

Abstract
Ligand binding to the glucocorticoid receptor (GR) results in receptor binding to glucocorticoid response elements (GREs) and the formation of transcriptional regulatory complexes. Equally important, these complexes are continuously disassembled, with active processes driving GR off GREs. We found that cochaperone p23-dependent disruption of GR-driven transcription depended on the ligand binding domain (LBD). Next, we examined the importance of the LBD and of ligand dissociation in GR-GRE dissociation in living cells. We showed in fluorescence recovery after photobleaching studies that dissociation of GR from GREs is faster in the absence of the LBD. Furthermore, GR interaction with a target promoter revealed ligand-specific exchange rates. However, using covalently binding ligands, we demonstrated that ligand dissociation is not required for receptor dissociation from GREs. Overall, these studies showed that activities impinging on the LBD regulate GR exchange with GREs but that the dissociation of GR from GREs is independent from ligand dissociation.
Organisms tune their activity to changing environmental conditions and throughout their development by the synthesis or demolition of the components required to optimize their function under the given circumstances. For an effective response to stimuli, cells need to react fast. Equally as important as responding to the occurrence, cells reverse their response upon a decrease or loss of stimuli. For example, steroid hormones are released into the bloodstream from the adrenal cortex and gonads and transduce their signals by interacting with their cognate intracellular receptors (IRs) in target organs. Hormone binding enables IRs to bind with high affinity to genomic response elements and nucleate the assembly of multiprotein regulatory complexes, resulting in the stimulation or repression of a receptor-specific subset of genes (40). Upon hormone withdrawal, these responses are rapidly reversed (29, 41).
In the absence of hormone, the glucocorticoid receptor (GR), a member of the IR superfamily, is part of a cytoplasmic aporeceptor complex (25). The ligand binding domain (LBD) of GR is associated with a molecular chaperone complex, which includes a dimer of hsp90, immunophilins, and p23. This complex serves to maintain GR in a conformation competent for high-affinity hormone binding (25-27). In vitro studies with purified components suggest that the interaction with cochaperone p23 stabilizes the GR-hsp90 complex and potentiates hormone binding by GR (26). Upon hormone binding, GR undergoes a conformational change (4, 34), translocates into the nucleus, and binds to genomic glucocorticoid response elements (GREs) (40). This conformational change includes alterations in the LBD of GR to create surfaces for interactions with coregulators (4), thus nucleating the assembly of multiprotein regulatory complexes.
The results obtained with photobleaching experiments in living cells suggest that the association of many transcription factors with their binding sites is highly transient. For example, GR and the coregulator glucocorticoid receptor interacting protein (GRIP) interact transiently with a genomic target gene and are actively displaced by energy-dependent processes (2, 20, 36). Several factors have been implicated in the active dissociation of GR from chromatin. These include chaperones p23 and hsp90 (12, 13, 19, 36), the proteasome (36), and SWI/SNF chromatin remodeling complexes (11, 22). Chaperones and SWI/SNF complexes displace receptors from DNA in vitro (11-13, 22). In addition, p23 and hsp90 localize to genomic response elements in a hormone-dependent manner in vivo and disrupt receptor-mediated transcriptional activation in vivo and in vitro (13). Together, these data suggest that chaperones are involved in an active disassembly of transcriptional regulatory complexes.
Similar to the interaction of GR with GREs, ligands interact dynamically with the glucocorticoid receptor in vivo; the synthetic glucocorticoid dexamethasone (Dex) dissociates with a half-time of ∼10 min, whereas the natural hormone cortisol dissociates with half-time of a few minutes (21). In contrast, purified hormone-receptor complexes are stable, consistent with crystallographic studies showing that the ligand is buried within the hydrophobic core of the LBD (4, 38). The rapid release of ligand in vivo suggests that active mechanisms facilitate ligand dissociation.
Ligand binding is an essential step that allows high-affinity binding of GR to GREs (3), and the withdrawal of hormone results in the loss of GR association at high-affinity binding sites (28, 41). However, it has not been determined whether hormone release is a prerequisite for GR release from GREs or for termination of transcriptional responses. Similarly, while GR-GRE dissociation may facilitate rapid responses to environmental cues, the biological function of receptor dynamics remains uncertain.
In this report, we examined the mechanisms controlling receptor dynamics by defining the functional domains that mediate receptor cycling, the potential role of cofactors, and the importance of ligand release for GR-GRE dissociation.
MATERIALS AND METHODS
Plasmids.
Gal4, Gal4-p23, 2×gal4-2×GRE-luciferase (13) (carrying two gal4 binding sites and two GREs), and p6R expression constructs for GR, N525 (amino acids 1 to 525), and 407C (amino acids 407 to 795) (14, 15) have been described elsewhere. Gal4-p23 L99W and gal4-p23 ANNA (F103A/W106A) were generated by site-directed mutagenesis using gal4-p23 as a template. The 2×gal4 2×MMTV-GRE (carrying two copies of the mouse mammary tumor virus GRE) reporter was generated by digesting the 2×gal4-2×GRE-luciferase reporter with PstI and SpeI to replace the two TAT GREs with the following: GGTCGACGAGGTCGTTACAAACTGTTCTGAGGTCGTTACAAACTGTTCTA (MMTV GREs are underlined). Green fluorescent protein (GFP)-GR (amino acids 1 to 795), GFP-407C or GFP-N525 constructs were generated by PCR amplification of rat GRα cDNA using primers that introduced Asp718I and XhoI restriction sites. PCR products were subcloned into the Asp718I-XhoI sites of pEGFP-C1 (BD Biosciences Clontech, Palo Alto, CA) to add an N-terminal GFP tag. Subsequently, GFP-GR, GFP-407C, and GFP-N525 were subcloned into the SmaI-NotI sites of pTRE-tight (Clontech, Palo Alto, CA).
Cell culture and generation of stable GFP-GR, GFP-N525, and GFP-407C lines.
To obtain GFP-GR, GFP-407C, or GFP-N525 cell lines, we transfected the relevant GR construct along with a puromycin resistance plasmid into the Tet-off murine mammary adenocarcinoma cell line 5858 (28). Cells expressing enhanced GFP-rat GR, GFP-407C, or GFP-N525 were selected in medium supplemented with 1.5 μg of puromycin/ml, and cell lines were derived from single-cell clones of GFP-positive cells. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum (FBS), 0.1 mM nonessential amino acids, 2 mM l-glutamine, 1 mM sodium pyruvate, 1 mg of G418/ml, 1.5 μg of puromycin/ml, and 10 μg/ml of tetracycline at 37°C in 5% CO2 in a humidified incubator.
U2OS treatments and transient transfections.
U2OS human osteosarcoma cells were maintained in DMEM (GIBCO) supplemented with 5% FBS. For reporter activity assays, cells were seeded into 24-well plates in DMEM-5% FBS at approximately 20,000 cells per well and transfected the following day in FBS-free DMEM using 0.8 μl of Lipofectamine and 1.6 μl of PLUS reagent (Invitrogen) per well according to manufacturer's instructions. Cells were transfected with indicated amounts of gal4 or gal4-p23 plasmids plus empty plasmid p6R to a total of 100 ng. In addition, transfections included 20 ng of p6R-GR, p6R-N525, or p6R-407C (14, 15) and 20 ng each of the lacZ and 2×gal4-2×GRE-luciferase reporters. After transfection (3 h), cells were refed with DMEM-5% FBS, allowed to recover for 3 h, and refed with DMEM-5% FBS containing 100 nM Dex or ethanol vehicle. Approximately 12 h later, cells were lysed in 100 μl per well of 1× lysis buffer (Pharmingen) and assayed for luciferase and β-galactosidase activity as described previously (17).
