

Abstract
2-Methoxyestradiol (2ME) is an estradiol metabolite with anti-tumor and anti-angiogenic properties. We studied the effect of 2ME on apoptosis of MCF-7 breast cancer cells and explored a combination therapy using 2ME and a polyamine analogue, bis(ethyl)norspermine (BE-3-3-3). Determination of viable cells on day 4 of treatment with 2ME/BE-3-3-3 combinations showed synergistic effects by Chou-Talalay analysis. APO-BRDU analysis showed that there was only 1.5 ± 0.5 % apoptosis at 200 nM 2ME and 3.7 ± 1.7 % in the presence of 2.5 μM BE-3-3-3. Combination of 200 nM 2ME and 2.5 μM BE-3-3-3 resulted in 52.2 ± 2.6 % apoptosis. Up to 90% of the cells underwent apoptosis in the presence of 1000 nM 2ME and 2.5 μM BE-3-3-3. Combination treatments resulted in total disruption of microtubules and depletion of putrescine, spermidine and spermine. In addition, phosphorylation of Akt and nuclear localization of cyclin D1 were altered by 2ME/BE-3-3-3 combination. Our results suggest an important strategy to induce apoptosis of breast cancer cells, with potential applications in therapy.
Keywords: Polyamine analogue, 2-methoxyestradiol, apoptosis, breast cancer cells
1. Introduction
2-Methoxyestradiol (2ME) is a metabolite of estradiol (E2) with growth inhibitory and apoptotic activity on several experimental models of cancer [1-4]. The mechanism of action of 2ME on normal and neoplastic breast epithelial cells is intriguing since increased production of 2-hydroxyestradiol, which is converted to 2ME by O-methylation, is linked to reduced breast cancer risk [5,6]. In contrast, the metabolic pathways that produce 4-hydroxyestradiol and 16α-hydroxyestrone have been linked to increased risk of breast and uterine cancers [7-9]. Relatively high serum concentrations (4000 pg/ml) of 2ME are found in pregnant women, possibly contributing to the protective effect of pregnancy on breast cancer [10].
At pharmacological concentrations (1 to 50 μM), 2ME is an anti-tumor and antiangiogenic agent, as demonstrated by several studies on pre-clinical cancer models [1-4]. These studies have also led to phase I and phase II clinical trials of 2ME [11,12]. 2ME is known to bind to the colchicine binding site of tubulin. 2ME depolymerizes microtubules in endothelial as well as tumor cells [13,14]. Dose-dependent mechanistic differences, such as G1 arrest or G2/M arrest of cancer cells have also been observed with 2ME [15]. In estrogen receptor (ER) -positive breast cancer cells growing in the presence of estradiol, 2ME exerted anti-proliferative effect, raising the possibility that 2ME could be utilized as an anti-estrogen in a subset of breast cancer [16,17]. 2ME also has a marked sensitivity toward cancer cells compared to normal cells [18]. However, the bioavailability of 2ME is poor so that serum levels in patients do not reach high concentrations required for apoptosis [11]. Therefore, we considered a combination therapy involving a polyamine analogue.
Polyamines --putrescine (H2N(CH2)4NH2), spermidine (H2N(CH2)4NH(CH2)3NH2) and spermine (H2N(CH2)3NH(CH2)4NH(CH2)3NH2-- are organic cations with multiple functions in cell growth and differentiation [19-22]. Polyamine levels are significantly higher in breast tumors compared to adjacent normal tissues [23]. Cellular polyamine levels are delicately regulated by biosynthetic enzymes (ornithine decarboxylase (ODC) and S-adenosylmethionine decarboxylase (SAMDC), catabolic enzymes (spermidine/spermine N1-acetyltransferase (SSAT), and polyamine oxidases) and by uptake/efflux pathways [20]. E2 increased ODC mRNA and enzyme activities in breast cancer cells [24]. We recently found that 2ME could reduce E2-induced increases in ODC activity and polyamine levels [16]. Among the polyamine analogues, bis(ethyl)norspermine (BE-3-3-3) is well characterized and has undergone phase I and phase II clinical trials [25-27].
While polyamine depletion compromises many cellular functions, it is possible that cell survival pathways might be altered by 2ME/BE-3-3-3 combinations. Akt signaling pathway is particularly important in imparting cellular resistance to chemotherapy [28,29]. About 35% of breast cancer patients have increased levels of phosphorylated Akt in their tumors and Akt phosphorylation is associated with poor prognosis for disease-free survival [30,31]. Pre-clinical and molecular biologic studies also show a link between tamoxifen resistance and Akt activation [32,33]. Previous studies showed that E2-induced Akt activation was inhibited by 2ME [16].
We investigated the anti-proliferative effects of 2ME, BE-3-3-3, and their combinations on MCF-7 cells growing in the presence of E2. Cell growth in the presence of E2 was examined because this model mimics the mitogenic effect of E2 as a controlling factor in the growth of ER-positive human breast cancer [34,35]. Combinations of BE-3-3-3 and 2ME showed synergistic anti-proliferative and apoptotic activity, compared to single agents. 2ME down-regulated E2-induced increase in ODC activity and polyamine levels in MCF-7 cells. Confocal microscopic studies showed progressive disruption of microtubules in cells treated with the combination. Our results further showed that 2ME/BE-3-3-3 combination decreased phosphorylation of Akt and reduced nuclear localization of cyclin D1.
2. Materials and Methods
2.1. Materials
MCF-7 cell line was obtained from American Type Culture Collection (Manassas, VA). E2 and 2ME were purchased from Steraloids, Inc. (Wilton, NH). 2ME was further purified by an HPLC method [36] and kindly provided by Dr. B.T. Zhu of the University of South Carolina, Columbia, SC. HPLC analysis of the re-purified 2ME showed a purity of >99%, and no E2, E1 or other metabolite was detected. Putrescine, spermidine, spermine, Dulbecco's modified Eagles medium (DMEM), phenol red-free DMEM, and fetal bovine serum (FBS), were purchased from Sigma Chemical Co. (St. Louis, MO). Antibiotics, trypsin, and other additives for cell culture medium were purchased from Invitrogen (Carlsbad, CA). Anti-α-tubulin and anti-β-actin antibodies were from Sigma. Cyclin D1 was purchased from Neomarker, Fremont, CA. Antibodies specific to p-Akt, total Akt and PTEN were from Cell SignalingTechnology, Beverly, CA.
2.2. Cell Culture
MCF-7 cells were maintained in DMEM containing 10% FBS supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 40 μg/ml gentamycin, 2 μg/ml insulin, 0.5 mM sodium pyruvate, 10 mM nonessential amino acids and 2 mM L-glutamine. MCF-7 cells were grown in phenol red-free DMEM for 1 week prior to all experiments [16]. This medium contained 10% FBS pretreated with dextran-coated charcoal (0.5% Norit A, and 0.05% Dextran T-70) to avoid the effects of serum-derived estrogenic compounds. T-47D cells were maintained in RPMI 1640 medium with serum and antibiotic supplements and grown in phenol red-free DMEM for one week prior to experiments. MDA-MB-231 cells were maintained in DMEM and was not changed to phenol red free medium prior to the experiments.
MCF-7 cells were plated in triplicate at a density of 5 × 104 cells/ml/well in 24-well plates. Cells were dosed 24 h after plating, with required concentrations of E2 and/or 2ME. Cells were re-dosed with a medium change 48 h after the first dose. After appropriate treatment periods, live cells were counted using the trypan blue exclusion method, using a hemocytometer.
