NaCl, 25 mM Mes (pH 6.0), 5 mM MgCl2, 2 mM DTT+ 0.5 mM Chaps. The histidine tag was cleaved from hTK1 by incubation with thrombin during dialysis. The protein was further purified by gel filtration using an S-200 column (GE Healthcare). Details on the overexpression and purification of TmTK will be described elsewhere (S.L. et al., unpublished).
Crystallization
hTK1 in complex with 1 mM of the bisubstrate inhibitor P1-(5′-adenosyl)P4-(5′-(2′-deoxy-thymidyl)) tetraphosphate (TP4A; Jena Biosciences) or 2 mM of the product thymidine 5′-monophosphate (TMP), and TmTK crystals in complex with TP4A were grown using the hanging-drop vapor diffusion method. For hTK1, the reservoir conditions were: 2.8 M NaCl, 0.1 M Mes (pH 6.0), 0.1 M NaH2PO4, 0.1 M KH2PO4. The crystallization solution was mixed in a 1:1 ratio with the pure protein at a concentration of 10 mg/ml. Crystals grew in two to three days to approximate dimensions of 300 μm × 300 μm × 50 μm. The reservoir conditions for TmTK were 65% 2-methyl-2,4-pentanediol (MPD), 0.1 M Na(OAc)2 (pH 5.0). Equal volumes of the precipitant and the protein (5 mg/ml) were mixed. The crystals grew in one week to approximately 150 μm × 150 μm × 100 μm.
Prior to data collection, hTK1 crystals were briefly incubated in the crystallization solution, plus 20% ethylene glycol as cryoprotectant, followed by flash freezing in liquid nitrogen. For the TmTK crystals the crystallization solution acted as a cryoprotectant.
Structure determination
The hTK1 crystals in complex with TMP were soaked in 5 mM dichloroethylenediamine platinum (II) (Hampton Research) for 24 h. The structure was determined with a single-wavelength anomalous diffraction experiment (SAD) at the inflection wavelength for platinum (1.072 Å). The X-ray diffraction data were reduced with the program XDS.25 The positions of two platinum atoms were determined by the program SOLVE.26 After density modification with RESOLVE,27 an initial experimental electron density map to 3.0 Å resolution was obtained and the phases of this map were extended to 2.3 Å using a native data set. The electron density map enabled us to build a model for hTK1 using the program O.28 The model was refined using the programs CNS29 and REFMAC.30 The model of hTK1 in complex with TMP (to be published elsewhere) was used to refine the structure of hTK1 in complex with TP4A. The TmTK structure was solved by the molecular replacement method using the CaTK structure as the search model (PDB ID 1XX6) with the program MOLREP.31 Structure Figures were generated using Molscript32 and Bobscript33 and rendered with RasterSD.34
In a previous hTK1 structure15 dTTP was observed bound to the nucleoside-binding site despite not being added to the crystallization mixture. The authors explain that this is due to dTTP remaining bound to the protein throughout the purification procedure. We have no evidence for dTTP binding to hTK1 in our preparations. We are confident that we observe the TP4 part of the TP4A molecule, and not a dTTP molecule, due to the excellent density of all four phosphate groups (Figure 1(e)). In addition, we successfully solved additional hTK1 structures that contain thymidine at the nucleoside-binding site (D.S.-P, unpublished data). As the affinity of thymidine to hTK1 is significantly lower than that of the bisubstrate inhibitor TP4A, this implies that if there is any dTTP present from the purification procedure, it does not hinder the formation of the desired (in this case, TP4A) complex.
Steady-state kinetics
The hTK1 enzymatic activity in the forward reaction using thymidine (kept constant at 100 μM) and ATP (varied between 2–1000 μM) as substrates was performed with a spectrophotometric coupled-enzyme assay that measures the decrease in absorbance at 340 nm associated with the oxidation of NADH.35,36 The buffer used to determine the catalytic rates contained 50 mM Tris-HCl (pH 7.5), 100 mM KC1, 5 mM MgCl2. The enzyme concentration was 300 nM. The enzymatic assay was performed in triplicate at 25 °C. Data were fit to the Hill equation using the enzyme kinetics module in SigmaPlot 2000.
Protein Data Bank accession codes
The atomic coordinates have been deposited with the RCSB Protein Data Bank and are available under accession 2ORV for hTK1 and 2ORW for TmTK.