FRAP and time-lapse microscopy.
To induce GFP-GR expression for imaging, cells were transferred to Lab-Tek II chambers (Nalge Nunc International) at approximately 40,000 cells per well 2 or 3 days prior to imaging and grown in medium without tetracycline. One day prior to imaging, medium was replaced with phenol red-free DMEM medium with 10% charcoal-stripped FBS. Cells were treated for 45 min at 37°C with Dex (100 nM), RU486 (10 nM), or Dex-21-mesylate (Dex-Mes; 1 μM). Alternatively, cells were treated with 1 μM corticosterone for 6 h in the presence or absence of 10 μM proteasome inhibitor MG-132 before fluorescence recovery after photobleaching (FRAP) analysis. FRAP analysis was carried out using a Zeiss 510 laser scanning confocal microscope with a 100× oil immersion objective (1.3 numerical aperture) and a 40-mW argon laser. The stage temperature was maintained at 37°C with an ASI 400 Air Stream incubator (Nevtek) or a heated stage. Five single-imaging scans were acquired prior to bleaching with a bleach pulse of 160 ms using 458-, 488-, and 514-nm laser lines at 100% laser power (laser output, 75%) without attenuation. Images of single z sections were collected at 0.5-s intervals using a 488-nm laser line with laser power attenuated to 0.2%. Fluorescence intensities in the regions of interest were analyzed, and FRAP recovery curves were generated using laser scanning microscopy software and Microsoft Excel as previously described (8). All of the quantitative data for FRAP recovery kinetics represent means ± standard error of the means (SEM) from at least 10 cells imaged in at least two independent experiments.
RNA FISH and immunofluorescence analysis.
Cells were grown on 22-mm square coverslips deposited at the bottom of a six-well plate; culture conditions were identical to those used for FRAP analysis. Cells were subjected to immunofluorescence analysis and then to RNA fluorescence in situ hybridization (FISH) to detect MMTV transcripts as described previously (28). Following treatment with ligands as described for FRAP analysis, cells were fixed with 2% paraformaldehyde for 20 min at room temperature. Coverslips were washed twice for 5 min with phosphate-buffered saline (PBS), permeabilized with 0.5% Triton X-100, and then incubated with primary antibody for 1 h at room temperature, followed by three washes with PBS. After incubation with secondary antibody for 1 h, coverslips were washed again with PBS and subsequently processed for RNA FISH by fixing with 5% formaldehyde and rinsing with 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). A digoxigenin-11-dUTP-labeled MMTV probe was prepared by using digoxigenin-nick translation mixture (Roche); the probe was denatured and hybridized with coverslips at 37°C overnight in hybridization buffer. After hybridization, coverslips were washed with 2× SSC and 4× SSC and incubated with anti-digoxigenin-rhodamine-conjugated secondary antibody (Roche) to detect the hybridized probe. GFP-GR constructs were detected using rabbit anti-GFP polyclonal antibody A11122 (Molecular Probes). The RNA FISH and immunofluorescence signals were quantified using MetaMorph software (Universal Imaging, Downingtown, PA.) after subtraction of the background nuclear fluorescence.
Quantitative real-time PCR analysis.
Total RNA was isolated from cells by using RNeasy mini kits (QIAGEN). Random-primed cDNA was prepared from 1 μg of total RNA by using a ProtoScript first-strand cDNA synthesis kit (New England Biolabs). One-fiftieth of the resultant cDNA was used per 50-μl reaction mixture containing 1.25 units of Taq DNA polymerase (Promega), 1.5 mM MgCl, a 300 nM concentration of each primer, 0.5 mM deoxynucleoside triphosphate mix, and 0.2× SYBR Green I dye (Molecular Probes) in 1× Taq buffer. Real-time PCR was performed in an Opticon-2 DNA Engine (MJ Research, Cambridge, MA) and analyzed by using the cycle threshold method (1a) and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as an internal control for data normalization. An MMTV cDNA fragment was amplified with the 5′-CGTGAGATTCGGCAGCATAAA-3′ and 5′-GACAGCACACATTTGCAGCTC-3′ primer pair. For normalization, a GAPDH cDNA fragment was amplified with the 5′-ATGGCCTTCCGTGTTCCTAC-3′ and 5′-CCTGCTTCACCACCTTCTTG-3′ primer pair.
Hormone binding assays.
For hormone binding assays, cells were seeded into 24-well plates in medium lacking tetracycline to induce GFP-GR expression for 2 days. One day before hormone binding was assayed, the medium was replaced with phenol red-free DMEM containing 10% charcoal-stripped FBS. Cells were pretreated with either vehicle (ethanol), Dex (1 μM), Dex-Cl (1 μM), or Dex-Mes (1 μM) for 45 min. Next, medium was aspirated, and cells were washed once with hormone-free medium for 1 min, followed by three washes for 5 min each with hormone-free medium. Subsequently, cells were treated for 45 min with 100 nM [3H]Dex in the presence or absence of a 10 μM (100-fold) excess of unlabeled Dex. After five washes with ice-cold PBS, ligand was extracted by adding 250 μl of ethanol for 45 min and quantified by liquid scintillation counting.
Gel Mes-GR.
Cells were grown, pretreated, and washed as described above for hormone binding assays with the following exceptions. The second hormone treatment was performed with 100 nM [3H]Dex-Mes in the presence or absence of excess cold Dex (10 μM), and cells were washed three times instead of five times with ice-cold PBS before cells were lysed in radioimmunoprecipitation assay buffer to prepare whole-cell extracts. Thirty percent of the cell extract coming from 1 well of a 24-well plate was resolved on a sodium dodecyl sulfate-polyacrylamide gel. The gel was fixed for 30 min (65:25:10, H2O:isopropanol:acetic acid), followed by a 30-min incubation with Amplify reagent (Amersham Biosciences). Gels were dried onto Whatman paper using a vacuum blotter and exposed to film at −80°C.
Synthesis and purification of Dex-Cl.
The 21-deoxy-21-chlorodexamethasone (Dex-Cl) was prepared essentially as described previously (7). To a solution of dexamethasone (83 mg) in pyridine (0.89 ml) at 0°C under N2 was added methanesulfonyl chloride (43.8 mg). After overnight stirring at 0°C, the resulting reaction mixture was poured onto ice water (11.1 ml). The resulting precipitate was collected by centrifugation, evaporated in vacuuo, and dissolved in ethylacetate. The ethylacetate solution containing a mixture of Dex-Mes and Dex-Cl (approximately 50:50 by thin-layer chromatography) was fractionated over a silica gel column using a 2:3 hexanes:ethylacetate solvent system to yield pure Dex-Cl. Liquid chromatography-mass spectroscopic data showed the predicated mass for Dex-Cl.