2.3. Chou-Talalay Analysis
For combination studies, ratio of the two drugs was set from the IC50 values for the inhibition of cell proliferation. Six doses of 2-ME and BE-3-3-3 were used from serial dilutions covering the IC50 of 2ME and BE-3-3-3. Percentage growth inhibition was calculated and the results plotted according to the median effect equation, fa/fu = (D/Dm)m, where fa is the fraction affected by dose D, fu is the fraction unaffected, D is the dose and Dm is the dose required for 50% growth inhibition and m is the coefficient of sigmoidicity [37]. Combination index (CI) was determined from the CI-isobologram method [38] using the computer program developed by Chou and Martin [39]. The three possibilities: CI < 1, CI = 1, and CI > 1, indicated synergy, additive effect, and antagonism, respectively [37,40].
2.4. APO-BRDU Assay for Apoptosis
MCF-7 cells (1 ×106) were plated in triplicate in 100 mm dishes and allowed to adhere for 24 h. Cells were treated with the appropriate concentrations of E2, 2ME or their combinations and re-dosed with a medium change 48 h after the first dose. Cells were harvested in PBS after 96 h of treatment and fixed in 1% paraformaldehyde. After two washes in PBS, cells were re-suspended in ethanol and stored at −20°C until further analysis. Percentage apoptosis was determined using the APO-BRDU assay kit from BD Biosciences (CA, USA). In this assay, the cell pellet was incubated with brominated deoxyuridine triphosphate (BRDU) and TdT (terminal deoxynucleotidyl transferase) enzyme for 24 h at 28 °C. BRDU incorporated into the 3'-hydroxyl termini of double- and single-stranded DNA was identified by staining the cells with a fluorescent labeled anti-BRDU monoclonal antibody, using flow cytometry (Epics Profile II Flow Cytometer, Beckman Coulter, Inc., Fullerton, CA).
2.5. Cell Cycle Analysis
MCF-7 cells (2 × 106 cells/well) were seeded in 100 mm dishes. Cells were dosed with required amount of 2ME/BE-3-3-3 after 24 h of plating. After the treatment period, cells were rinsed with 1 ml of phosphate buffered saline (PBS). Cells were harvested by adding a citrate buffer (40 mM sodium citrate, 250 mM sucrose, 5% DMSO). Solution A (10 mM Tris.HCl (7.6), containing 50 μg trypsin inhibitor, 10 μg ribonuclease A in 3.4 mM trisodium citrate, 0.1% Nonidet P-40, and 15 mM spermine tetrahydrochloride) was added to the cells and incubated for 10 minutes at 22 °C. Propidium iodide (42 μg/100 ml) solution in citrate buffer was added and cell cycle analysis was conducted by flow-cytometry.
2.6. Confocal Microscopy
Cells (5 × 104 cells/well) were seeded onto Labtek chamber-slide 4-well plates. Cells were allowed to adhere for 48 h before dosing with required concentrations of 2ME/BE-3-3-3. After the treatment period, cells were fixed in ice-cold methanol for 5 min and permeabilized using 0.1% Triton in PBS for 5 min. Cells were subsequently washed with PBS (3 to 5 min) and blocked with 5% goat serum in PBS for 30 min. For microtubule structure, cells were incubated with mouse monoclonal anti-α-tubulin antibody (1:2000) for 2 h. For cyclin D1 staining, cells were incubated with anti-cyclin D1 primary antibody (1:100). Cells were then washed with PBS before incubating with appropriate secondary antibodies conjugated with Alexa Fluor 488 for 1 h (1:400). Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI, 1 nM) solution for 5 min. Cells were then washed with PBS and mounted with an anti-fading agent (Gel Mount, sigma) and cover slips, before confocal microscopic imaging. Cells were imaged by confocal microscopy using a Zeiss LSM-510 laser scanning microscope (Carl Zeiss, Thornwood, NY, U.S.A.).
2.7. Western Blot Analysis
Cell lysate was prepared according to the procedure previously described (16,41). Briefly, monolayers of MCF-7 cells (3.5 × 106/100 mm dish) were washed twice with PBS and lysed by the addition of ice-cold lysis buffer (150 mM Tris.HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 2 mM EDTA, 50 mM sodium fluoride, 0.2% SDS, 100 mM sodium vanadate, 2 mg/ml leupeptin, aprotonin, and pepstatin, and 1 mM phenylmethylsulfonyl fluoride). Thirty-microgram protein (by the Bradford protein assay) was diluted in 2 X SDS-PAGE Laemmli buffer (150 mM Tris base (pH 6.8), 30% glycerol, 4% SDS, 7.5 mM dithiothreitol, 0.01% bromophenol blue) and separated on a 10% SDS-polyacrylamide gel. After electrophoresis, separated proteins were transferred to PVDF Polyscreen membrane. After blocking the nonspecific binding sites with 5% nonfat milk in Tris-buffered saline, containing 0.1% Tween-20, the membrane was immunoblotted with a 1:200 dilution of the primary antibody. Antibodies specific to cyclin D1, p-Akt, total Akt, PTEN, and β-actin proteins were used in this study. Protein bands were visualized using a horseradish peroxidase conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and SuperSignal West Peco Chemiluminescent Substrate (Pierce, Woburn, MA). Same membranes were used sequentially to detect several proteins by stripping and reblotting. To verify equal protein loading, membranes were re-blotted with an anti-β-actin monoclonal antibody.
2.8. Polyamine Assay
Polyamine levels were quantified by a previously described procedure using a Hitachi L-7000 HPLC unit (24). MCF-7 cells were plated at a density of 3 × 106 cells/100 mm dish, and treated with E2, 2ME, BE-3-3-3 or their combinations for 48 h. Cells were homogenized in 8% sulfosalicylic acid and centrifuged at 5,000 × g. Polyamines were converted to dansyl derivatives and separated on a C18 analytical column with acetonitrile-water gradient, and quantified by a fluorescence detector. 1,6-Diaminohexane was used as the internal standard.
2.8. ODC Activity Assay
MCF-7 cells (3 × 106 cells/dish) were plated in 100 mm dishes. Cells were treated with E2, 2ME, BE-3-3-3 or their combinations. ODC assay was performed as described previously (24). Briefly, cell pellets were sonicated on ice for 10 seconds in 1 ml of Tris buffer (10 mM Tris.HCl, pH 7.4, 2.5 mM dithiothreitol). After centrifugation, supernatants were used for the assay. Ornthine mix containing 6.5 μCi/ml of 14C-ornithine and 22 mM unlabeled ornithine in 10 mM Tris.HCl (pH 7.4) and pyridoxal-5-phosphate (2 mM) were added to tubes fitted with rubber stoppers with center wells (Kontess Glass Company, Vineland, NJ). Hyamine hydroxide was used to trap 14CO2and radioactivity determined using a scintillation counter. ODC activity was calculated as nmols/mg protein/hour.
2.9. Statistical Analysis
All experiments were repeated at least 3 times. All blots shown are representative of 3 separate experiments with mean of fold changes in intensities reported in the text. Statistical difference between control and treatment groups was determined by one way analysis of variance (ANOVA) followed by Dunnet's test or Tukey's test (GraphPad Prism Software program, San Diego, CA). A P value of < 0.05 was considered to be statistically significant.
3. Results
3.1. Cell Growth
We first determined the dose-dependent effects of 2ME on the growth of MCF-7 cells growing in the presence of 10 nM E2. Cells were seeded on 24-well culture plates and dosed with different concentrations of 2ME. Cell growth was determined on day 4 of treatment by trypan blue exclusion assay. Figure 1A shows that 2ME inhibited the growth of MCF-7 cells with an IC50 of 180 nM. Figure 1B shows the effect of BE-3-3-3 on the growth of MCF-7 cells. Our results showed that MCF-7 cells were growth inhibited by BE-3-3-3 with an IC50 of 1.6 μM.