Supplementary Material
Acknowledgments
We are grateful to Michael Godsey and the staff of SERCAT for help in data collection. This work was supported in part by NIH grant AI046943 (to D. S.-R, A.L. and M.K.) and GM69958 (S.L.). M.K. was also supported by the Deutsche Forschungsge-meinschaft and the Max-Planck-Society. We thank Elisabetta Sabini for help in construction of clones, and Ludmilla Shuvalova for access to the robotic crystallization screening instrument and for helpful suggestions.
Abbreviations used
- hTK1
human thymidine kinase 1
- TmTK
Thermotoga maritima thymidine kinase
- CaTK
Clostridium acetobutylicum thymidine kinase
- TP4A
P1-(5′-adenosyl)P4-(5′-(2′-deoxy-thymidyl)) tetraphosphate
- dTTP
deoxythymidine triphosphate
- AppNHp
adenosine-5′-[(β, γ)-imido]triphosphate
Footnotes
Supplementary Data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2007.02.104
Edited by M. Guss
References
- 1.Spyrou G, Reichard P. Dynamics of the thymidine triphosphate pool during the cell cycle of synchronized 3T3 mouse fibroblasts. Mutat Res. 1988;200:37–43. doi: 10.1016/0027-5107(88)90069-3. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson S, Thelander L, Akerman M. Allosteric regulation of calf thymus ribonucleoside diphosphate reductase. Biochemistry. 1979;18:2948–2952. doi: 10.1021/bi00581a005. [DOI] [PubMed] [Google Scholar]
- 3.Boucher PD, Im MM, Freytag SO, Shewach DS. A novel mechanism of synergistic cytotoxicity with 5-fluorocytosine and ganciclovir in double suicide gene therapy. Cancer Res. 2006;66:3230–3237. doi: 10.1158/0008-5472.CAN-05-3033. [DOI] [PubMed] [Google Scholar]
- 4.Bello LJ. Regulation of thymidine kinase synthesis in human cells. Exp Cell Res. 1974;89:263–274. doi: 10.1016/0014-4827(74)90790-3. [DOI] [PubMed] [Google Scholar]
- 5.Tommasi S, Pfeifer GP. Constitutive protection of E2F recognition sequences in the human thymidine kinase promoter during cell cycle progression. J Biol Chem. 1997;272:30483–30490. doi: 10.1074/jbc.272.48.30483. [DOI] [PubMed] [Google Scholar]
- 6.Chang ZF, Huang DY, Chi LM. Serine 13 is the site of mitotic phosphorylation of human thymidine kinase. J Biol Chem. 1998;273:12095–12100. doi: 10.1074/jbc.273.20.12095. [DOI] [PubMed] [Google Scholar]
- 7.Li CL, Lu CY, Ke PY, Chang ZF. Perturbation of ATP-induced tetramerization of human cytosolic thymidine kinase by substitution of serine-13 with aspartic acid at the mitotic phosphorylation site. Biochem Biophys Res Commun. 2004;313:587–593. doi: 10.1016/j.bbrc.2003.11.147. [DOI] [PubMed] [Google Scholar]
- 8.Ke PY, Chang ZF. Mitotic degradation of human thymidine kinase 1 is dependent on the anaphase-promoting complex/cyclosome-CDHl-mediated pathway. Mol Cell Biol. 2004;24:514–526. doi: 10.1128/MCB.24.2.514-526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ke PY, Kuo YY, Hu CM, Chang ZF. Control of dTTP pool size by anaphase promoting complex/cyclosome is essential for the maintenance of genetic stability. Genes Dev. 2005;19:1920–1933. doi: 10.1101/gad.1322905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobrovolsky VN, Bucci T, Heflich RH, Desjardins J, Richardson FC. Mice deficient for cytosolic thymidine kinase gene develop fatal kidney disease. Mol Genet Metab. 2003;78:1–10. doi: 10.1016/s1096-7192(02)00224-x. [DOI] [PubMed] [Google Scholar]
- 11.Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, et al. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc Natl Acad Sci USA. 2004;101:3154–3159. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welin M, Kosinska U, Mikkelsen NE, Carnrot C, Zhu C, Wang L, et al. Structures of thymidine kinase 1 of human and mycoplasmic origin. Proc Natl Acad Sci USA. 2004;101:17970–17975. doi: 10.1073/pnas.0406332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kenny LM, Vigushin DM, AL-Nahhas A, Osman S, Luthra SK, Shousha S, et al. Quantification of cellular proliferation in tumor and normal tissues of patients with breast cancer by [18F] fluorothymidine-positron emission tomography imaging: evaluation of analytical methods. Cancer Res. 2005;65:1014–1012. doi: 10.1158/0008-5472.CAN-04-4297. [DOI] [PubMed] [Google Scholar]