Hormone dissociation assays.
For hormone dissociation assays, cells were grown as described for hormone binding assays and treated for 45 min with 100 nM [3H]Dex plus or minus a 200-fold excess of cold Dex. After hormone binding, a 200-fold excess of unlabeled Dex (20 μM) was added for the time indicated to determine hormone dissociation. Cells were washed three times with ice-cold PBS; the ligand was then extracted by adding 250 μl of ethanol for 45 min and quantified by liquid scintillation counting.
RESULTS
Cochaperone p23-mediated reduction of GR-regulated transcription depended on the GR LBD.
Chaperones perform multiple functions in cells, which hinders genetic analysis of their effects in a particular context of interest. For example, the cochaperone p23 is involved in the folding of numerous cellular proteins (10), thus complicating the analysis of its specific role in dissociation of GR from DNA. To circumvent this problem, we selectively elevated the local concentration of p23 near a GRE-linked reporter gene in cells expressing endogenous levels of the wild-type proteins. Specifically, the reporter contains two Gal4 binding sites linked to two GREs (linked response element [LRE]) (Fig. 1A), and candidate factors to be assessed for GR dissociation activity were constructed as fusions to the Gal4 DNA binding domain (DBD). The LRE reporter was used in mammalian cells to demonstrate a role for p23 and hsp90 in the disassembly of regulatory complexes in vivo (13). Using this artificial “tethering” strategy provides a means to study in vivo the role of p23 in the promoter context specifically. Therefore, we used the LRE reporter to determine the effect of deletions of GR domains on the ability of p23 to disrupt GR-mediated transcription. We chose U2OS human osteosarcoma cells, which lack endogenous GR but support the function of ectopically introduced GR. Thus, these cells can be used to assess the activities of mutant receptors in a GR-null background.
FIG. 1.
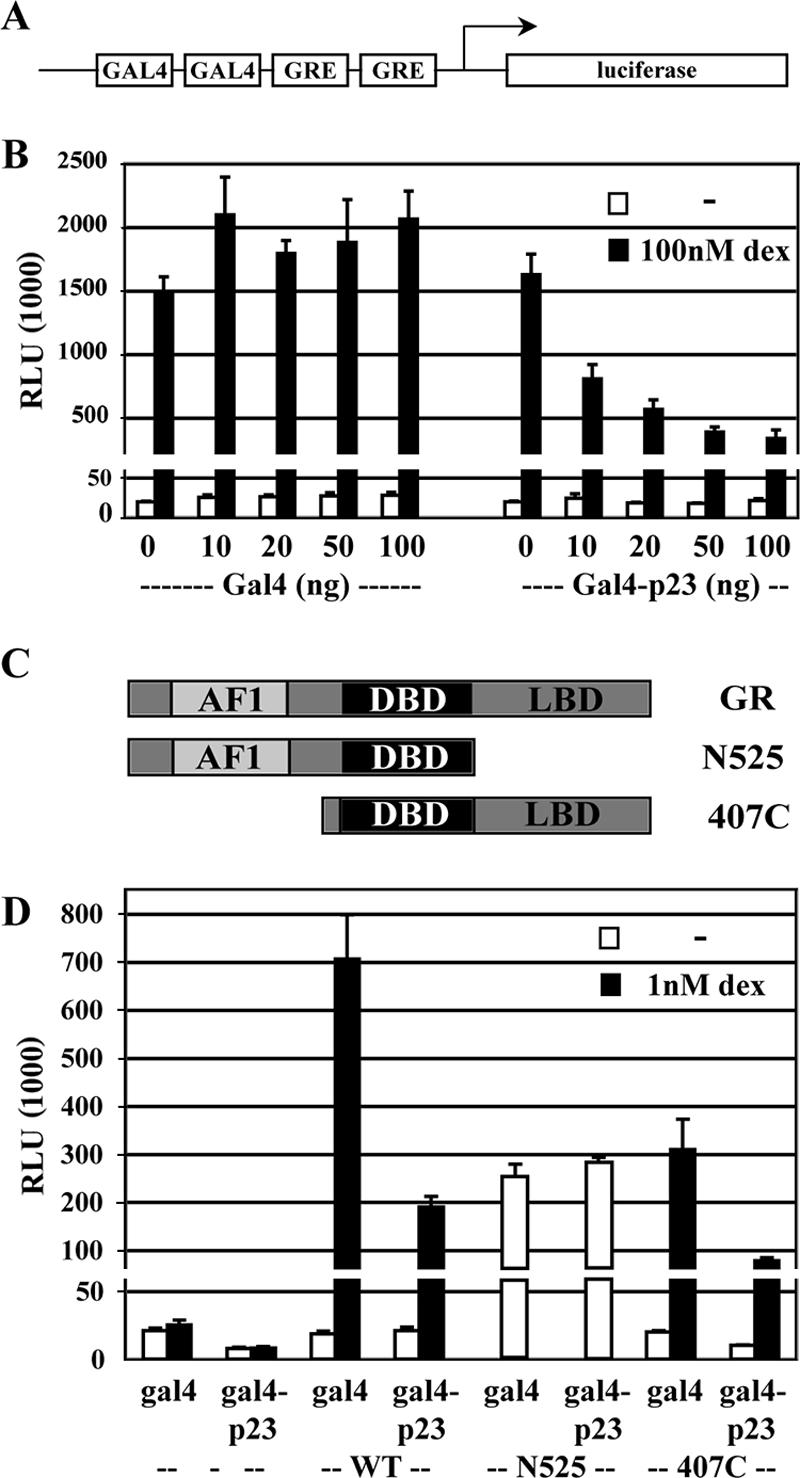
Cochaperone p23-mediated reduction of GR-driven transcription depends on the LBD of GR. (A) U2OS cells were transiently transfected with LRE reporter that consists of two gal4 binding sites and two GREs that drive the expression of a luciferase reporter gene. (B) In addition to the LRE, cells were transfected with an expression construct for GR and increasing amounts of expression constructs for the Gal4 DNA binding domain or Gal4-p23. (C) GR expression constructs used in this study: full-length GR, GR lacking the LBD (N525), and GR lacking the N-terminal activation domain (407C). (D) The effect of p23 on the activity of receptor deletion mutants was determined by transfecting the appropriate receptor or vector control (−) with LRE reporter and 100 ng of Gal4 or Gal4-p23. Cells were treated overnight with Dex (black) or vehicle (white). Reporter activity was measured, normalized to β-galactosidase activity, and expressed as relative luminescence units (RLUs). Averages of three independent experiments ± SEM. are shown. The y axis is broken to visualize reporter activity in the absence of Dex.
First, we tested whether we could reproduce the effects of p23 on GR-dependent transcription in U2OS cells. As reported previously (13), expression of increasing amounts of Gal4-p23 reduced GR-mediated transcriptional activation of the LRE reporter (13), consistent with a role for p23 in GR-GRE dissociation (Fig. 1B) although we cannot rule out the possibility that p23 acts as a corepressor. Next, we mapped the GR domains essential for this activity using GR truncation mutations either missing the amino-terminal 407 amino acids (407C) or the carboxy-terminal LBD (N525) (Fig. 1C). As reported (15, 37), 407C retained hormone-dependent transcriptional activation, whereas the N525 deletion mutant was not hormone regulated (data not shown). We found that transcriptional activation by N525 was unaffected by increased amounts of p23 whereas 407C was as sensitive as full-length GR (Fig. 1D). Interestingly, in vitro DNA binding assays indicate that p23-mediated dissociation of the thyroid hormone receptor, an IR related to GR, from DNA depends on the LBD (12). Taken together, these results indicate that disruption of GR-dependent transcriptional activation by p23 requires the LBD, suggesting that p23 interacts with the LBD to displace GR from GREs.