Figure 1.
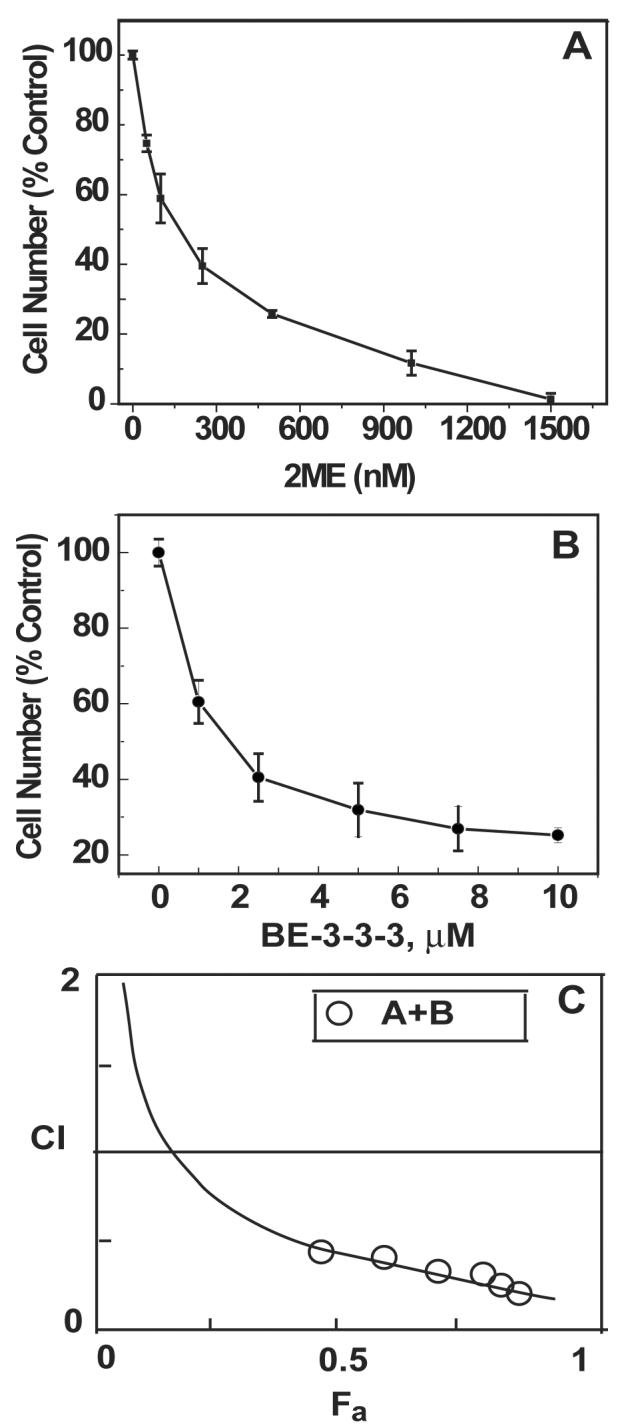
Dose-dependent effect of 2ME (A) and BE-3-3-3 (B) on cell proliferation and the combination index (CI) plot (C). Experiments were conducted in the presence of 10 nM E2. Cell number was determined on day 4 of treatment by trypan blue exclusion assay. For combination experiments, six doses of 2-ME and BE-3-3-3 were used from serial dilutions covering the IC50 values. CI analysis was conducted as described in Materials and Methods. CI were < 1 for all combinations. Data are ± SEM from three separate experiments.
We next examined possible synergistic growth inhibitory interactions between 2-ME and BE-3-3-3 using the Chou-Talalay method [38]. The IC50 values of 2ME and BE-3-3-3 were used to design experiments to determine synergistic anti-proliferative interaction between these compounds. Six doses of the agents, ranging from 25 to 300 nM 2ME and 200 to 2,400 nM BE-3-3-3, were used for combination studies. The combination index (CI) plot is given in Figure 1C. Values representing fractional inhibition and dose-reduction are presented in Table 1 of Supplementary Material. Our results suggest that as much as 14-fold reduction in the dose of BE-3-3-3 might be possible due to combination with 2ME. As shown in Figure 1C, CI values range from 0.25 to 0.5, indicating strong synergism in the growth inhibitory effect.
3.2. Apoptosis
Apoptosis of MCF-7 cells was examined in the presence of 2ME and BE-3-3-3. Cells growing in the presence of E2 were treated with 0, 200, 500, and 1000 nM 2-ME with or without 1, 2.5 and 5 μM BE-3-3-3. Cells were harvested after 4 days of treatment. Figure 2 shows the effects of combination treatments on apoptosis of MCF-7 cells. Combination of 2ME with 1 μM BE-3-3-3 did not increase apoptosis compared to that of cells treated with 2ME alone. In contrast, cells treated with 200, 500, and 1000 nM 2ME showed a remarkable increase in apoptosis in combination with 2.5 μM or 5 μM BE-3-3-3. Indeed, 52 and 78% of cells underwent apoptosis in the presence of 200 and 500 nM 2ME, respectively, when combined with 2.5 μM BE-3-3-3. Apoptosis was less than 5% when cells were treated with these concentrations of 2ME or BE-3-3-3 as single agents. Increase in apoptosis was also seen in cells treated with 5 μM BE-3-3-3 in combination with 2ME, although this concentration of BE-3-3-3 also induced significant levels of apoptosis as a single agent.
Figure 2.
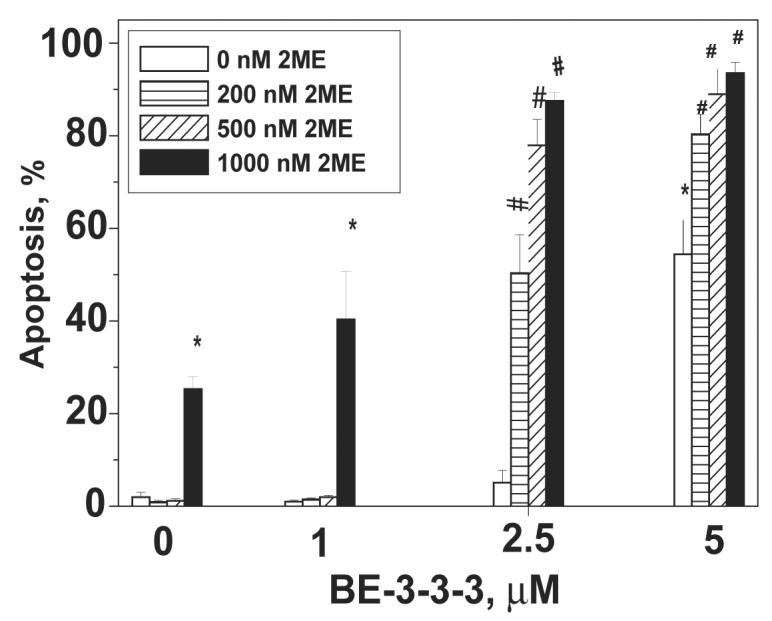
Apoptosis of MCF- 7 cells in the presence of 2ME/BE-3-3-3 combinations. Cells were treated with 2ME, BE-3-3-3, or their combinations, 24 h after plating. All of the groups also received 10 nM E2. Apoptosis was quantified using APOP-BRDU kit using flow cytometry. Data are mean ± SE from three separate experiments. * Statistically significant compared to the control (P < 0.01). #Statistically significant compared to groups treated with 2ME or BE-3-3-3 as single agents (P < 0.01).