- 14.Barthel H, Perumal M, Latigo J, He Q, Brady F, Luthra SK, et al. The uptake of 3′-deoxy-3′-[18F]fluorothymidine into L5178Y tumours in vivo is dependent on thymidine kinase 1 protein levels. Eur J Nucl Med Mol Imaging. 2005;32:257–263. doi: 10.1007/s00259-004-1611-0. [DOI] [PubMed] [Google Scholar]
- 15.Birringer MS, Claus MT, Folkers G, Kloer DP, Schulz GE, Scapozza L. Structure of a type II thymidine kinase with bound dTTP. FEES Letters. 2005;579:1376–1382. doi: 10.1016/j.febslet.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 16.Kosinska U, Carnrot C, Eriksson S, Wang L, Eklund H. Structure of the substrate complex of thymidine kinase from Ureaplasma urealyticum and investigations of possible drug targets for the enzyme. FEES J. 2005;272:6365–6372. doi: 10.1111/j.1742-4658.2005.05030.x. [DOI] [PubMed] [Google Scholar]
- 17.Bone R, Cheng YC, Wolfenden R. Inhibition of thymidine kinase by P1-(adenosine-S′)-P5-(thymidine-5′)-pentaphosphate. J Biol Chem. 1986;261:5731–5735. [PubMed] [Google Scholar]
- 18.Birringer MS, Perozzo R, Kut E, Stillhart C, Surber W, Scapozza L, Folkers G. High-level expression and purification of human thymidine kinase 1: quaternary structure, stability, and kinetics. Protein Expr Purif. 2006;47:506–515. doi: 10.1016/j.pep.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Barion S, Franchi M, Gallori E, Di Giulio M. The first lines of divergence in the Bacteria domain were the hyperthermophilic organisms, the Thermotogales and the Aquificales, and not the mesophilic Planctomycetales. Biosystems. 2007;81:13–19. doi: 10.1016/j.biosystems.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Mongodin EF, Hance IR, Deboy RT, Gill SR, Daugherty S, Huber R, et al. Gene transfer and genome plasticity in Thermotoga maritima, a model hyperthermophilic species. J Bacterial. 2005;187:4935–4944. doi: 10.1128/JB.187.14.4935-4944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munch-Petersen B, Tyrsted G, Cloos L. Reversible ATP-dependent transition between two forms of human cytosolic thymidine kinase with different enzymatic properties. J Biol Chem. 1993;268:15621–15625. [PubMed] [Google Scholar]
- 22.Berenstein D, Christensen JF, Kristensen T, Hofbauer R, Munch-Petersen B. Valine, not methionine, is amino acid 106 in human cytosolic thymidine kinase (TK1). Impact on oligomerization, stability, and kinetic properties. J Biol Chem. 2000;275:32187–32192. doi: 10.1074/jbc.M005325200. [DOI] [PubMed] [Google Scholar]
- 23.Sabini E, Ort S, Monnerjahn C, Konrad M, Lavie A. Structure of human dCK suggests strategies to improve anticancer and antiviral therapy. Nature Struct Biol. 2003;10:513–519. doi: 10.1038/nsb942. [DOI] [PubMed] [Google Scholar]
- 24.Derewenda ZS, Vekilov PG. Entropy and surface engineering in protein crystallization. Acta Crystallog sect D. 2006;62:116–124. doi: 10.1107/S0907444905035237. [DOI] [PubMed] [Google Scholar]
- 25.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallog. 1993;24:795–800. [Google Scholar]
- 26.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Act Crystallog sect D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallog sect D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallog sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallog sect D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallog. 1997;30:1022–1025. [Google Scholar]
- 32.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallog. 1991;24:946–950. [Google Scholar]
- 33.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graph Model. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 34.Merritt EA, Murphy ME. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallog sect D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 35.Schelling P, Folkers G, Scapozza L. A spectrophotometric assay for quantitative determination of kcat of herpes simplex virus type 1 thymidine kinase substrates. Anal Biochem. 2001;295:82–87. doi: 10.1006/abio.2001.5191. [DOI] [PubMed] [Google Scholar]
- 36.Agarwal KC, Miech RP, Parks RE., Jr Guanylate kinases from human erythrocytes, hog brain, and rat liver. Methods Enzymol. 1978;51:483–90. doi: 10.1016/s0076-6879(78)51066-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.