GFP-GR truncation mutants localize to MMTV array in vivo.
We reasoned that if displacement of GR from GREs by p23 depends on the LBD, the deletion of this domain might affect GR-GRE dissociation dynamics in vivo. To investigate the role of GR domains in GR-GRE dissociation, we generated stable cell lines expressing tetracycline-repressible GFP-GR, GFP-N525, or GFP-407C. We used a murine cell line that contains approximately 200 integrated copies of the MMTV long terminal repeat (MMTV array); each long terminal repeat includes multiple GREs, so the entire MMTV array contains ∼103 GREs (20). The MMTV array enables the direct visualization of GFP-GR binding to the MMTV array by live-cell microscopy; notably, FRAP studies of GR have demonstrated that the receptor exchange is rapid at GREs (20). Western blotting showed that each of the three GFP constructs was expressed and that expression of the constructs was tightly regulated by tetracycline, with higher expression levels for GFP-GR than for endogenous GR (Fig. 2A). In the absence of ligand, GFP-GR was primarily cytoplasmic, with low levels of nuclear GR and low basal levels of MMTV transcription, as determined by quantitative RNA FISH and quantitative real-time PCR (Fig. 2B and C). Upon hormone treatment, GFP-GR and GFP-407C localized to the nucleus, and transcription from the MMTV array was induced (Fig. 2C). Furthermore, ligand treatment resulted in a brighter and larger nuclear spot for GFP-GR and GFP-407C that colocalized with the MMTV RNA FISH signal (Fig. 2C). The GFP-N525 truncation of GR, which lacks the LBD, constitutively localized to the nucleus as previously described (30), and transcription of the MMTV array was induced upon GFP-N525 expression. Nuclear GFP-N525 also colocalized with the MMTV transcripts, demonstrating that GFP-N525 is targeted to the MMTV promoter (Fig. 2C). In summary, we found that the GFP-GR fusions activated transcription and localized to the MMTV array, thus allowing us to study the role of GR domains in GR-GRE dissociation.
FIG. 2.

Analysis of MMTV-array cell lines expressing GFP-GR truncation mutants. (A) Tetracycline-regulated expression of GFP-GR constructs. Total cell extracts from cells grown in the presence or absence of tetracycline were probed by Western blotting for the expression of the GFP-GR constructs with anti-GFP and anti-GR antibodies. Equal loading was verified by anti-actin antibody. (B) Transcriptional activity of GFP-GR-, GFP-407C-, or GFP-N525-expressing cells treated as indicated with either vehicle (−), 100 nM Dex, 1 μM Dex-Mes, or 10 nM RU486 was determined by quantifying MMTV RNA FISH intensity (solid bars) and by quantitative real-time PCR of mRNA (numbers on the graph are mean values ± SEM). (C) GFP-GR, GFP-N525, and GFP-407C localized to the MMTV promoter array as determined by combined RNA FISH and immunofluorescence microscopy.
Role of receptor domains in GFP-GR mobility.
To test the functional role of GR domains on the kinetics of GR-GRE dissociation, we photobleached the three GFP-GR constructs specifically bound at the MMTV array to determine their association and dissociation kinetics (Fig. 3A). Additionally, we examined the mobility of bulk GR by photobleaching GFP-GR residing in the nucleoplasm. In the absence of ligand, the recovery of nucleoplasmic GFP-GR was rapid and complete, with a half-maximal time for fluorescence recovery (t1/2) of 0.43 ± 0.04 s (Fig. 3B and Table 1). Treatment with Dex resulted in a marked decrease in the rate of nucleoplasmic GR recovery (t1/2 of 1.36 ± 0.15 s), suggesting that liganded GR interacts with structures (such as chromatin) that reduce its mobility. The fluorescence recovery of GFP-GR at the MMTV array was slower (t1/2 of 1.60 ± 0.09 s) than the recovery rates observed for nucleoplasmic GFP-GR (Fig. 3B). Next, we determined FRAP rates of GR deletion mutants lacking either the LBD or the amino terminus. We found that deletion of the amino terminus resulted in recovery rates (t1/2 of 1.34 ± 0.10 s) at the array that were slightly faster than for wild-type GFP-GR (Fig. 3C). However, deletion of the LBD resulted in faster recovery rates at the array (t1/2 of 0.69 ± 0.06 s) than those observed for wild-type GR but slower than those of unliganded GR (Fig. 3B and C). Mirroring the results found at the MMTV array, fluorescence recovery rates in the nucleoplasm were fastest for N525 and slower for 407C, with the slowest recovery for wild-type GR (Fig. 3D).
FIG. 3.

Increased GR mobility in the absence of the ligand binding domain. (A) FRAP analysis of GFP-GR in live cells. Single z section images were collected before photobleaching and during recovery at indicated times. Cells were treated with 100 nM Dex for 45 min (array and nucleoplasm) or left untreated (no Dex and nucleoplasmic bleach). White circles indicate bleached areas. (B) Quantitative FRAP analysis of GFP-GR at the array and in the nucleoplasm in cells treated with 100 nM Dex for 30 to 60 min or left untreated (−Dex). Quantitative FRAP analysis at the array (C) or elsewhere in the nucleus (D) for GFP-GR (100 nM Dex), GFP-N525, and GFP-407C (100 nM Dex). All FRAP recovery kinetic data represent averages ± SEM from at least 10 cells imaged in two independent experiments. WT, wild type.
TABLE 1.
Half-times of dissociation for various treatments and glucocorticoid receptor mutants
| Receptor | Treatment | GR localization | t1/2 (s)a |
|---|---|---|---|
| GFP-GR | Nucleoplasm | 0.425 ± 0.040 (n = 15) | |
| GFP-GR | DEX | Nucleoplasm | 1.357 ± 0.153 (n = 11) |
| GFP-GR | DEX | Array | 1.597 ± 0.088 (n = 15) |
| GFP-GR | DEX-mes | Nucleoplasm | 0.917 ± 0.089 (n = 17) |
| GFP-GR | DEX-mes | Array | 1.404 ± 0.100 (n = 19) |
| GFP-GR | RU486 | Nucleoplasm | 0.798 ± 0.069 (n = 11) |
| GFP-GR | RU486 | Array | 0.819 ± 0.064 (n = 17) |
| GFP-GR | DEX-Cl | Nucleoplasm | 0.985 ± 0.089 (n = 15) |
| GFP-GR | Corticosterone | Nucleoplasm | 0.829 ± 0.119 (n = 6) |
| GFP-GR | Corticosterone + MG-132 | Nucleoplasm | 1.207 ± 0.144 (n = 7) |
| GFP-N525 | Nucleoplasm | 0.560 ± 0.035 (n = 12) | |
| GFP-N525 | Array | 0.691 ± 0.058 (n = 24) | |
| GFP-407C | Nucleoplasm | 0.453 ± 0.038 (n = 10) | |
| GFP-407C | DEX | Nucleoplasm | 0.956 ± 0.067 (n = 12) |
| GFP-407C | DEX | 1.340 ± 0.095 (n = 18) |
Values are means ± SEM. n, number of cells assayed.