3.3. Cell Cycle
2ME has been reported to induce accumulation of cells in G2/M phase for a number of cell types and G1 or S phase arrest in some cases [1,15,42]. We therefore examined the effect of 2ME, We conducted cell cycle analysis at 48, 72 and 96 h of treatment. However, 72 h of treatment provided maximal changes in the percentages of distribution of cells. Table 1 shows the percentages of distribution of cells in G1, S and G2/M phases of the cell cycle. E2 treatment induced a significant decrease of cells in G1 phase and a concomitant increase in S phase, as expected from its growth stimulatory effects. 2ME at 200 and 500 nM concentrations showed a trend toward increase of cells in G1 phase, compared to the E2 treatment group. However, the increase in G1 phase cells was significant at 1000 nM 2ME. BE-3-3-3 treatment resulted in significant accumulation of cells in G1 phase at 2.5 and 5 μM concentrations. There was also a significant decrease in S phase cells. Combinations of 2ME with BE-3-3-3 maintained the accumulation of cells in G1 phase.
Table 1.
Cell Cycle distribution of MCF-7 cells treated with 2ME/BE-3-3-3
| Treatments | G1 Phase | S Phase | G2/M Phase |
|---|---|---|---|
| Control | 67.2 ± 3.06 | 15.8 ± 2 | 12.8 ± 1.3 |
| E2 | 55.9 ± 1.9# | 22.2 ± 0.5# | 15.3 ± 1.3 |
| 200 nM 2ME | 60.25 ± 1.26 | 20.25 ± 0.6 | 14.2 ± 0.9 |
| 500 nM 2ME | 61.4 ± 1.36 | 19.7 ± 0.7 | 14.2 ± 0.9 |
| 1000 nM 2ME | 63.7 ± 1.8* | 16.9 ± 1.2 | 12.7 ± 0.7 |
| 2.5 μM BE-3-3-3 | 69.1 ± 2.5** | 14.6 ± 1.8** | 12.2 ± 0.4 |
| 5 μM BE-3-3-3 | 72.9 ± 2.1** | 11.7 ± 1.4** | 11.6 ± 0.8 |
| 200 nM 2ME + 2.5 μM BE-3-3-3 | 69.8 ± 1.9** | 13.6 ± 1.9** | 11.1 ± 0.2 |
| 500 nM 2ME + 2.5 μM BE-3-3-3 | 68.9 ± 1.2** | 12.8 ± 1.6** | 11.6 ± 0.4 |
| 1000 nM 2ME + 2.5 μM BE-3-3-3 | 67.8 ± 0.7** | 11.9 ± 1.3** | 11.6 ± 0.8 |
| 200 nM 2ME + 5 μM BE-3-3-3 | 73.3 ± 1.06** | 11.2 ± 1.0** | 11.2 ± 1 |
| 500 nM 2ME + 5 μM BE-3-3-3 | 76 ± 1.3** | 9.25 ± 1.2** | 10.5 ± 0.8 |
| 1000 nM 2ME + 5 μM BE-3-3-3 | 76.4 ± 1.06** | 7.8 ± 1.1** | 10 ± 0.7 |
All samples except the control also contained 10 nM E2. Cells were treated for 72 h with appropriate concentrations of E2 , 2ME and BE-3-3-3 with re-dosing at 48 h. Total number of cells will also include a sub-G1 fraction accounting for 5 to 10% of the cells.
Indicates statistical significance compared to the control (P < 0.05).
Indicates statistical significance compared to E2-treated samples (P < 0.05).
Indicates statistical significance compared to E2-treated samples (P < 0.01). Data are mean ± SE from three separate experiments.
3.4. Cyclin D1 Levels and its Nuclear Localization
Since we observed G1 arrest due to 2ME/BE-3-3-3 treatment, we examined cyclin D1 levels by Western blots. There was no statistically significant change in cyclin D1 protein level at 24 h of treatment with 2ME, BE-3-3-3 or their combinations (results not shown). Recent studies indicate that nuclear/cytoplasmic shuttling of cyclin D1 is important for cell cycle progression [43]. We therefore examined the effect of 2ME and BE-3-3-3 on the nuclear/cytoplasmic localization of cyclin D1 protein by immunofluorescence microscopy. Cells were treated with 200 nM 2ME and 2.5 μM BE-3-3-3 for 24 h. Cells were examined under a confocal microscope and the number of cells with cyclin D1 either in the cytoplasmic or nuclear compartments were quantified. Our results show that E2 treatment increased the number of cells with nuclear cyclin D1 (Figure 3). Treatment of cells with 200 nM 2ME or 2.5 μM BE-3-3-3 did not show significant changes in the number of cells with nuclear cyclin D1. In contrast, combination treatment with 2ME and BE-3-3-3 resulted in a significant decrease in the number of cells with cyclin D1 in the nucleus. These results indicate that growth inhibitory/apoptotic effects of 2ME/BE-3-3-3 combination on MCF-7 might involve the exclusion of cyclin D1 from nucleus.
Figure 3.

Effect of E2, 2ME and BE-3-3-3 on the nuclear localization of cyclin D1 in MCF-7 cells. Cells were plated in Labteck chamber slides and treated with E2, or E2 + 2ME, or E2 + BE-3-3-3 or E2 + 2ME + BE-3-3-3 for 24 h. Immunocytochemical staining was conducted using anti-cyclin D1 antibody and Alexa-488-labeled secondary antibody (Right Panels). Nuclei were stained with DAPI (Left Panels). Cells with high nuclear cyclin D1 were scored and plotted. Approximately 75 cells each from three separate triplicate experiments were quantified. *Indicates statistically significant compared to the control. # Indicates statistically significant compared to E2-treated group (P < 0.05).
3.5. Akt Pathway
Since E2 is known to induce Akt phosphorylation as a part of its ability to stimulate cell growth and prevent apoptosis [44,45], we next examined the effect of 200 nM 2ME and 2.5 μM BE-3-3-3 on the level of Akt phosphorylation. Treatment with E2 resulted in increased levels of Akt phosphorylation 24 h after its addition (Figure 4). Treatment with 200 nM 2ME resulted in 30 to 40% decrease in Akt phosphorylation. Phospho-Akt levels were not altered by 2.5 μM BE-3-3-3. Combination treatment resulted in 85% reduction of phospho-Akt levels compared to the E2-treatment group. Total Akt protein level was determined by re-probing the blot with an antibody that recognized total Akt protein. BE-3-3-3 and 2ME/BE-3-3-3 combination reduced total Akt protein levels by 10 to 15% compared to E2-treatment group. Since phosphorylation of Akt is also controlled by upstream phosphatase and tensin homologue deleted from chromosome 10 (PTEN) [29,45], we examined PTEN levels by re-probing the same blot with an antibody that recognized PTEN. Our results showed that PTEN levels did not change with treatment. In addition, re-probing with anti-β-actin antibody showed equal levels of protein in control and treatment groups, indicating equal protein loading on the gel and transfer to membrane. These results suggest that 2ME/BE-3-3-3 combination has the capacity to down-regulate Akt phosphorylation, thereby contributing to apoptosis sensitivity.
Figure 4.