The FRAP results demonstrated that the LBD and, to a smaller extent, the amino terminus influenced the exchange rates of GR with GREs. The effect of deletion of the amino terminus was small, whereas the deletion of the LBD resulted in faster GR dynamics. The influence of the receptor domains on nucleoplasmic receptor dynamics paralleled those observed at the array, emphasizing the importance of the LBD. These results suggest that the mechanisms controlling receptor mobility at the array and in the rest of the nucleus may be similar.
Ligand-specific GFP-GR mobilities.
Our findings demonstrated the importance of the LBD and of ligand binding on GR mobility. Consistent with the importance of events at the LBD, several studies have demonstrated ligand-specific GR mobilities in the nucleoplasm (31, 32) and at the array using a GR mutant with increased ligand affinity (36). To investigate the effects of specific ligands on wild-type GFP-GR mobility at the array and in the nucleoplasm, we tested the following ligands: Dex, a high affinity agonist; RU486, a high affinity antagonist; and Dex-Mes, which binds covalently to GR. Treatment with each ligand resulted in nuclear translocation of GR and induction of transcription from the MMTV array as demonstrated by indirect immunofluorescence microscopy and quantitative FISH (Fig. 2B and 4A). The levels of transcriptional induction were similar for Dex-Mes and Dex, whereas a significant but lower level of transcription induction was detected with antagonist RU486 (Fig. 2B). When we treated cells with each of these ligands, a subnuclear GFP-GR fluorescence signal colocalized with the MMTV RNA FISH signal, demonstrating the recruitment of GR to the array (Fig. 4A). Next, we tested the effect of the ligands on GR-GRE exchange at the MMTV array. FRAP experiments showed that RU486 treatment resulted in faster fluorescence recovery at the array (t1/2 of 0.82 ± 0.06 s) than the recovery observed with Dex (Fig. 4B). Dex-Mes treatment induced MMTV transcription at levels comparable to Dex and resulted in GR dynamics similar to those for Dex (t1/2 of 1.40 ± 0.10 s for Dex-Mes and 1.60 ± 0.09 s for Dex) (Fig. 4B). Nucleoplasmic GR mobility was faster for RU486 and Dex-Mes than for Dex (Fig. 4C), while GR-GRE dissociation was fastest for RU486 and displayed comparable rates for Dex-Mes and Dex. In summary, we found that the mobility of GR is affected by the nature of the ligand bound to GR. This further supports the importance of the LBD in regulating GR-GRE dissociation.
FIG. 4.

Ligand-specific GR mobility. (A) Immunofluorescence (middle) and RNA FISH (top) of GFP-GR-expressing cells untreated (−) or treated with 100 nM Dex, 1 μM Dex-Mes, or 10 nM RU486; an overlay of the FISH and GFP-GR signals is shown at the bottom. Quantitative FRAP analysis at the array (B) or in the nucleoplasm (C) for cells treated as indicated for panel A. All quantitative data values in FRAP recovery kinetics represent averages ± SEM. from at least 10 cells imaged in two independent experiments. We chose saturating ligand concentrations for Dex and Dex-Mes because the slowest receptor dynamics are typically observed at saturating ligand concentrations (31). For RU486 we used ligand concentrations that result in maximal slow-down of nucleoplasmic receptor dynamics (31). WT, wild type.
GR dissociation from GREs is independent of ligand dissociation.
Ligand binding increases the affinity of GR for GREs and is accompanied by a decrease in receptor mobility. Conversely, hormone withdrawal results in the cessation of activation and loss of receptors associated with GREs. To test if hormone dissociation is required for GR-GRE dissociation, we used Dex-Mes, which covalently binds GR. Dex-Mes is a mesylate derivative of Dex that forms a thioether bond with cysteine 656 in the LBD of GR (33). To determine the efficiency of covalent binding by Dex-Mes, we preincubated cells for 45 min with 1 μM Dex-Mes, ethanol, or 1 μM Dex and subsequently determined the proportion of receptors available to bind [3H]Dex in a whole-cell binding assay. We found that after Dex treatment, approximately 70% of the receptor lost its ligand during the washes and was available for subsequent [3H]Dex binding (Fig. 5A). In contrast to Dex, preincubation with 1 μM Dex-Mes prevented >99% of GR from subsequent [3H]Dex binding (Fig. 5A). In parallel experiments, we found that preincubation with Dex-Mes blocked covalent receptor binding with [3H]Dex-Mes, whereas binding with Dex allowed subsequent [3H]Dex-Mes binding by GR after hormone wash-out as assayed by fluorography (Fig. 5B).
FIG. 5.

Covalent binding of Dex-Mes and Dex-Cl to GR. (A) The number of bound ligand molecules per cell was determined after pretreatment for 45 min with vehicle, 1 μM Dex, 1 μM Dex-Mes, or 1 μM Dex-Cl, followed by three washes for 5 min each with ligand-free medium and subsequent treatment with 100 nM [3H]Dex in the presence or absence of a 100-fold excess of cold Dex. Specific activity and the number of cells per well were used to calculate the number of bound [3H]Dex molecules per cell. Specific binding (below chart) was determined by subtracting nonspecific binding (open bars) in the presence of excess unlabeled Dex from total binding in the presence of 100 nM [3H]Dex (black bars). (B) Fluorograph of covalently labeled GR by [3H]Dex-Mes. Cells were pretreated and washed as described for panel A, followed by a 45-min treatment with 100 nM [3H]Dex-Mes. Protein extracts from whole cells were chromatographed on sodium dodecyl sulfate-polyacrylamide gels and analyzed by fluorography. (C) Synthesis of Dex-Cl (right) and Dex-Mes (middle) from Dex (left). (D) Localization of GFP-GR of untreated cells (top) or cells treated for 45 min with 1 μM Dex-Cl (bottom). (E) Transcriptional activation from the MMTV array is shown as relative mRNA abundance ± SEM for untreated and treated cells as determined by quantitative PCR following a 2-h treatment with 1 μM Dex-Cl.
If receptor dissociation from DNA required ligand dissociation, we would predict that covalent ligands would render chromatin-bound GR immobile. By in vivo FRAP microscopy, however, no immobile receptor population was found for Dex-Mes, either at the array or in the nucleoplasm (Fig. 4B and C and 6A); in fact, the recovery was faster than the recovery observed for Dex. As a control, we treated the cells with proteasome inhibitor MG-132 and corticosterone, since proteasome activity is required for nuclear mobility of a fraction of GR (6, 31, 36). As expected, proteasome inhibition resulted in an immobile receptor population (approximately 20%) (Fig. 6A and B). Together, these results suggest that GR mobility is independent of ligand exchange.
FIG. 6.