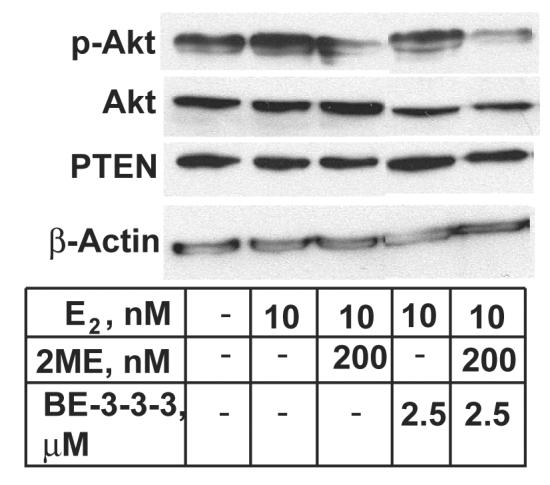
Effect of E2, 2ME and BE-3-3-3 on Akt phosphorylation levels in MCF-7 cells. Cells were treated with E2, or E2 + 2ME/BE-3-3-3 combinations for 24 h. Cells were harvested and the lysate was analyzed by sequential Western blots using antibodies specific to phospho-Akt, total Akt, PTEN and β-actin. Similar results were obtained from 3 separate experiments.
3.6. Microtubule Structure
We next determined the effect of 2ME, BE-3-3-3 and their combination on microtubule structure using confocal microscopy. Figure 5 shows representative confocal images of microtubule structures at 24, 48 and 96 h of treatment. Minor changes in the microtubule structure were observed for 200 and 1000 nM 2ME at 24 h of treatment. BE-3-3-3 treatment also induced minor changes in microtubules at this time point. Combination treatments showed further loss of microtubules at the 24 h time point. At the 48 h time point, cells treated with combinations showed a remarkable disruption and loss of microtubules compared to the control. BE-3-3-3 also caused a reduction of microtubules at 48 and 96 h of treatment. At 96 h of treatment, there was a considerable loss of microtubules in cells treated with 200 nM 2ME or 2.5 μM BE-3-3-3. However, microtubules were completely destroyed in cells treated with 500 nM 2ME + 2.5 μM BE-3-3-3 or 1000 nM 2ME + 2.5 μM BE-3-3-3. These results indicate that apoptosis induced by 2ME/BE-3-3-3 combinations involves a severe loss of microtubule structure.
Figure 5.

Effects of 2ME, BE-3-3-3, and their combinations on microtubules in MCF-7 cells. Cells were plated in Labteck chamber wells. All of the groups were treated with 10 nM E2. After treatment with 2ME, BE-3-3-3 or their combinations cells were fixed and processed for immunofluorescence using anti-α-tubulin antibody and secondary antibody conjugated with Alexa-488. Control and treatment groups were processed at 24, 48, or 96 h after the addition of E2 and drugs. By 96 h, control cells growing in the presence of E2 appeared as large colonies, so that individual cells could not be imaged for microtubules as it was done for 24 h treatment group. Similar results were obtained in three separate experiments.
3.7. Polyamine Pathway
In order to examine whether apoptotic concentrations of 2-ME and BE-3-3-3 can produce additive or synergistic depletion of polyamines, we determined polyamine levels in cells treated with these compounds individually and in combination. Table 2 shows changes in polyamine levels induced by 2ME and BE-3-3-3 after 48 h of treatment. E2-induced increase in polyamine levels were down-regulated by 200 nM 2ME and this was not further reduced by 500 or 1000 nM 2ME. BE-3-3-3 at 2.5 μM concentration, reduced spermidine and spermine levels significantly, whereas decrease in putrescine level was not significant. However, at 5 μM BE-3-3-3, putrescine, spermidine, and spermine levels decreased significantly. In general, combinations of 2ME and BE-3-3-3 yielded more severe depletion of putrescine, spermidine and spermine compared to individual treatments, with some exceptions, such as putrescine at 200 nM 2ME + 2.5 μM BE-3-3-3. In this case, putrescine level was suppressed to the level of control without E2, but no further reduction was found. In contrast, spermidine and spermine levels were reduced by 60 and 41%, respectively, in the presence of 200 nM 2ME + 2.5 μM BE-3-3-3, compared to the control group without E2. Higher concentrations of 2ME (500 and 1000 nM) and BE-3-3-3 (2. 5 or 5 μM) produced progressive depletion of all three polyamines by 48 h of treatment.
Table 2.
Effects of 2ME and BE-3-3-3 on polyamine levels in MCF-7 cells
| Treatment | Putrescine | Spermidine | Spermine | ||
|---|---|---|---|---|---|
| E2 | 2ME | BE-3-3-3 | nmoles/mg/protein | ||
| nM | nM | μM | |||
| 0 | 0 | 0 | 1.32 ± 0.12 | 9.02 ± 1.8 | 6.5 ± 0.4 |
| 10 | 0 | 0 | 1.7 ± 0.10# | 12 ± 0.5# | 11.7 ±1.3# |
| 10 | 200 | 0 | 1.0 ± 0.15* | 6.3 ± 0.98* | 7.5 ± 1.5* |
| 10 | 500 | 0 | 1.1 ± 0.17* | 8.3 ± 0.48* | 8.2 ± 1.1* |
| 10 | 1000 | 0 | 1.2 ± 0.18* | 6 ± 1.0* | 6.8 ± 1.0* |
| 10 | 0 | 2.5 | 1.42 ± 0.16 | 6.1 ± 1.1* | 5.2 ± 0.5* |
| 10 | 0 | 5 | 0.4 ± 0.04* | 2.8 ± 0.7* | 2.3 ± 0.4* |
| 10 | 200 | 2.5 | 1.3 ± 0.3* | 3.6 ± 0.6** | 3.8 ± 1.0** |
| 10 | 500 | 2.5 | 0.88 ± 0.06* | 2.15 ± 0.38** | 2.3 ± 0.05** |
| 10 | 1000 | 2.5 | 0.54 ± 0.08** | 1.38 ± 0.24** | 2.6 ± 0.5** |
| 10 | 200 | 5 | 0.45 ± 0.06* | 1.7 ± 0.46* | 2.4 ± 0.5* |
| 10 | 500 | 5 | 0.48 ± 0.09* | 1.09 ± 0.20** | 2.67 ± 0.8* |
| 10 | 1000 | 5 | 0.21 ± 0.05** | 1 ± 0.05** | 2.04 ± 0.4* |
Cells were treated for 48 h with appropriate concentrations of E2, 2ME and BE-3-3-3 and harvested.
Indicates statistically significant compared to the control (P < 0.05).
Indicates statistically significant compared to E2-treated samples (P < 0.05).
Indicates statistical significance compared to 2-ME and BE-3-3-3 treatment groups (P < 0.05). Data are mean ± SE from three separate experiments.
Determination of ODC activity at 48 h of treatment showed that ODC activity was down-regulated with increasing concentrations of 2ME (Figure 6). BE-3-3-3 also reduced E2-induced ODC activity. In cells treated with 200 nM 2ME and 2.5 μM BE-3-3-3, there was further reduction of ODC activity compared to that in the presence of single agents. Determination of SSAT activity showed 3- to 5-fold increase in the presence of BE-3-3-3, but there was no further increase due to 2ME/BE-3-3-3 combination (results not shown). These results indicate that down-regulation of ODC activity is associated to polyamine depletion and overall effects of 2ME and BE-3-3-3.
Figure 6.

Effect of E2, 2ME, and BE-3-3-3 on ODC enzyme activity in MCF-7 cells. Cells were treated with E2, or E2 + 2ME/BE-3-3-3 combinations for 48 h. Cells were harvested and ODC activity determined using 14C-ornithine by measurement of 14CO2 released. Data are mean ± SE from 3 separate experiments. #Statistically significant compared to the control (P < 0.01) *Statistically significant compared to E2-treatment group (P < 0.01). &Statistically significant compared to individual treatment groups (P < 0.05).