GFP-GR mobility independent of ligand dissociation. (A) Single z section images were collected before photobleaching (Prebleach), directly after (Bleach), or 15 s after photobleaching. Cells were treated with 1 μM Dex-Mes for 45 min (top) or with a 10 μM concentration of the proteasome inhibitor MG-132 for 6 h, followed by a 1-h treatment with corticosterone (bottom). White circles indicate the photobleached area. (B) Quantitative FRAP analysis for nuclear GFP-GR of cells grown for 6 h in the presence or absence of 10 μM MG-132 following a 1-h treatment with 1 μM corticosterone or for cells treated for 45 min with Dex-Cl (1 μM). (C) Hormone dissociation was determined by adding a 200-fold excess of unlabeled Dex for various times to cells treated with 100 nM [3H]Dex for 45 min. Specific binding was determined by subtracting nonspecific binding in the presence of excess unlabeled Dex from total binding in the presence of 100 nM [3H]Dex. The count at 0 min after adding excess unlabeled Dex was set at 100 (0% dissociation); 100% dissociation indicates a complete loss of specific binding. WT, wild type.
To further test this hypothesis, we synthesized a second ligand, predicted to bind GR covalently, Dex-Cl (7). We reasoned that Dex-Cl would form a thioether bond with cysteine 656 of GR; however, Dex-Cl has not been tested for covalent receptor binding or for its function as a GR ligand. We synthesized and purified Dex-Cl (Fig. 5C) as described previously (7) and tested if Dex-Cl could serve as a GR ligand. Indeed, GFP-GR localized to the nucleus of cells treated with Dex-Cl (Fig. 5D) even at low nanomolar concentrations (data not shown), and Dex-Cl treatment resulted in the activation of MMTV transcription (Fig. 5E). Moreover, Dex-Cl covalently bound GR, albeit less efficiently than Dex-Mes (∼80% for Dex-Cl versus >99% for Dex-Mes) (Fig. 5A and B). The binding, nuclear translocation, and transcriptional activation all indicated that Dex-Cl is a bona fide GR ligand. Next, we tested GR mobility for Dex-Cl-bound GR by FRAP analysis. Similar to our findings with Dex-Mes, treatment with Dex-Cl did not render a fraction of nucleoplasmic GR immobile (Fig. 6B). These results are consistent with the idea that GR mobility is independent of ligand dissociation.
To compare the kinetics of GR-GRE dissociation with those of GR-Dex dissociation, we determined the half-time of ligand dissociation in the cell line we used for in vivo FRAP studies. Cells expressing GFP-GR were treated for 45 min with [3H]Dex followed by addition of a 200-fold excess of unlabeled Dex; we monitored ligand dissociation as the release of specifically bound [3H]Dex from the intact cells. We found that GR-Dex dissociation occurs at a rate that is much slower (t1/2 of ∼20 min) than the rate of receptor dissociation from GREs (<2 s, >600-fold slower) (compare Fig. 3A and 6C). We conclude that ligand exchange is not required for either GR dissociation from GREs or nucleoplasmic GR mobility.
DISCUSSION
Role of chaperones and receptor domains in GR dynamics.
Nuclear proteins including steroid hormone receptors move rapidly and exchange quickly with multiple target sites in the nucleus (16, 24). Molecular chaperones in the cytoplasm facilitate high-affinity ligand-binding by GR and also function in the nucleus as nuclear mobility factors for steroid hormone receptors (9). The folding of GR is mediated through interactions of the chaperone complex with the LBD; however, the exact interaction surface of the LBD is unknown. The findings described here show that disruption of GR-mediated transcription by cochaperone p23 depended on the presence of the LBD of the receptor. These findings are in agreement with the observation (12) that the effects of p23 on GR activities in vivo are mediated by the LBD and with in vitro studies demonstrating that thyroid receptor displacement from DNA depend on the LBD (12). Although we do not understand mechanistically how p23 displaces the receptor, we suggest that perhaps the chaperones induce a conformational change when interacting with liganded receptor, analogous to the conformational change that facilitates ligand binding by GR.
In a simple scenario, p23 displaces GR from GREs through its interaction with the LBD, and deletion of the LBD would result in slower exchange of the receptor at a target promoter. However, when we tested GR mobility at the MMTV array, exchange rates for GR lacking the LBD were faster. This suggests that the effect of removal of the LBD on receptor mobility is complex. For example, the LBD may contribute to the intrinsic affinity of the full-length receptor for DNA (5), although studies with purified receptor suggest that the intrinsic affinity of the DBD of GR is comparable to that of full-length receptor (15). Furthermore, studies in vitro and in vivo have shown that the destabilizing effect of p23 can be countered by receptor binding to GRIP (12, 13), a transcriptional coregulatory factor that interacts with the GR LBD. Similarly, the interaction of GR with HMGB1 decreases the mobility of GR (1). In support of the notion that specific interactions at the LBD are important for GR mobility, several point mutations that affect the interaction of GR with cofactors result in increased GR mobilities (18). Moreover, the proteasome contributes to increased GR mobility and, similar to our findings for p23, the effects of the proteasome depend on the LBD (18). Hence, removal of the LBD affects several interactions, some destabilizing GR-GRE association (e.g., p23 and the proteasome) and some stabilizing the association (e.g., GRIP). Similar to what has been proposed based on nucleoplasmic GR mobility (18), we propose that the balance of stabilizing and destabilizing factors serves as a key determinant of the residency time of receptor on DNA, and removal of the LBD appears to shift this balance in favor of dissociation of GR from DNA.
Furthermore, the role of chaperones in GR mobility might be more complex. For example, hsp90 can displace receptor from DNA in vitro, but inhibition of hsp90 activities in vivo could result in either faster GR mobility (36) or slower mobility (C. Elbi and G. Hager, unpublished data), depending on the geldanamycin concentration used to inhibit hsp90. Although p23 and hsp90 appear to act in concert in vivo, data with purified components have demonstrated that p23 can displace nuclear receptors from DNA in the absence of hsp90 (13). We found in the LRE assay that p23 mutations that disrupt its interaction with hsp90 (23, 39) did not completely disrupt p23-dependent reduction of transcriptional activation by GR (data not shown) in agreement with a role for p23 independent of hsp90.
In conclusion, we found that large truncations of GR differentially affected receptor mobility. Future experiments with more subtle GR mutants that disrupt specific interactions and influence receptor mobility might help to dissect the contributions of individual factors to receptor dynamics on chromatin.
Ligand binding and GR dynamics.
The data presented confirm findings by others (31, 36) that ligand binding results in reduced GR mobility, emphasizing the importance of the LBD in stabilizing the interaction of GR with chromatin. We found ligand-specific mobilities for GR at the array and in the nucleoplasm. Although different, the GR mobilities in the nucleoplasm paralleled those observed at the array for each of the ligands, suggesting that similar mechanisms control mobility at the array and in the nucleoplasm. One simple model is that receptor mobilities in the nucleoplasm and at the array reflect nonspecific and specific DNA binding, respectively.