We have been testing the effect of 2ME and BE-3-3-3 on E2-induced cell proliferation, using MCF-7 cells as a model system. Expression of ERα in MCF-7 cells enables these cells to respond E2, stimulating its proliferation [16]. In order to examine whether effects of 2ME and BE-3-3-3 is reproducible in another ER-positive cell line, we examined the effects of 2ME and BE-3-3-3 individually and in combination on apoptosis of T-47D cell line. This cell line has both ERα and ERβ and is growth stimulated by E2 [16]. These results confirmed the increased apoptotic activity of 2ME/BE-3-3-3 combinations compared to that of single agents (Figure 1 of the Supplementary Material). We also examined the anti-proliferative activity of 2ME/BE-3-3-3 combinations on an ER-negative cell line MDA-MB-231 (Figure 2 of the Supplementary Material) These results indicated that breast cancer cell proliferation stimulated by serum factors and other mitogenic agents was also sensitive to 2ME/BE-3-3-3 combination. Thus, our observation of synergistic interaction between 2ME and BE-3-3-3 is not unique to ER-positive cell line; instead these agents are acting on common growth regulatory pathways available on both subtypes of breast cancer cell lines.
4. Discussion
Our results show synergism in the interaction of 2ME and BE-3-3-3 in inducing apoptosis of MCF-7 cells. 2ME/BE-3-3-3 combinations reduced the level of Akt phosphorylation and the number of cells with nuclear localization of cyclin D1, compared to E2-treated control cells. Treatment with 2ME/BE-3-3-3 combinations also led to a remarkable loss of microtubules, compared to single agents. 2ME suppressed E2-induced increases in ODC activity and polyamine levels. Combinations of 2ME and BE-3-3-3 produced progressive decreases in intracellular levels of putrescine, spermidine, and spermine. These results indicate that down-regulation of Akt phosphorylation, enhanced disruption of microtubules, and polyamine depletion are associated with synergistic apoptosis of MCF-7 cells in the presence of 2ME and BE-3-3-3.
2ME is known to bind to colchicine binding site of tubulin dimers and cause depolymerization of microtubules [13,14]. Recent studies show that selection of MDA-MB-231 cells for 2ME resistance resulted in a mutation of β-tubulin gene, indicating the importance of tubulin binding in the mechanism of action of 2ME [2]. Polyamine depletion exacerbated 2ME effects on tubulin depolymerization because neutralization of negatively charged sites is important in the dynamic process of tubulin polymerization and de-polymerization [46,47]. Disruption of microtubules could initiate signaling pathways that result in dephosphorylation of Akt, and initiation of apoptosis [48,49].
Effects of 2ME at pharmacological concentrations demonstrated G2/M arrest in some cell types, although G1 and S phase arrests are also reported [1,15,42]. Microtubule-disrupting agents are generally considered to exert G2/M arrest; however, in some cell types, cells exit mitosis without undergoing cytokineses [50]. These cells then enter an abnormal G1 phase and accumulate in G1 phase, probably due to a microtubule sensitive G1 check-point. Our results show significant increase in cells in G1 phase of the cell cycle in the presence of 1000 nM 2ME or 2.5 to 5 μM BE-3-3-3, with a corresponding decrease in S phase cells. In the case of melanoma cells, G1 arrest in the presence BE-3-3-3 was also observed in a previous report [51]. Combinations did not further increase the percentage of cells in G1 phase compared to treatment with single agents, although G1 arrest was maintained. Thus, our results support a model of apoptosis of MCF-7 cells, associated with G1 phase arrest and microtubule disruption.
Our results show that 2ME/BE-3-3-3 combination reduced the level of Akt phosphorylation compared to individual treatment groups. Akt phosphorylation in response to E2 has been reported to be occurring through Ras signaling and activation of PI3K [44,52]. Pre-clinical and molecular biologic studies show that Akt activation has a key role in the resistance of breast cancer cells to tamoxifen and other forms of chemotherapy [31-34]. Down-regulation of Akt pathway appears to sensitize breast cancer cells to apoptosis. Phosphorylation of Akt and the activation of down-stream kinases may lead to nuclear localization of cyclin D1 [43,45]. E2 increased the number of cells containing nuclear cyclin D1, as expected from the increased activation of Akt. As single agents, 2ME and BE-3-3-3 did not significantly change the number of cells containing nuclear cyclin D1. However, combination treatment resulted in a significant decrease in the number of cells with nuclear localization of cyclin D1. These results indicate that cyclin D1 re-distribution is important to the anti-proliferative effects of 2ME and BE-3-3-3.
ODC activity was down-regulated by 2ME at 200 nM concentration. BE-3-3-3 also reduced ODC activity, and the combinations further reduced ODC activity compared to individual treatment groups. Polyamine levels also showed consistent decreases in combination treatment groups. Other studies showed down-regulation of spermidine synthase in 2ME- treated multiple myleoma cells [53]. Thus, factors other than down-regulation of ODC activity might contribute to polyamine depletion.
In ER-positive breast cancer cells, 2ME down-regulated estrogenic functions such as Akt phosphorylation and activation of polyamine pathway and this might lead to synergistic anti-proliferative action of BE-3-3-3. However, the synergistic effect of 2ME/BE-3-3-3 combination was not exclusive to ER-positive cells. Therefore, other pathways in the action of 2ME and BE-3-3-3 might also be relevant. One such pathway is the generation of reactive oxygen species during the degradation of polyamines as well as during the metabolism of 2ME [54,55]. Combinations of 2ME and BE-3-3-3 might therefore reduce Akt activation linked to redox regulation. In ER-positive cells ER is reported to be involved in the redox regulation of Akt [56]. Combinations of 2ME and BE-3-3-3 need to be further explored for utilization in breast cancer therapy as they are linked to a number of pathways in cell growth regulation and apoptosis.