Ligand binding is an essential step for high-affinity binding of GR to GREs. However, it was previously unclear whether hormone release is a prerequisite for GR release from GREs. An alternative mechanism for receptor clearance from DNA is proteasomal degradation of chromatin-bound receptor. However, we do not favor proteasomal GR degradation as the primary explanation for receptor clearance as [3H]Dex-Mes-bound GR is stable in cells throughout a 1-h washout period (data not shown), demonstrating that GR protein turnover is slow relative to the many rounds of receptor-DNA binding. Our data with covalently bound ligands suggest that receptor release from chromatin is independent of ligand dissociation.
In agreement with the finding that receptor clearance is independent of ligand dissociation, Freeman and Yamamoto (12) found no indication for ligand loss during receptor dissociation in vitro. Similarly, SWI/SNF can displace covalently ligand-bound GR from chromatin in vitro (11). Finally, receptor dynamics and ligand dissociation appear to occur on vastly different time scales; GR is cleared from chromatin with half-times of only several seconds whereas the half-time for Dex dissociation was ∼20 min, approximately 600 times slower than receptor exchange. Therefore, we propose that ligand dissociation is not essential for either nucleoplasmic mobility or for receptor dynamics at GREs.
If receptor clearance from DNA is independent of ligand release, what is the function of dynamic ligand association? It was shown previously that GR covalently bound to ligand displays reduced transcriptional activity (35), suggesting that ligand exchange may be important for transcriptional activation by GR. However, in the cell line used in our studies, Dex-Mes induces transcription as well as Dex. Thus, transcriptional activation does not rely strictly on ligand exchange, although we cannot exclude a role for ligand exchange in the regulation of some genes. Perhaps the most obvious function for ligand exchange is to respond to changes in hormone levels. Indeed, serum levels of circulating glucocorticoids fluctuate, e.g., in response to food intake, about three- to fivefold over 2- to 4-h periods throughout the day. Local availability of biologically active hormone at target organs may show even greater and more rapid fluctuations, for example, by local conversion of cortisol to biologically inactive cortisone by type 2 11β-hydroxysteroid dehydrogenase. Ligand exchange would provide a means to respond to these fluctuations.
Rapid receptor exchange function.
The biological function of the dynamic interaction of nuclear receptors with chromatin remains unclear. Notably, ligand binding enhances the affinity of GR for many binding sites, and, with limited receptor numbers, increased nucleoplasmic mobility would facilitate scanning the genome rapidly for high-affinity binding sites at which GR interacts productively. In addition to scanning the chromatin for binding sites, a dynamic interaction at a functional GRE might serve other functions. Effective transcriptional regulation involves multiple steps, including chromatin modification, remodeling, recruitment of the basal transcription machinery, and initiation and elongation of transcription. These processes must occur in a temporally organized manner. For example, recruitment of the basal transcription machinery typically requires prior chromatin remodeling and modification events to poise a promoter for transcriptional initiation. Highly dynamic receptor binding could provide a means to allow events like chromatin modifications, remodeling, and recruitment of the basal transcription machinery to take place in a specific order. Supporting the significance of nuclear mobility in transcriptional regulation, GR bound to ligands with low transcriptional activities is more mobile than GR bound to ligands with high activities (18). Furthermore, receptor mutants with decreased transcriptional activity are more mobile than wild-type receptors (18). Finally, the transient interaction of GR with chromatin may serve to rapidly respond to environmental changes.
In conclusion, our data establish the importance of the LBD of GR in the exchange of the receptor with DNA. We propose that the LBD functions as a “switch” that influences chromatin binding and dissociation by GR. However, the function of this switch does not require the release of ligand for GR release from the GRE. With multiple factors impinging on the LBD, some stabilizing and some destabilizing the interaction of GR with chromatin, the balance of these factors influences the dynamics of the GR-chromatin interaction.
Acknowledgments
We thank Marc Diamond, Abigail Kroch, Fred Schaufele, and Alex So for reviewing the manuscript and Samantha Cooper for computational assistance. Imaging was carried out in the Fluorescence Imaging Facility, Laboratory of Receptor Biology and Gene Expression, National Cancer Institute.
S.H.M. is supported by a postdoctoral fellowship from the Leukemia and Lymphoma Society. This work is supported by grants from the NIH to K.R.Y. and in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Published ahead of print on 29 January 2007.
REFERENCES
- 1.Agresti, A., P. Scaffidi, A. Riva, V. R. Caiolfa, and M. E. Bianchi. 2005. GR and HMGB1 interact only within chromatin and influence each other's residence time. Mol. Cell 18:109-121. [DOI] [PubMed] [Google Scholar]
- 1a.Applied Biosystems. 1997. ABI 7700 user's bulletin no. 2. Applied Biosystems, Foster City, CA.
- 2.Becker, M., C. Baumann, S. John, D. A. Walker, M. Vigneron, J. G. McNally, and G. L. Hager. 2002. Dynamic behavior of transcription factors on a natural promoter in living cells. EMBO Rep. 3:1188-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker, P. B., B. Gloss, W. Schmid, U. Strahle, and G. Schutz. 1986. In vivo protein-DNA interactions in a glucocorticoid response element require the presence of the hormone. Nature 324:686-688. [DOI] [PubMed] [Google Scholar]
- 4.Bledsoe, R. K., V. G. Montana, T. B. Stanley, C. J. Delves, C. J. Apolito, D. D. McKee, T. G. Consler, D. J. Parks, E. L. Stewart, T. M. Willson, M. H. Lambert, J. T. Moore, K. H. Pearce, and H. E. Xu. 2002. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110:93-105. [DOI] [PubMed] [Google Scholar]
- 5.Dahlman-Wright, K., A. P. Wright, and J. A. Gustafsson. 1992. Determinants of high-affinity DNA binding by the glucocorticoid receptor: evaluation of receptor domains outside the DNA-binding domain. Biochemistry 31:9040-9044. [DOI] [PubMed] [Google Scholar]
- 6.Deroo, B. J., C. Rentsch, S. Sampath, J. Young, D. B. DeFranco, and T. K. Archer. 2002. Proteasomal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol. Cell. Biol. 22:4113-4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunkerton, L. V., F. S. Markland, and M. P. Li. 1982. Affinity-labelling corticoids. I. Synthesis of 21-chloroprogesterone, deoxycorticosterone 21-(1-imidazole) carboxylate, 21-deoxy-21-chloro dexamethasone, and dexamethasone 21-mesylate, 21-bromoacetate, and 21-iodoacetate. Steroids 39:1-6. [DOI] [PubMed] [Google Scholar]
- 8.Elbi, C., D. A. Walker, M. Lewis, G. Romero, W. P. Sullivan, D. O. Toft, G. L. Hager, and D. B. DeFranco. 2004. A novel in situ assay for the identification and characterization of soluble nuclear mobility factors. Sci. STKE 2004:pl10. [DOI] [PubMed] [Google Scholar]
- 9.Elbi, C., D. A. Walker, G. Romero, W. P. Sullivan, D. O. Toft, G. L. Hager, and D. B. DeFranco. 2004. Molecular chaperones function as steroid receptor nuclear mobility factors. Proc. Natl. Acad. Sci. USA 101:2876-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Felts, S. J., and D. O. Toft. 2003. p23, a simple protein with complex activities. Cell Stress Chaperones 8:108-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fletcher, T. M., N. Xiao, G. Mautino, C. T. Baumann, R. Wolford, B. S. Warren, and G. L. Hager. 2002. ATP-dependent mobilization of the glucocorticoid receptor during chromatin remodeling. Mol. Cell. Biol. 22:3255-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman, B. C., S. J. Felts, D. O. Toft, and K. R. Yamamoto. 2000. The p23 molecular chaperones act at a late step in intracellular receptor action to differentially affect ligand efficacies. Genes Dev. 14:422-434. [PMC free article] [PubMed] [Google Scholar]
- 13.Freeman, B. C., and K. R. Yamamoto. 2002. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296:2232-2235. [DOI] [PubMed] [Google Scholar]
- 14.Godowski, P. J., D. Picard, and K. R. Yamamoto. 1988. Signal transduction and transcriptional regulation by glucocorticoid receptor-LexA fusion proteins. Science 241:812-816. [DOI] [PubMed] [Google Scholar]
- 15.Godowski, P. J., S. Rusconi, R. Miesfeld, and K. R. Yamamoto. 1987. Glucocorticoid receptor mutants that are constitutive activators of transcriptional enhancement. Nature 325:365-368. [DOI] [PubMed] [Google Scholar]
- 16.Hager, G. L., C. Elbi, and M. Becker. 2002. Protein dynamics in the nuclear compartment. Curr. Opin. Genet. Dev. 12:137-141. [DOI] [PubMed] [Google Scholar]
- 17.Iniguez-Lluhi, J. A., D. Y. Lou, and K. R. Yamamoto. 1997. Three amino acid substitutions selectively disrupt the activation but not the repression function of the glucocorticoid receptor N terminus. J. Biol. Chem. 272:4149-4156. [DOI] [PubMed] [Google Scholar]
- 18.Kino, T., S. H. Liou, E. Charmandari, and G. P. Chrousos. 2004. Glucocorticoid receptor mutants demonstrate increased motility inside the nucleus of living cells: time of fluorescence recovery after photobleaching (FRAP) is an integrated measure of receptor function. Mol. Med. 10:80-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, J., and D. B. DeFranco. 1999. Chromatin recycling of glucocorticoid receptors: implications for multiple roles of heat shock protein 90. Mol. Endocrinol. 13:355-365. [DOI] [PubMed] [Google Scholar]
- 20.McNally, J. G., W. G. Muller, D. Walker, R. Wolford, and G. L. Hager. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287:1262-1265. [DOI] [PubMed] [Google Scholar]
- 21.Munck, A., and R. Foley. 1976. Kinetics of glucocorticoid-receptor complexes in rat thymus cells. J. Steroid Biochem. 7:1117-1122. [DOI] [PubMed] [Google Scholar]
- 22.Nagaich, A. K., D. A. Walker, R. Wolford, and G. L. Hager. 2004. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol. Cell 14:163-174. [DOI] [PubMed] [Google Scholar]
- 23.Oxelmark, E., R. Knoblauch, S. Arnal, L. F. Su, M. Schapira, and M. J. Garabedian. 2003. Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J. Biol. Chem. 278:36547-36555. [DOI] [PubMed] [Google Scholar]
- 24.Phair, R. D., P. Scaffidi, C. Elbi, J. Vecerova, A. Dey, K. Ozato, D. T. Brown, G. Hager, M. Bustin, and T. Misteli. 2004. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 24:6393-6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pratt, W. B. 1993. The role of heat shock proteins in regulating the function, folding, and trafficking of the glucocorticoid receptor. J. Biol. Chem. 268:21455-21458. [PubMed] [Google Scholar]
- 26.Pratt, W. B., and D. O. Toft. 2003. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. 228:111-133. [DOI] [PubMed] [Google Scholar]
- 27.Pratt, W. B., and D. O. Toft. 1997. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18:306-360. [DOI] [PubMed] [Google Scholar]
- 28.Rayasam, G. V., C. Elbi, D. A. Walker, R. Wolford, T. M. Fletcher, D. P. Edwards, and G. L. Hager. 2005. Ligand-specific dynamics of the progesterone receptor in living cells and during chromatin remodeling in vitro. Mol. Cell. Biol. 25:2406-2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reik, A., G. Schutz, and A. F. Stewart. 1991. Glucocorticoids are required for establishment and maintenance of an alteration in chromatin structure: induction leads to a reversible disruption of nucleosomes over an enhancer. EMBO J. 10:2569-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savory, J. G., B. Hsu, I. R. Laquian, W. Giffin, T. Reich, R. J. Hache, and Y. A. Lefebvre. 1999. Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol. Cell. Biol. 19:1025-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaaf, M. J., and J. A. Cidlowski. 2003. Molecular determinants of glucocorticoid receptor mobility in living cells: the importance of ligand affinity. Mol. Cell. Biol. 23:1922-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaaf, M. J., L. J. Lewis-Tuffin, and J. A. Cidlowski. 2005. Ligand-selective targeting of the glucocorticoid receptor to nuclear subdomains is associated with decreased receptor mobility. Mol. Endocrinol. 19:1501-1515. [DOI] [PubMed] [Google Scholar]
- 33.Simons, S. S., Jr., J. G. Pumphrey, S. Rudikoff, and H. J. Eisen. 1987. Identification of cysteine 656 as the amino acid of hepatoma tissue culture cell glucocorticoid receptors that is covalently labeled by dexamethasone 21-mesylate. J. Biol. Chem. 262:9676-9680. [PubMed] [Google Scholar]
- 34.Simons, S. S., Jr., F. D. Sistare, and P. K. Chakraborti. 1989. Steroid binding activity is retained in a 16-kDa fragment of the steroid binding domain of rat glucocorticoid receptors. J. Biol. Chem. 264:14493-14497. [PubMed] [Google Scholar]
- 35.Simons, S. S., Jr., and E. B. Thompson. 1981. Dexamethasone 21-mesylate: an affinity label of glucocorticoid receptors from rat hepatoma tissue culture cells. Proc. Natl. Acad. Sci. USA 78:3541-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stavreva, D. A., W. G. Muller, G. L. Hager, C. L. Smith, and J. G. McNally. 2004. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol. Cell. Biol. 24:2682-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szapary, D., M. Xu, and S. S. Simons, Jr. 1996. Induction properties of a transiently transfected glucocorticoid-responsive gene vary with glucocorticoid receptor concentration. J. Biol. Chem. 271:30576-30582. [DOI] [PubMed] [Google Scholar]
- 38.Williams, S. P., and P. B. Sigler. 1998. Atomic structure of progesterone complexed with its receptor. Nature 393:392-396. [DOI] [PubMed] [Google Scholar]
- 39.Wochnik, G. M., J. C. Young, U. Schmidt, F. Holsboer, F. U. Hartl, and T. Rein. 2004. Inhibition of GR-mediated transcription by p23 requires interaction with Hsp90. FEBS Lett. 560:35-38. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto, K. R. 1985. Steroid receptor regulated transcription of specific genes and gene networks. Annu. Rev. Genet. 19:209-252. [DOI] [PubMed] [Google Scholar]
- 41.Zaret, K. S., and K. R. Yamamoto. 1984. Reversible and persistent changes in chromatin structure accompany activation of a glucocorticoid-dependent enhancer element. Cell 38:29-38. [DOI] [PubMed] [Google Scholar]