In summary, our results demonstrate synergistic apoptosis of breast cancer cells due to combination treatment with 2ME and BE-3-3-3. Increased apoptosis was associated with enhanced microtubule disruption as well as increased polyamine depletion. Akt phosphorylation was also down-regulated in the combination group, compared to individual treatment groups, concomitant with a decrease in nuclear localization of cyclin D1. Our study demonstrates interactions between the mechanistic pathways involved in the action of 2ME and BE-3-3-3 in breast cancer cells. Combinations of 2ME and BE-3-3-3 might allow the use of low doses of BE-3-3-3 and alleviate problems due to low bioavailability of 2ME and achieve favorable therapeutic index.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
This work was supported by NIH Grants CA42439, CA80163, and CA73058 from the National Cancer Institute, and ES05022 from the National Institute of Environmental Health Sciences (NIEHS Center Excellence). We thank Dr. Bao T. Zhu of University of South Carolina, Columbia, South Carolina for providing HPLC purified 2-methoxyestradiol for our experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mooberry SL. New insights into 2-methoxyestradiol, a promising antiangiogenic and antitumor agent. Curr. Opin. Oncol. 2003;15:425–430. doi: 10.1097/00001622-200311000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Gokmen-Polar Y, Escuin D, Walls CD, Soule SE, Wang Y, Sanders KL, Lavallee TM, Wang M, Guenther BD, Giannakakou P, Sledge GW., Jr. β-Tubulin mutations are associated with resistance to 2-methoxyestradiol in MDA-MB-435 cancer cells. Cancer Res. 2005;65:9406–9414. doi: 10.1158/0008-5472.CAN-05-0088. [DOI] [PubMed] [Google Scholar]
- 3.Han GZ, Liu ZJ, Shimoi K, Zhu BT. Synergism between the anticancer actions of 2-methoxyestradiol and microtubule-disrupting agents in human breast cancer. Cancer Res. 2005;65:387–393. [PubMed] [Google Scholar]
- 4.Ricker JL, Chen Z, Yang XP, Pribluda VS, Swartz GM, Van Waes C. 2-Methoxyestradiol inhibits hypoxia-inducible factor 1alpha, tumor growth, and angiogenesis and augments paclitaxel efficacy in head and neck squamous cell carcinoma. Clin. Cancer Res. 2004;10:8665–8673. doi: 10.1158/1078-0432.CCR-04-1393. [DOI] [PubMed] [Google Scholar]
- 5.Fowke JH, Qi D, Bradlow HL, Shu XO, Gao YT, Cheng JR, Jin F, Zheng W. Urinary estrogen metabolites and breast cancer: differential pattern of risk found with pre-versus post-treatment collection. Steroids. 2003;68:65–72. doi: 10.1016/s0039-128x(02)00116-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhu BT, Conney AH. Is 2-methoxyestradiol an endogenous estrogen metabolite that inhibits mammary carcinogenesis? Cancer Res. 1998;58:2269–2277. [PubMed] [Google Scholar]
- 7.Zheng W, Xie DW, Jin F, Cheng JR, Dai Q, Wen WQ, Shu XO, Gao YT. Genetic polymorphism of cytochrome P450-1B1 and risk of breast cancer. Cancer Epidemiol. Biomarkers Prev. 2000;9:147–150. [PubMed] [Google Scholar]
- 8.Liehr JG, Ricci MJ, Jefcoate CR, Hannigan EV, Hokanson JA, Zhu BT. 4-Hydroxylation of estradiol by human uterine myometrium and myoma microsomes: implications for the mechanism of uterine tumorigenesis. Proc. Natl. Acad. Sci. USA. 1995;92:9220–9224. doi: 10.1073/pnas.92.20.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo J, Hasan LM, Balogh G, Guo S, Russo IH, Estrogen and its metabolites are carcinogenic agents in human breast epithelial cells. J. Steroid Biochem Mol. Biol. 2003;87:1–25. doi: 10.1016/s0960-0760(03)00390-x. [DOI] [PubMed] [Google Scholar]
- 10.Berg D, Sonsalla R, Kuss E. Concentrations of 2-methoxyoestrogens in human serum measured by a heterologous immunoassay with an 125I-labelled ligand. Acta Endocrinol (Copenh) 1983;103:282–288. doi: 10.1530/acta.0.1030282. [DOI] [PubMed] [Google Scholar]
- 11.Sweeney C, Liu G, Yiannoutsos C, Kolesar J, Horvath D, Staab MJ, et al. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin. Cancer Res. 2005;11:6625–6633. doi: 10.1158/1078-0432.CCR-05-0440. [DOI] [PubMed] [Google Scholar]
- 12.Lakhani NJ, Sarkar MA, Venitz J, Figg WD. 2-Methoxyestradiol, a promising anticancer agent. Pharmacotherapy. 2003;23:165–712. doi: 10.1592/phco.23.2.165.32088. [DOI] [PubMed] [Google Scholar]
- 13.D'Amato RJ, Lin CM, Flynn E, Folkman J, Hamel E. 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicine site. Proc. Natl. Acad. Sci. USA. 1994;91:3964–3968. doi: 10.1073/pnas.91.9.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesanno R, Nawroth PP, Schwelgerer L. The endogenous oestrogen metabolite 2-methoxyestradiol inhibits angiogenesis and suppresses tumor growth. Nature. 1994;368:237–239. doi: 10.1038/368237a0. [DOI] [PubMed] [Google Scholar]
- 15.Golebiewska J, Rozwadowski P, Spodnik JH, Knap N, Wakabayashi T, Wozniak M. Dual effect of 2-methoxyestradiol on cell cycle events in human osteosarcoma 143B cells. Acta Biochim Pol. 2002;49:59–65. [PubMed] [Google Scholar]
- 16.Vijayanathan V, Venkiteswaran S, Nair SK, Verma A, Thomas TJ, Zhu BT, Thomas T. Physiological levels of 2-methoxyestradiol interfere with nongenomic signaling of 17β-Estradiol in human breast cancer cells. Clinical Cancer Res. 2006;12:2038–2048. doi: 10.1158/1078-0432.CCR-05-2172. [DOI] [PubMed] [Google Scholar]
- 17.Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- 18.Liu ZJ, Zhu BT. Concentration-dependent mitogenic and antiproliferative actions of 2-methoxyestradiol in estrogen receptor-positive human breast cancer cells. J. Steroid Biochem Mol. Biol. 2004;88:265–275. doi: 10.1016/j.jsbmb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Thomas T, Balabhadrapathruni S, Gallo MA, Thomas TJ. Development of polyamine analogs as cancer therapeutic agents. Oncol. Res. 2002;13:123–135. [PubMed] [Google Scholar]
- 20.Gerner EW, Meyskens FL., Jr. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4:781–792. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y, Pledgie A, Casero RA, Jr, Davidson NE. Molecular mechanisms of polyamine analogs in cancer cells. Anticancer Drugs. 2005;16:229–241. doi: 10.1097/00001813-200503000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Dolan ME, Fleig MJ, Feuerstein BG, Basu HS, Luk GD, Casero RA, Jr, Marton LJ. Effect of 1,19-bis(ethylamino)-5,10,15-triazanonadecane on human tumor xenografts. Cancer Res. 1994;54:4698–4702. [PubMed] [Google Scholar]
- 23.Canizares F, Salinas J, de las Heras M, Diaz J, Tovar I, Martinez P, Penafiel R. Prognostic value of ornithine decarboxylase and polyamines in human breast cancer: correlation with clinicopathologic parameters. Clin Cancer Res. 1999;5:2035–41. [PubMed] [Google Scholar]
- 24.Thomas T, Thomas TJ. Estradiol control of ornithine decarboxylase mRNA, enzyme activity, and polyamine levels in MCF-7 breast cancer cells: therapeutic implications. Breast Cancer Res. Treat. 1994;29:189–101. doi: 10.1007/BF00665680. [DOI] [PubMed] [Google Scholar]
- 25.Bernacki RJ, Oberman EJ, Seweryniak KE, Atwood A, Bergeron RJ, Porter CW. Preclinical antitumor efficacy of the polyamine analogue N1, N11-diethylnorspermine administered by multiple injection or continuous infusion. Clin. Cancer Res. 1995;1:847–857. [PubMed] [Google Scholar]
- 26.Hahm HA, Ettinger DS, Bowling K, Hoker B, Chen TL, Zabelina Y, Casero RA., Jr Phase I study of N1),N11-diethylnorspermine in patients with non-small cell lung cancer. Clin Cancer Res. 2002;8:684–690. [PubMed] [Google Scholar]
- 27.Wolff AC, Armstrong DK, Fetting JH, Carducci MK, Riley CD, Bender JF, Casero RA, Jr, Davidson NE. A Phase II study of the polyamine analog N1,N11-diethylnorspermine (DENSpm) daily for five days every 21 days in patients with previously treated metastatic breast cancer. Clin. Cancer Res. 2003;9:5922–5928. [PubMed] [Google Scholar]
- 28.Kumar R, Hung MC. Signaling intricacies take center stage in cancer cells. Cancer Res. 2005;65:2511–2515. doi: 10.1158/0008-5472.CAN-05-0189. [DOI] [PubMed] [Google Scholar]
- 29.Thompson JE, Thompson CB. Putting the rap on Akt. J. Clin. Oncol. 2004;22:4217–4226. doi: 10.1200/JCO.2004.01.103. [DOI] [PubMed] [Google Scholar]
- 30.Tokunaga E, Kimura Y, Oki E, Ueda N, Futatsugi M, Mashino K, et al. Akt is frequently activated in HER2/neu-positive breast cancers and associated with poor prognosis among hormone-treated patients. Int. J. Cancer. 2005;118:284–289. doi: 10.1002/ijc.21358. [DOI] [PubMed] [Google Scholar]
- 31.Stal O, Perez-Tenorio G, Akerberg L, Olsson B, Nordenskjold L. Skoog, Rutqvist LE. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. 2003;5:R37–44. doi: 10.1186/bcr569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1:707–717. [PubMed] [Google Scholar]
- 33.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou DS, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor a: a new model for anti-estrogen resistance. J. Biol. Chem. 2001;276:9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 34.Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr. Top. Med. Chem. 2006;6:181–202. [PubMed] [Google Scholar]
- 35.Thomas T, Gallo MA, Thomas TJ. Estrogen receptors as targets for drug development for breast cancer, osteoporosis and cardiovascular diseases. Curr. Cancer Drug Targets. 2004;4:483–99. doi: 10.2174/1568009043332880. [DOI] [PubMed] [Google Scholar]
- 36.Lee AJ, Kosh JW, Conney AH, Zhu BT. Characterization of the NADPH-dependent metabolism of 17β-estradiol to multiple metabolites by human liver microsomes and selectively expressed human cytochrome P450 3A4 and 3A5. J. Pharmacol. Exp. Ther. 2001;298:420–432. [PubMed] [Google Scholar]
- 37.Chou TC. The median-effect principle and combination index for quantitation of synergism and antagonism. In: Chou TC, Rideout DC, editors. Synergism and Antagonism in Chemotherapy. Academic press; San Diego: 1991. pp. 61–102. [Google Scholar]
- 38.Chou TC, Talalay P. Quantitiative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Ad. Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 39.Chou TC, Martin N. A computer program for quantitation of synergism and antagonism in drug combinations, and the determination of IC50 and ED50 and LD50 values. ComboSyn; Paramus, NJ: 2005. CompuSyn for Drug Combinations: PC Software and User's Guide. [Google Scholar]
- 40.Chou TC, Motzer RJ, Tong Y, Bosl GJ. Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: a rational approach to clinical protocol design. J, Natl Cancer Inst. 1994;86:1517–1524. doi: 10.1093/jnci/86.20.1517. [DOI] [PubMed] [Google Scholar]
- 41.Lewis JS, Thomas TJ, Pestell RG, Albanese C, Gallo MA, Thomas T. Differential effects of 16α-hydroxyestrone and 2-methoxyestradiol on cyclin D1 involving the transcription factor ATF-2 in MCF-7 breast cancer cells. J. Mol. Endocrinol. 2005;34:91–105. [Google Scholar]
- 42.Ray G, Dhar G, Van Veldhuizen PJ, Banerjee S, Saxena NK, Sengupta K, Banerjee SK. Modulation of cell-cycle regulatory signaling network by 2-methoxyestradiol in prostate cancer cells is mediated through multiple signal transduction pathways. Biochemistry. 2006;45:3703–3713. doi: 10.1021/bi051570k. [DOI] [PubMed] [Google Scholar]
- 43.Chen B, Pan H, Zhu L, Deng Y, Pollard JW. Progesterone inhibits the estrogen-induced phosphoinositide 3-kinase-->AKT-->GSK-3β-->cyclin D1-->pRB pathway to block uterine epithelial cell proliferation. Mol. Endocrinol. 2005;19:1978–1990. doi: 10.1210/me.2004-0274. [DOI] [PubMed] [Google Scholar]
- 44.Fernando RI, Wimalasena J. Estradiol abrogates apoptosis in breast cancer cells through inactivation of BAD: Ras-dependent nongenomic pathways requiring signaling through ERK and Akt. Mol Biol Cell. 2004;15:3266–3284. doi: 10.1091/mbc.E03-11-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol. Cell Biol. 2003;23:6139–6149. doi: 10.1128/MCB.23.17.6139-6149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolff J. Promotion of microtubule assembly by oligocations: cooperativity between charged groups. Biochemistry. 1998;37:10722–9. doi: 10.1021/bi980400n. [DOI] [PubMed] [Google Scholar]
- 47.Nogales E. Structural insight into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 2001;30:397–420. doi: 10.1146/annurev.biophys.30.1.397. [DOI] [PubMed] [Google Scholar]
- 48.Flusberg DA, Numaguchi Y, Ingber DE. Cooperative control of Akt phosphorylation, bcl-2 expression, and apoptosis by cytoskeletal microfilaments and microtubules in capillary endothelial cells. Mol. Biol. Cell. 2001;12:3087–3094. doi: 10.1091/mbc.12.10.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mollinedo F, Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 2003;8:413–450. doi: 10.1023/a:1025513106330. [DOI] [PubMed] [Google Scholar]
- 50.Blajeski AL, Phan VA, Kottke TJ, Kaufmann SH. G(1) and G(2) cell-cycle arrest following microtubule depolymerization in human breast cancer cells. J. Clin. Invest. 110:91–99. doi: 10.1172/JCI13275. 20020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kramer DL, Vujcic S, Diegelman P, Alderfer J, Miller JT, Black JD, Bergeron RJ, Porter CW. Polyamine analogue induction of the p53-p21WAF1/CIP1-Rb pathway and G1 arrest in human melanoma cells. Cancer Res. 1999;59:1278–1286. [PubMed] [Google Scholar]
- 52.Sun M, Paciga JE, Feldman RI, Yuan Z, Coppola D, Lu YY, Shelley SA, Nicosia SV, Cheng JQ. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res. 2001;61:5985–5991. [PubMed] [Google Scholar]
- 53.Chauhan D, Li G, Auclair D, Hideshima T, Richardson P, Podar K, et al. Identification of genes regulated by 2-methoxyestradiol (2ME2) in multiple myeloma cells using oligonucleotide arrays. Blood. 2003;101:3606–3614. doi: 10.1182/blood-2002-10-3146. N. [DOI] [PubMed] [Google Scholar]
- 54.Pledgie A, Huang Y, Hacker A, Zhang Z, Woster PM, Davidson NE, Casero RA., Jr. Spermine oxidase SMO(PAOh1), Not N1-acetylpolyamine oxidase PAO, is the primary source of cytotoxic H2O2 in polyamine analogue-treated human breast cancer cell lines. J. Biol. Chem. 2005;280:39843–39851. doi: 10.1074/jbc.M508177200. [DOI] [PubMed] [Google Scholar]
- 55.Gao N, Rahmani M, Dent P, Grant S. 2-Methoxyestradiol-induced apoptosis in human leukemia cells proceeds through a reactive oxygen species and Akt-dependent process. Oncogene. 2005;24:3797–3809. doi: 10.1038/sj.onc.1208530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Urata Y, Ihara Y, Murata H, Goto S, Koji T, Yodoi J, Inoue S, Kondo T. 17Beta-estradiol protects against oxidative stress-induced cell death through the glutathione/glutaredoxin-dependent redox regulation of Akt in myocardiac H9c2 cells. J. Biol. Chem. 2006 May;281:13092–13102. doi: 10.1074/jbc.M601984200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.